

Abstract
Naturally occurring membranolytic antimicrobial peptides (AMPs) are rarely cell-type selective and highly potent at the same time. Template-based peptide design can be used to generate AMPs with improved properties de novo. Following this approach, 18 linear peptides were obtained by computationally morphing the natural AMP Aurein 2.2d2 GLFDIVKKVVGALG into the synthetic model AMP KLLKLLKKLLKLLK. Eleven of the 18 chimeric designs inhibited the growth of Staphylococcus aureus, and six peptides were tested and found to be active against one resistant pathogenic strain or more. One of the peptides was broadly active against bacterial and fungal pathogens without exhibiting toxicity to certain human cell lines. Solution nuclear magnetic resonance and molecular dynamics simulation suggested an oblique-oriented membrane insertion mechanism of this helical de novo peptide. Temperature-resolved circular dichroism spectroscopy pointed to conformational flexibility as an essential feature of cell-type selective AMPs.
The global increase in the prevalence of resistant bacteria is a threat to modern society, and the discovery of novel antibiotics that have not yet become susceptible to resistance is an urgent need.1 Membranolytic antimicrobial peptides (AMPs) could be a partial alternative to the current target-specific antibiotic agents.2 A large variety of AMPs from natural sources have shown promising results as potential antibiotics with a direct mode of action on prokaryotic cell membranes as well as on mammalian cancer cells.3,4 Their direct interaction with membrane structures without the need for target proteins or receptors makes them less susceptible to a rapid onset of bacterial resistance and renders them potentially active against resistant strains.5 More than 3000 naturally occurring AMPs have been described and compiled in databases.6−8 These sequence collections offer a basis for template-based design and statistical analysis to develop novel AMPs.9 Several studies have revealed steep sequence–activity relationships between highly active and completely inactive peptides, with the possibility of a single-amino acid substitution abrogating antimicrobial activity (Table S1).10−12
Natural AMPs adopt a broad range of secondary structures and folds.2 We hereafter focus on short cationic α-helical AMPs from the Anura order, namely, the Aurein and Citropin families.13,14 Amino acid substitutions in an Aurein AMP confirmed that subtle changes in the peptide sequence can have a profound impact on the peptide’s activity.15 These antimicrobial activity changes correlated with a difference in structural flexibility between the studied peptides, specifically in their terminal portions. Similarly, a conformationally restricted analogue of an influenza virus fusion protein was less potent in promoting lipid mixing, a surrogate for the ability to lyse bacterial membranes. The flexibility of the fusion peptide was essential for the peptide’s ability to destabilize the host membrane and promote transfection.16 In a related study, a diarylethene moiety was incorporated into the backbone of membrane-interacting peptides.17 By reversible isomerization of the linker, rigid and flexible peptide conformations were induced, and differences in their interactions with lipid membranes could be studied by biophysical methods. These previous studies suggest that the conformational degrees of freedom (“flexibility”) of the amino acid residues may critically affect the activity and mechanism of action.
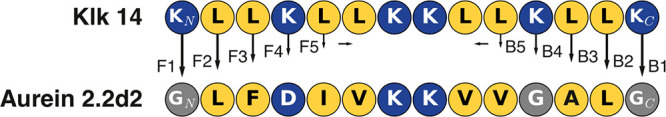
To gain deeper insight into the relationship between the amino acid sequence and the antimicrobial activity of AMPs, we performed a detailed analysis of the influence of sequence variation on the antimicrobial activity of an amphipathic α-helical semisynthetic AMP from the Aurein peptide family.15 On the basis of published solution nuclear magnetic resonance (NMR) structures of Aureins, one can assume an inducible overall α-helical secondary structure of many, if not all, members of the Aurein family. As not all Aureins are active AMPs, the question of whether a peptide’s amino acid sequence pattern or slight changes in secondary structure are causative factors for antimicrobial activity arises. To investigate which of these features accounts for antimicrobial or cytotoxic activity, we performed sequence “morphing” with the aim of conserving an overall helical peptide structure while modifying the amino acid sequence, changing one residue at a time. As starting points, we selected two template peptides, both encompassing 14 residues: the C-terminally shortened Aurein 2.2 [Au2.2d2, sequence of GLFDIVKKVVGALG; Protein Data Bank (PDB) entry 5mxl]15,18 and the synthetic model AMP Klk14 (sequence of KLLKLLKKLLKLLK).19 These cationic peptides were selected as they both can form amphipathic helices with a central twin-lysine motif (Figure 1) and possess a common pharmacophore sequence pattern (PHHPHHPPHHPHHP, where P is a polar residue and H is a hydrophobic residue). By sequence morphing in both directions (from the N- to C-terminus and from the C- to N-terminus), we generated 18 chimeras of Au2.2d2 and Klk14 peptides. A schematic of the morphing approach is shown in Figure 1.
Figure 1.

Schematic representation of the morphing process. (a) Idealized helical structures [constructed with the “fab” command in PyMol (http://www.pymol.org)] for both templates. (b) Helical wheel plots of both template sequences showing amphipathicity quantified as hydrophobic moments [values and arrows, plotted with modlAMP (http://modlamp.org)].26 As both helices share the same orientation of their hydrophobic faces, sequential changes should not affect the overall amphipathicity. (c) Stepwise residue replacement between the two template sequences in forward (N- to C-terminus, denoted F) and backward (C- to N-terminus, denoted B) directions. Hydrophobic residues are colored yellow, and polar ones blue.
Template-based design approaches have already been pursued in AMP research. For example, Han et al. optimized the cationic helical AMP myxinidin by substituting selected residues with Arg, Lys, or Trp and thereby increased activity and selectivity.20 Similarly, the palindromic antimicrobial peptide Pa-MAP2 was designed on the basis of Pa-MAP by Franco and co-workers, revealing a well-organized α-helix with flexible termini and profound interactions with phospholipids.21 Abdel Monaim et al. optimized the depsipeptide Teixobactin to obtain a cationic AMP.22 In contrast to these single-template design approaches, our method allows us to simultaneously morph one AMP into another AMP while retaining an overall helical structure of the intermediates. This concept enabled the systematic investigation of the sequence space between two known cationic helical AMPs and resulted in the discovery of relevant features for AMP activity and selectivity.
Methods
Peptide Synthesis, Analytics, and Purification
Peptide sequences were synthesized by 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis23,24 on automated parallel peptide synthesizers (Symphony and Prelude, Gyros Protein Technologies) using 200 mM Rink amide 4-methyl benzhydrylamine (MBHA) resin (AAPPTec) (0.52 mmol g–1) as a solid support, leading to C-terminally amidated peptides. All amino acids were purchased from AAPPTec and dissolved in dimethylformamide (DMF) (Sigma-Aldrich). Peptides were treated with 20% pyrrolidine (Acros Organics) in DMF (v/v) to deprotect the resins and the amino acids from Fmoc groups. Amino acids were subsequently coupled to the resin-bound peptides using 200 mM O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) (Protein Technologies) and 400 mM N-methylmorpholine (NMM) (Fisher Chemical) as coupling reagents. After coupling of the terminal amino acids, peptides were washed with DMF and dichloromethane (DCM) (Sigma-Aldrich) and cleaved from the resin by a mixture of 95% trifluoroacetic acid (TFA) (ABCR), 2.5% triisopropylsilane (TIS) (Sigma-Aldrich), and 2.5% doubly distilled H2O (ddH2O) (v/v/v). Peptides were precipitated with diisopropyl ether (Fluka) at −20 °C overnight, centrifuged (for 10 min at −10 °C and 3000 rpm), and resuspended for four cycles in ice-cold diisopropyl ether and dried under an ambient atmosphere for 3 days. The crude peptides were purified using reverse-phase preparative high-performance liquid chromatography (HPLC) (Shimadzu) on a Nucleodur C18 HTec column (150 mm × 21 mm, 5 μm, 110 Å) with a linear 5 to 70% acetonitrile (Sigma-Aldrich) in water gradient containing 0.1% formic acid (Merck Millipore) and a flow rate of 0.5 mL min–1 (at 40 °C and 120 mbar). Detection was performed by ultraviolet (UV) spectroscopy at a wavelength of 210 nm. The purity of the peptides was analyzed by UV detection and electrospray mass detection using analytical reverse-phase HPLC-MS on a Nucleodur C18 HTec column (150 mm × 3 mm, 5 μm, 110 Å) and conditions identical to those of preparative HPLC. Purified peptides were lyophilized at 0.03 mbar and −85 °C using an Alpha 2-4 LDplus Freeze-Dryer (Christ).
Antimicrobial Growth Inhibition
For all intermediate sequences, the ability to inhibit the growth of GFP-expressing Staphylococcus aureus SH1000 was measured. Minimal inhibitory concentrations (MICs) are listed in Table 1. S. aureus SH1000 expressing GFP was a gift from P. Hill (The University of Nottingham, Nottingham, U.K.) and was cultured on tryptic soy agar (Sigma-Aldrich) containing 10 μg mL–1 chloramphenicol. To monitor bacterial growth, 1 × 106 bacteria were incubated in 200 μL of nutrient broth containing 10 μg mL–1 chloramphenicol and a 50 μM peptide solution (or serial dilutions thereof) in transparent Nunclon Edge 96-well plates (SIFIN GmbH). Plates were shaken for 20 h at 37 °C in a model M200 PRO Quad4Monochromator-based multimode reader (Tecan, Anif, Austria). Loading the plate moats with 4 × 3 mL of 0.1% agarose reduced the extent of evaporation of the culture medium. Growth was monitored by detecting the fluorescence signal of GFP (excitation at 485 nm/9 nm, emission at 535 nm/20 nm) and OD600 every hour. Antimicrobial screening of the key ESKAPE bacterial pathogens S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Escherichia coli as well as the fungal pathogens Cryptococcus neoformans and Candida albicans was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (U.K.) and The University of Queensland (Australia).25 Full details of the applied methods are provided as Supporting Information.
Table 1. Peptide Sequences with Experimentally Determined Minimal Inhibitory Concentrations (MICs) on Bacteria and Fungi (left part) and Toxicity Results on Human Cells (right part) in Micromolara.
| peptide | sequence | S. aureus SH1000 | S. aureus ATCC43300* | E. coli ATCC25922* | K. pneumoniae ATCC700603* | P. aeruginosa ATCC19606* | A. baumanii ATCC27853* | C. albicans ATCC90028* | C. neoformans H99, ATCC208821* | hRBC (MHC) | HDMEC (CC50) | HEK293 (CC50) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Klk14 | KLLKLLKKLLKLLK | 10 | 19 | 19 | >19 | >19 | 9 | 9 | 1 | 8 | 3 | 10 |
| 1 | GLLKLLKKLLKLLK | 33 | >19 | >19 | >19 | >19 | 1 | 12 | 1 | 8 | 102 | 12 |
| 2 | GLFKLLKKLLKLLK | 33 | >20 | >20 | >20 | >20 | >20 | >20 | >20 | 125 | 2 | >19 |
| 3 | GLFDLLKKLLKLLK | 100 | ||||||||||
| 4 | GLFDILKKLLKLLK | 100 | ||||||||||
| 5 | GLFDIVKKLLKLLK | 100 | ||||||||||
| 6 | GLFDIVKKVLKLLK | 3.3 | >20 | >20 | >20 | >20 | >20 | >20 | >20 | 8 | 85 | |
| 7 | GLFDIVKKVVKLLK | 50 | >20 | >20 | >20 | >20 | >20 | >20 | >20 | 32 | 236 | |
| 8 | GLFDIVKKVVGLLK | 50 | >21 | >21 | >21 | >21 | >21 | >21 | >21 | 32 | 28 | |
| 9 | GLFDIVKKVVGALK | 33 | >22 | >22 | >22 | >22 | >22 | >22 | >22 | 2 | 103 | |
| Au2.2d2 | GLFDIVKKVVGALG | 50 | >23 | >23 | >23 | >23 | >23 | >23 | >23 | 16 | 64 | |
| 10 | KLFDIVKKVVGALG | >100 | ||||||||||
| 11 | KLLDIVKKVVGALG | >100 | ||||||||||
| 12 | KLLKIVKKVVGALG | 100 | ||||||||||
| 13 | KLLKLVKKVVGALG | 33 | >22 | >22 | >22 | >22 | >22 | >22 | >22 | >250 | 533 | |
| 14 | KLLKLLKKVVGALG | 10 | 22 | 11 | 22 | >22 | 5 | 22 | 11 | 125 | 67 | >21 |
| 15 | KLLKLLKKLVGALG | 10 | 5 | 3 | 5 | 21 | 1 | 21 | 3 | 32 | 25 | 12 |
| 16 | KLLKLLKKLLGALG | 10 | 3 | 1 | 5 | 21 | <1 | 11 | 3 | 8 | 8 | 7 |
| 17 | KLLKLLKKLLKALG | 33 | 10 | 20 | >20 | >20 | 1 | 20 | 3 | 8 | 4 | >20 |
| 18 | KLLKLLKKLLKLLG | >100 |
Hemolysis and HDMEC cytotoxicity were tested only for peptides active at 50 μM on S. aureus SH1000 and HEK 293 cytotoxicity for peptides active against resistant strains (*).25
Cytotoxicity Assay
Determination of the concentration at 50% cytotoxicity (CC50) of human embryonic kidney cells (HEK 293) was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust and The University of Queensland.25 Full details of the applied methods are provided as Supporting Information. The CC50 for HDMEC was determined using Reaction Biology Corp.’s HDMEC assay on a fee-for-service basis.
Hemolysis Assay
Fresh human blood, obtained from healthy donors (Blutspende Zürich, Zurich, Switzerland), was centrifuged for 10 min (800g and 4 °C) to harvest the erythrocytes. These were then washed three times with phosphate-buffered saline (PBS) and resuspended in PBS to attain a final erythrocyte concentration of 5% (v/v). Fifty microliters of the erythrocyte solution was incubated with 50 μL of a serial dilution of peptides in PBS (three technical replicates) for 1 h at 37 °C in a round-bottom 96-well plate (Greiner Bio-One, Frickenhausen, Germany). One hundred microliters of PBS was added to every well after incubation. After centrifugation (10 min at 800g and 4 °C), the supernatant was transferred to a flat-bottom 96-well plate (TPP AG, Trasadingen, Switzerland), and hemoglobin release was measured as the absorbance at 540 nm using an infinite M1000 plate reader (Tecan, Männedorf, Switzerland). Controls for zero hemolysis and 100% hemolysis consisted of erythrocytes in PBS and 1% Triton X-100, respectively. PBS served only as a blank reference. The percentage of hemolysis was calculated from the measured absorbance as follows:
| 1 |
The minimal hemolytic concentration (MHC) was defined as the concentration leading to ≤5% hemolysis.
Linear and Circular Dichroism Spectroscopy
A Chirascan spectrometer (Applied Photophysics, Leatherhead, U.K.) was used to measure both linear dichroism (LD) and circular dichroism (CD) peptide spectra. CD was recorded using a 1 mm path length quartz cuvette (type 110-QS, Helma Analytics). Peptide concentrations of 30 μM were measured in both water and 50% 2,2,2-trifluoroethanol (99.8% extra pure, Acros Organics). Near-UV CD measurements were performed in triplicate with a scanning range of 185–260 nm and a step size of 1 nm for 1 s nm–1. At every measured temperature, the sample was equilibrated for 5 min before the measurement was taken. With the help of Pro-Data Viewer software (Applied Photophysics, version 4.2.15), triplets were averaged, and the baseline was subtracted and smoothed (window size of 4). Analysis of CD spectra was performed with help of the modlAMP Python package.26 The helical content was calculated using the DichroWeb service portal27 by applying the CONTIN algorithm with reference set 3.28 LD spectra were recorded in the presence of large unilamellar vesicles (LUVs) with diameters of 400–500 nm, consisting of 60% 1-palmitoyl-2-oleyl-sn-glycero-3-phospho(1′-rac-glycerol) (POPG) and 40% cardiolipin (CL). The chosen peptide:lipid ratio was 1:10 (100 μM:1 mM), and vesicle deformation for peptide alignment was achieved with a shear rate of 3960 s–1, using a spinning quartz Couette cell (Starna Scientific Ltd.). Reduced LD spectra were calculated by dividing the LD signal by the sample’s absorbance under isotropic (nonspinning) conditions, as shown in eq 2.
| 2 |
Nuclear Magnetic Resonance Spectroscopy
The secondary structures of Klk14, peptide 6, and peptide 14 were elucidated by high-resolution liquid-state NMR spectroscopy in 30% 2,2,2-trifluoroethanol-d2 (TFE-d2). Approximately 10 mg of peptide lyophilizate was dissolved in 350 μL of H2O, and the pH was changed to 4.5 with 1% hydrochloric acid (≤37% hydrochloric acid fuming, Sigma-Aldrich, Steinheim, Germany), aiding the complete dissolution of the sample. Finally, 150 μL of TFE-d2 (d, 98%) (Cambridge Isotope Laboratories, Inc., Tewksbury, MA) was added, yielding a sample concentration of approximately 10 mM. Measurements were performed on a Bruker Avance III 500 MHz spectrometer at 291 K. Two-dimensional TOCSY (mixing time of 80 ms), NOESY (mixing times of 50, 100, 150, and 250 ms), and double-quantum-filtered COSY were carried out using standard pulse sequences and presaturation for water suppression.
Molecular Dynamics (MD) Simulations
MD simulations over 100 ns were performed with the GROMACS molecular dynamics package, version 5.1.2,29,30 with the AMBER99SB-ILDN force field.31 The starting structures of peptides (amidated C-terminus) were generated by projecting the three-dimensional coordinates onto an idealized α-helix with the help of the MOE program (Molecular Operating Environment, 2015.10; Chemical Computing Group, Montreal, QC). The obtained structures were relaxed as isolated systems by energy minimization to a root-mean-square deviation (RMSD) threshold of 0.0001 kcal mol–1 Å–2 in MOE (AMBER10:EHT force field, steepest gradient). The relaxed peptides were centered in a cubic box with periodic boundary conditions, and the box size was adjusted to obtain a minimal distance of 1 nm between the peptide and box border. The box was solvated (SPC/E water model),32 and Cl– or Na+ ions were added to obtain a neutral system charge. We used the SETTLE algorithm33 to keep water molecules rigid and the LINCS algorithm34 (fourth-order, one iteration) to enforce constraints of all solute bonds. The system was first subjected to energy minimization (steepest decent method, threshold of 1000 kJ mol–1 nm–1) to remove bad initial contacts. Further equilibration comprised a 50 ps simulation with leapfrog integration of the NVT ensemble with a modified Berendsen thermostat35 coupled to 300 K with a time step of 5 fs. This was followed by another 50 ps run with leapfrog integration of the NPT ensemble with a Parrinello–Rahman barostat36 to obtain an average system pressure of 1 bar. The Verlet cutoff scheme37 with cutoff values of 1 nm was used for both short-range electrostatic and van der Waals interactions, while particle mesh Ewald (PME)38 (grid spacing of 0.16 nm, interpolation order of 4) accounted for long-range electrostatic interactions. The time step of the simulation was set to 2 fs during the production runs of 100 ns with a leapfrog integrator. Secondary structure assignments were computed with the DSSP algorithm.39 A peptide’s helical content was expressed as the number of residues in a helical conformation, averaged over the production run trajectory while omitting the first 5 ns to reduce the impact of the forced initial α-helical peptide conformation.
Molecular Dynamics Simulations in TFE and Water
A simulation box of 125 2,2,2-trifluoroethanol molecules (parameters for AMBER99SB-ILDN according to Gerig40 and Fioroni et al.41) and 525 water molecules (SPC/E water model),32 representing a mixture of approximately 50% (v/v) at 20 °C, was equilibrated to remove residual ordering of the solvent molecules by NVT and NPT with periodic boundary conditions for 100 ps each. The resulting box then served as a template for solvating the peptides. After solvation, the MD simulations for TFE and water followed the same protocol as described above for the water-only systems. All calculations were performed on a cluster of 64 AMD Opteron 6376 processors.
Preparation of Giant Vesicles
Giant vesicles (GVs) that were 10–20 μm in diameter were prepared by swelling dried lipid membranes as previously described.42 In brief, 1 mL of an aqueous solution of 1% (w/v) agarose was added to a 250 mL round-bottom glass flask and distributed by slowly turning the flask. The agarose was dried under a gentle stream of nitrogen on a hot plate at 50 °C. Five hundred microliters of a 1 μM lipid solution, consisting of 20 μL of a stock solution of POPC and a 10.8 μM DiD dye solution in 536 μL of a MeOH/CHCl3 mixture (1:1), was then added on top of the agarose film and distributed by gentle slewing. The flask was again placed under a gentle stream of nitrogen for at least 30 min to remove all residual solvent. GVs were then grown by finally placing the flask in an oven at 50 °C with 4 mL of preheated ddH2O slowly added along the sidewall. After 2 h, the swollen giant liposomes inside the residual liquid were harvested with a glass pipet. The membrane dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) was obtained from Fluorochem Ltd. (Hadfield, U.K.), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Three examples of GVs produced by the described procedure can be found in Figure S1. Large unilamellar vesicles (LUVs) used for LD spectroscopy were produced by the same swelling method (without dye) but subsequently extruded 21 times through a 400 nm pore size Whatman Cyclopore polycarbonate and polyester membrane (Sigma-Aldrich, St. Louis, MO).
Microfluidic Chip Experiments
Two-layer microfluidic chips composed of PDMS (polydimethylsiloxane, Sylgard 184) (Dow Corning, Midland, MI) were designed as described previously.42 In brief, the bottom (fluid) layer accommodates 60 microchambers with vesicle traps in the center to capture single GVs. Chambers can be closed by actuation of a valve moving down the top (pressure) layer. The chip master form was fabricated using SU-8 2015 obtained from Microchem (Newton, MA) and processed to a height of 20 μm by spin-coating, baking steps, exposure to a UV light source through a foil mask, development, and silanization. From this master, microfluidic chips composed of PDMS were fabricated with multilayer soft lithography and bonded to a glass slide using a plasma cleaner (PDC-32 G, Harrick, NY). To prepare chips for the experiment, ddH2O was pipetted into the inlet reservoirs and compressed into the chip by centrifugation at 2000 rpm for 5 min to avoid air bubbles. A filtered 4% (w/v) BSA solution (Sigma-Aldrich) in PBS (Gibco Life Technologies, Paisley, U.K.) was then pumped into the chip channels by a Nanojet syringe pump (10 μL min–1 for 10 min, followed by a 10 min incubation) to coat the internal surfaces for the prevention of unspecific peptide or GV binding. Then, GVs were injected into the chip at a rate of 1–10 μL min–1 for 30 min before 2 bar of pressure was applied to the donut-shaped valves in the upper layer to enclose the trapped vesicles. Afterward, the channels were flushed with 100 μM peptide with 50 μM calcein (Acros Organics, Morris Plains, NJ) in ddH2O at a rate of 10 μL min–1 for 5 min. The flow rate was then reduced to 0.5 μL min–1, and the vesicle-containing compartments were opened. Upon contact with the peptide, calcein influx and rupturing of the vesicles were monitored through microscopic imaging.
Light Microscopy
The microfluidic chips were imaged as previously described.42 Briefly, chips were mounted on the stage of an inverted microscope (IX71, Olympus) equipped with a 100×/1.30 NA oil objective. The fluorophores were excited with a SpectraX-6-LCR-SA LED system (Lumencore, Beaverton, OR). Different optical filter sets were used for imaging of (i) calcein [474 nm/27 nm BrightLine HC (excitation) and 515 nm HC Quadband (emission)] and (ii) DiD [635 nm/18 nm BrightLine HC (excitation) and 730 nm HC Quadband (emission)]. In both cases, we used the dichroic mirror BrightLine Quadband BS 409/493/573/652 nm. Images of GVs were recorded with an EMCCD camera (iXon Ultra DU-897U, Andor, Belfast, Northern Ireland). For the imaging of vesicle rupture, a high-speed camera (Phantom Miro M110 camera, Vision Research) was used.
Results and Discussion
The 18 designed peptides as well as the two templates were synthesized using Fmoc solid-phase peptide synthesis23,24 on a robotic synthesizer and purified by preparative LC-MS (Table S4). These linear peptides were synthesized with amidated C-termini to increase the general membrane affinity.43−45 The two templates (Au2.2d2 and Klk14) and 11 of the 18 designs showed growth inhibitory activity at a MIC of ≤50 μM against the nonresistant S. aureus SH1000 strain (Table 1). This inhibitory activity proportion of 61% is high in comparison to those from related studies.46−48 A potential explanation for this observation could be the low MIC of the starting template Klk14, in combination with the shared pharmacophore sequence pattern of the generated peptides, allowing all of them to form distinct amphipathic helices (helical wheel plots of all peptides in Figure S10). The active peptides were further tested against five multiresistant bacteria (S. aureus, E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii) (see Table 1 for details on the ATCC strains) from the ESKAPE panel49 and resistant strains of the fungi C. albicans and C. neoformans. Five of the designs and the Klk14 template inhibited the growth of one or more resistant strains at a MIC of ≤22 μM (Table 1).
The naturally occurring AMPs Aurein and Citropin contain an N-terminal GL (Gly-Leu) residue motif that is conserved in all members of this peptide family. Changing this motif led to a complete loss of activity, e.g., peptide 10 (Table 1). In comparison, changing residues 9–13 retained or even increased the activity, e.g., peptides 6–9. We therefore hypothesize that substituting residues in sequence positions with high Shannon information50 affects antimicrobial activity more than changes in less information-rich residues (Figure S9 and Tables S5 and S6).
To be suitable for drug development, AMPs must not kill human cells. Cytotoxic concentrations leading to 50% cell death (CC50) were determined on human dermal microvascular endothelial cells (HDMECs) and human embryonic kidney cells (HEK 293). Hemolysis of human erythrocytes (hRBCs) was determined as the minimal hemolytic concentration (MHC) leading to 5% lysis. The results are listed in Table 1. We identified peptide 14 as a selective AMP active against six resistant strains, with a calculated therapeutic index of 12.5 (fraction of the MHC for hRBC over the MIC for S. aureus SH1000). Peptides 6 and 15 also exhibited higher activity on bacteria than on human cells, albeit with calculated therapeutic indices of <3. The broadly active and selective peptide 14 is the center of the C- to N-terminal morphing series (Figure 1), incorporating the cationic N-terminal half of Klk14 and the hydrophobic C-terminal portion of Au2.2d2. Sequences 14–16 reveal that the Val9 → Leu9 and Val10 → Leu10 transitions led to a pronounced increase in membranolytic activity against both resistant strains and human cells. Val and Leu side chains share the same pharmacophore, have similar hydrophobicity values, and are generally considered similar.51 One explanation for this steep activity increase might thus be found in the different helix-stabilizing properties of these two residues.52
For verification of α-helical structure, we performed CD spectroscopy of all peptides in both water and a 50% TFE/water mixture (v/v) at room temperature (Figure 2). The templates and all designs displayed characteristic spectra of α-helices in 50% TFE, whereas all spectra in pure water suggested random coil structures. This observation corroborates our design hypothesis of the intermediate sequences adopting α-helical structures in a hydrophobic environment (50% TFE).
Figure 2.

Circular dichroism spectra of all peptides. All measurements were performed under equal conditions with a peptide concentration of 33 μM. The maximum at 190 nm and the minima at 208 and 222 nm are characteristic of α-helical secondary structures. Every line represents the average of three technical replicates under the indicated condition (blue, water; red, 50% TFE in water).
Because these CD spectra did not explain the observed differences in peptide activity and selectivity, we hypothesized that the structural stability of the α-helices could be a selectivity-determining factor. Therefore, we recorded CD at increasing temperatures in a 50% TFE/water mixture (v/v), using the α-helical stability at elevated temperatures as a surrogate for structural flexibility. The changes in the CD signal at three typical α-helical wavelengths (192, 208, and 222 nm) between the lowest (30 °C) and highest (75 °C) temperature were compared to the activity and selectivity of all peptides that inhibited S. aureus SH1000 (Table 2 and Figure S6). The chosen temperatures do not have a physiological meaning but were selected for the sole purpose of determining α-helical stability. We observed that increasing relative differences in CD spectra recorded at low and high temperatures suggest greater peptide selectivity (comparing S. aureus SH1000 vs HDMEC and hRBC). This result points to increased structural flexibility at elevated temperatures as a discriminative feature of cell-type selective AMPs and potentially explains why the Val → Leu modification impacts AMP selectivity. Measuring CD at high (although physiologically nonrelevant) temperatures may be a suitable method for determining AMP selectivity.
Table 2. Correlation Analysis of Peptide Activity with Relative Differences in CD Spectra at Increasing Temperaturesa.
| S. aureus MIC | HDMEC CC50 | hRBC MHC | |
|---|---|---|---|
| ∂ 192 nm | 0.082 | 0.608 | 0.773 |
| ∂ 208 nm | 0.038 | 0.609 | 0.770 |
| ∂ 222 nm | 0.096 | 0.537 | 0.739 |
Signal differences (∂) at three characteristic wavelengths comparing spectra recorded at 30–75 °C were correlated with peptide minimal inhibitory concentrations (MICs) on S. aureus SH1000, cytotoxicity on human dermal microvascular epithelial cells (HDMEC CC50), and the minimal hemolytic concentration of human red blood cells (hRBC MHC). Numbers are Pearson product–moment correlation coefficients ranging from 0 (no correlation) to 1 (perfect correlation).
Hydrophobicity has been suggested to be critically important for the activity of model peptides against Gram-positive bacteria.53,54 In contrast, peptides 13–16 have the same global Eisenberg hydrophobicity value of 0.23 and a hydrophobic moment of 0.61 but differ in their selectivity (Table 1). However, the calculated global flexibility (modlAMP flexibility scale26) of peptides 13–17 is correlated to their hemolytic activity (Pearson r2 = 0.74; p value for noncorrelation of 0.16), with the more flexible peptides (peptides 13–15) being less hemolytic, but apparently does not correlate with the peptides’ antimicrobial activity (Table 3).
Table 3. Comparison of the Global Flexibility [modlAMP flexibility scale26 (Table S7)] to the Hemolytic Activity (MHC) of Peptides 13–17a.
| peptide | MHC (μM) | flexibility |
|---|---|---|
| 13 | >250 | 0.527 |
| 14 | 125 | 0.521 |
| 15 | 32 | 0.515 |
| 16 | 8 | 0.509 |
| 17 | 8 | 0.488 |
A peptide’s flexibility is calculated by averaging the flexibility values of the individual amino acids. The flexibility scale ranges from 0 to 1 (see Table S7 for individual amino acids).
To further investigate structural flexibility and learn about the potential mechanism of action of the peptides, we performed MD simulations for all peptides in water and a 50% TFE/water mixture (v/v) over 100 ns. MD confirmed the CD measurements in that all peptides adopted helical structures in the TFE simulations, whereas they unfolded during simulations in pure water. The designs incorporating the C-terminus of Au2.2d2 seemed to partially unfold over time [i.e., peptide 14 (Figure S3)]. In contrast, the peptides containing the N-terminus of Au2.2d2 did not show this behavior [i.e., peptide 4 (Figure S2)]. A simulation snapshot of peptide 14 shows its C-terminus “snorkeling” into the TFE layer, suggesting that the designed peptides enter membranes by the adoption of oblique oriented α-helices (Figure 3). This mechanism of membrane invasion by amphibian AMPs has been previously described.55 Profile plots of hydrophobicity and hydrophobic moments can also hint at oblique orientation. Figure 4 reveals the increase in hydrophobicity toward the C-terminus of peptide 14 and the concurrent decrease in the helical hydrophobic moment, suggesting an unstructured hydrophobic tail that can penetrate a lipid bilayer. These calculations support the observed peptide “snorkeling” in the MD simulations.
Figure 3.
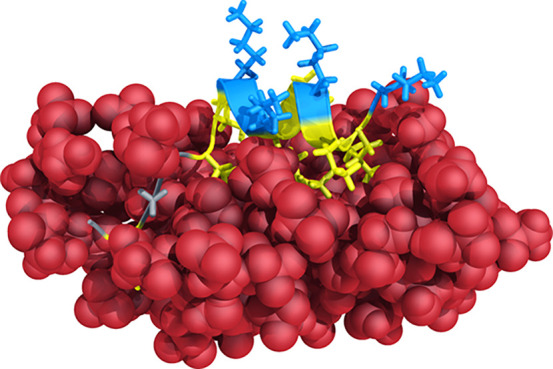
Molecular dynamics simulation snapshot of peptide 14 with its C-terminal end (left) snorkeling into the TFE layer (red spheres). This observation supports the hypothesis of membrane invasion by the adoption of oblique oriented α-helices.
Figure 4.

Hydropathy plot of peptide 14. The gray line with the corresponding left y-axis shows the Eisenberg hydrophobicity, and the dashed black line with the corresponding right y-axis shows the calculated hydrophobic moment. The linear hydrophobicity gradient increases toward the C-terminus. The results suggest that the more flexible hydrophobic C-terminal part initiates membrane insertion.
The kinetics and mechanism of action were further investigated using giant vesicles (diameters of 10–20 μm) in a microfluidic chamber. As the α-helices of all our sequences are too short (14 residues) to span lipid bilayers, the formation of barrel-stave pores is impossible. Video sequences recorded for peptide 6 (the most active peptide on S. aureus SH1000) imply a fast, carpet-forming membranolytic event55 upon the accumulation of peptide molecules on the vesicle surface (Figure 5, Video S1, and Video S2). Consequently, LD spectroscopy was performed to obtain preliminary information about the relative orientation of templates Au2.2d2 and Klk14, as well as peptides 6 and 14 in model membranes. The LD spectra obtained showed marked differences between Au2.2d2 and the three other peptides, with 6 and 14 resembling the Klk14 template (Figure S7). The spectrum of Au2.2d2 is clearly distinct from the LD spectra of the other three peptides. However, these findings cannot be clearly related to the observed differences in growth inhibitory or cytotoxic activity, or to the recorded CD spectra.
Figure 5.
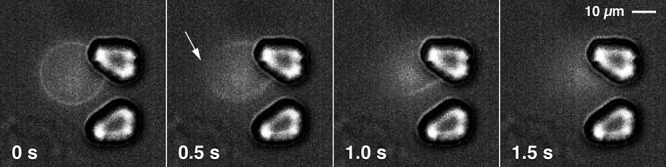
Snapshots of a video sequence of a giant POPC vesicle (diameter of 20 μm) with membrane-incorporated DiD dye, floating in the microfluidic chip chamber in the presence of 50 μM peptide 6, the most active peptide on S. aureus SH1000. The flow was from left to right at a rate of 0.5 μL min–1. Two micropillars acted as a hydrodynamic trap for the vesicle. The white arrow indicates the position at which the lipid membrane of the vesicle started to rupture upon exposure to peptide 6.
To verify the observed relationship between peptide flexibility and cell selectivity and potentially illuminate differences in LD spectra, we performed solution NMR spectroscopy with Klk14, as well as peptides 6 and 14, in a 30% TFE/water mixture. The structure of Au2.2d2 in a 30% TFE/water mixture was already known from a previous study.14 Structural ensembles of the 10 lowest-energy structures of these four peptides are shown in Figure 6. Terminal flexibility observed for peptide 14 in MD simulations could partially be confirmed by NMR. Compared to peptide 6, the C-terminus of peptide 14 is indeed more flexible, confirmed by the respective Ramachandran plots (Figure S8). Overall, Klk14 shows the fewest nuclear Overhauser effect (NOE)-derived NMR constraints in this measurement. A decrease in the number of observed NOEs together with line broadening is typical for increased exchange phenomena due to flexibility. One can observe such a drop in the number of residual constraints for the terminal, hydrophobic portion (sequence of GALG) of peptide 14 relative to its central part and to the corresponding residues of peptide Klk14. Both the MD snapshot shown in Figure 3 and the MD trajectory of peptide 14 simulated in TFE (Figure S2) are in agreement with the NMR result. These findings provide a potential explanation for the different orientation of Au2.2d2 in contact with lipid vesicles (which was observed during the LD experiment), as this peptide was previously found to have higher helical stability in terms of NMR constraints.14 With regard to the differences in TFE percentage between the experimental NMR conditions (30% TFE) and the MD simulations (50% TFE), studies by Fioroni et al. have shown that secondary structure-stabilizing TFE cluster formation reaches a maximum at approximately 30% TFE and water, obviating large differences relative to that in 50% TFE used in the MD simulation.41
Figure 6.

Solution NMR results. (a) Structural ensemble of the 10 lowest-energy structures of templates Au2.2d2 (PDB entry 5MXL) and Klk14 (PDB entry 6HNG), as well as of peptide 6 (PDB entry 6HNE) and peptide 14 (PDB entry 6HNH). Hydrophobic residues are colored yellow, and polar ones blue. Gly is colored gray. (b) Nuclear Overhauser effect (NOE) statistics plotted against the corresponding residues. Blue bars represent intraresidue NOEs, yellow bars sequential restraints, and orange bars medium-range NOEs (1 < |i – j| < 5). Restraints for Au2.2d2 can be found in ref (15). Structures were modeled from spectra recorded in a 30% TFE/water mixture.
In summary, systematic “morphing” of the natural helical AMP Aurein Au2.2d2 into the synthetic helical AMP Klk14 resulted in chimeric sequences exhibiting general antimicrobial activity and selectivity against several antibiotic-resistant bacteria and fungi. Helical amphipathicity turned out to be a necessary but insufficient condition for the membranolytic activity of these designs. For the new AMPs that kill S. aureus SH1000, we detected a positive correlation between the peptides’ structural flexibility at elevated temperatures and their cell-type selectivity. In agreement with the results from biophysical measurements, MD simulations suggested that active peptides with higher selectivity toward bacteria possess a greater proportion of flexible areas than less selective AMPs. The NMR structures obtained for peptides 6 and 14 additionally confirmed the more pronounced flexibility of the C-terminal part of the more selective peptide 14. Together, these results support the working hypothesis of partial structural flexibility playing a key role in the cell-type selectivity of AMPs.
Single changes of certain amino acids led to a loss of antibacterial activity, even when the pharmacophore features of the original and the substituted residues were preserved. With current AMP prediction models being unable to forecast such steep activity cliffs, the systematic transformation approach presented here offers a suitable, template-based optimization strategy for helical AMPs.
The results of this study can be linked to previous work. For example, Cheng et al. investigated the relative importance of a hydrophobic residue in position 13 of Aurein 2.2 for peptide structure and membranolytic activity.10 Accordingly, the exact nature of the residue appears to be less important, as long as it is hydrophobic. To better distinguish between amino acids of the same pharmacophore type, Senes et al. developed an empirical potential of insertion of protein into lipid bilayers (Ez).56 The Ez potential determines the free energy profile for each residue of the peptide sequence as a continuous function of the depth of insertion into the lipid membrane. Global potential can then be determined via a reverse Boltzmann statistical approach to convert propensities into pseudo-energies of membrane insertion. Giguère et al. presented learning algorithms with generic string kernels to capture patterns responsible for peptide bioactivity.57 A de novo-designed sequence (WWKRWKKLRRIFLML) exhibited MICs of 4 μM against both E. coli and S. aureus.58 Combining the calculated sequence motifs by Giguère et al., the preferred location and membrane depth propensities of Trp residues in AMPs,57,59 and the results of the study presented here, one could propose a “universal” membranolytic sequence. Accordingly, such a minimalist helical AMP sequence would need to have one or several anchoring Trp or Tyr residues in the N-terminal part, repetitive cationic amino acids to form an amphipathic secondary structure, and a hydrophobic C-terminus to, at least initially, interact with the hydrophobic core of the membrane. In fact, it has been suggested that a majority of α-helical AMPs are candidates for obliquely oriented helix formation.60 For several AMPs (Aurein 1.2, Citropin 1.1, Maculatin 1.1, and Carein 1.1), such an oblique membrane insertion at an angle of approximately 50° was demonstrated by solid-state NMR and oriented CD spectroscopy.59 Taken together, the findings presented here should foster future AMP design studies toward more selective and highly active α-helical AMPs, based on partial peptide flexibility as a driving factor.
Acknowledgments
The authors thank S. Haller, P. Rabatscher, and J. A. Fuchs for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00565.
Supporting data and methodological details about the computational activity predictions, molecular dynamics simulations, CD spectroscopy, solution-phase NMR, peptide flexibility analysis, and CO-ADD antimicrobial and antifungal screening (PDF)
Video sequence (version 1) of the interaction of peptide 6 with a giant POPC vesicle (AVI)
Video sequence (version 2) of the interaction of peptide 6 with a giant POPC vesicle (AVI)
Author Present Address
⊥ G.G.: Quantitative Biology Center, Eberhard Karls University Tübingen, 72076 Tübingen, Germany.
Author Contributions
A.T.M. designed peptide sequences. A.T.M., G.G., and C.N. performed peptide syntheses. A.T.M. and G.P. performed activity assays. A.T.M. and S.B. performed microfluidics assays. A.T.M. ran molecular dynamics simulations. B.P. recorded NMR spectra. M.B. performed NMR analysis. J.A.H., P.S.D., K.-H.A., S.W., and G.S. designed and supervised the study. A.T.M. and G.S. wrote the manuscript.
This research was supported by ETH Research Grant ETH-01 12-1 (to G.S.), the Swiss National Science Foundation (Grant 200021_157190 to G.S. and J.A.H. and Grant CRSII2_160699 to G.S.), and the Austrian Science Fund (FWF Grants P_24074 and P_24315 to S.W.).
The authors declare the following competing financial interest(s): G.S. is a co-founder of inSili.com LLC, Zurich, Switzerland, and M.B. is a paid employee of Novartis AG, Basel, Switzerland.
Supplementary Material
References
- World Health Organization (2015) Worldwide Country Situation Analysis: Response to Antimicrobial Resistance. Geneva.
- Fjell C. D.; Hiss J. A.; Hancock R. E. W.; Schneider G. (2012) Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discovery 11 (1), 37–51. 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- da Cunha N. B.; Cobacho N. B.; Viana J. F. C.; Lima L. A.; Sampaio K. B. O.; Dohms S. S. M.; Ferreira A. C. R.; de la Fuente-Núñez C.; Costa F. F.; Franco O. L.; Dias S. C. (2017) The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discovery Today 22 (2), 234–248. 10.1016/j.drudis.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet G.; Müller A. T.; Hiss J. A.; Schneider G. (2016) Membranolytic anticancer peptides. MedChemComm 7 (12), 2232–2245. 10.1039/C6MD00376A. [DOI] [Google Scholar]
- Rios A. C.; Moutinho C. G.; Pinto F. C.; Del Fiol F. S.; Jozala A.; Chaud M. V.; Vila M. M. D. C.; Teixeira J. A.; Balcão V. M. (2016) Alternatives to overcoming bacterial resistances: state-of-the-art. Microbiol. Res. 191, 51–80. 10.1016/j.micres.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44 (D1), D1087–D1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghu F. H.; Gopi L.; Barai R. S.; Ramteke P.; Nizami B.; Idicula-Thomas S. (2014) CAMP: Collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 42 (D1), D1154–D1158. 10.1093/nar/gkt1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera-Mendoza L.; Marrero-Ponce Y.; Tellez-Ibarra R.; Llorente-Quesada M. T.; Salgado J.; Barigye S. J.; Liu J. (2015) Overlap and diversity in antimicrobial peptide databases: Compiling a non-redundant set of sequences. Bioinformatics 31 (15), 2553–2559. 10.1093/bioinformatics/btv180. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Lin Y.-C.; Koch C. P.; Pillong M.; Perna A. M.; Reutlinger M.; Hiss J. A. (2013) Adaptive peptide design. Chimia 67 (12), 859–863. 10.2533/chimia.2013.859. [DOI] [PubMed] [Google Scholar]
- Solstad R. G.; Johansen C.; Stensvåg K.; Strøm M. B.; Haug T. (2020) Structure-activity relationship studies of shortened analogues of the antimicrobial peptide EeCentrocin 1 from the sea urchin Echinus esculentus. J. Pept. Sci. 26 (2), e3233. 10.1002/psc.3233. [DOI] [PubMed] [Google Scholar]
- Cheng J. T. J.; Hale J. D.; Kindrachuk J.; Jessen H.; Elliott M.; Hancock R. E. W.; Straus S. K. (2010) Importance of residue 13 and the C-terminus for the structure and activity of the antimicrobial peptide aurein 2.2. Biophys. J. 99 (9), 2926–2935. 10.1016/j.bpj.2010.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strøm M. B.; Haug B. E.; Skar M. L.; Stensen W.; Stiberg T.; Svendsen J. S. (2003) The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 46 (9), 1567–1570. 10.1021/jm0340039. [DOI] [PubMed] [Google Scholar]
- Rozek T.; Wegener K. L.; Bowie J. H.; Olver I. N.; Carver J. A.; Wallace J. C.; Tyler M. J. (2000) The antibiotic and anticancer active aurein peptides from the Australian bell frogs Litoria aurea and Litoria raniformis. Eur. J. Biochem. 267 (17), 5330–5341. 10.1046/j.1432-1327.2000.01536.x. [DOI] [PubMed] [Google Scholar]
- Wegener K. L.; Wabnitz P. A.; Carver J. A.; Bowie J. H.; Chia B. C. S.; Wallace J. C.; Tyler M. J. (1999) Host defence peptides from the skin glands of the Australian Blue Mountains tree-frog Litoria citropa. Solution structure of the antibacterial peptide citropin 1.1. Eur. J. Biochem. 265 (2), 627–637. 10.1046/j.1432-1327.1999.00750.x. [DOI] [PubMed] [Google Scholar]
- Stutz K.; Müller A. T.; Hiss J. A.; Schneider P.; Blatter M.; Pfeiffer B.; Posselt G.; Kanfer G.; Kornmann B.; Wrede P.; Altmann K.-H.; Wessler S.; Schneider G. (2017) Peptide–membrane interaction between targeting and lysis. ACS Chem. Biol. 12 (9), 2254–2259. 10.1021/acschembio.7b00504. [DOI] [PubMed] [Google Scholar]
- Bertocco A.; Formaggio F.; Toniolo C.; Broxterman Q. B.; Epand R. F.; Epand R. M. (2003) Design and function of a conformationally restricted analog of the influenza virus fusion peptide. J. Pept. Res. 62 (1), 19–26. 10.1034/j.1399-3011.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- Babii O.; Afonin S.; Schober T.; Komarov I. V.; Ulrich A. S. (2017) Flexibility vs rigidity of amphipathic peptide conjugates when interacting with lipid bilayers. Biochim. Biophys. Acta, Biomembr. 1859 (12), 2505–2515. 10.1016/j.bbamem.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Xia Y.; Li D.; Du Q.; Liang D. (2014) Relationship between peptide structure and antimicrobial activity as studied by de novo designed peptides. Biochim. Biophys. Acta, Biomembr. 1838 (12), 2985–2993. 10.1016/j.bbamem.2014.08.018. [DOI] [PubMed] [Google Scholar]
- Han H. M.; Gopal R.; Park Y. (2016) Design and membrane-disruption mechanism of charge-enriched AMPs exhibiting cell selectivity, high-salt resistance, and anti-biofilm properties. Amino Acids 48 (2), 505–522. 10.1007/s00726-015-2104-0. [DOI] [PubMed] [Google Scholar]
- Migliolo L.; Felício M. R.; Cardoso M. H.; Silva O. N.; Xavier M.-A. E.; Nolasco D. O.; de Oliveira A. S.; Roca-Subira I.; Vila Estape J.; Teixeira L. D.; Freitas S. M.; Otero-Gonzalez A. J.; Gonçalves S.; Santos N. C.; Franco O. L. (2016) Structural and functional evaluation of the palindromic alanine-rich antimicrobial peptide Pa-MAP2. Biochim. Biophys. Acta, Biomembr. 1858 (7), 1488–1498. 10.1016/j.bbamem.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Abdel Monaim S. A. H.; Ramchuran E. J.; El-Faham A.; Albericio F.; de la Torre B. G. (2017) Converting teixobactin into a cationic antimicrobial peptide (AMP). J. Med. Chem. 60 (17), 7476–7482. 10.1021/acs.jmedchem.7b00834. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85 (14), 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Chan W. C., and White P. D. (2000) Fmoc Solid Phase Peptide Synthesis, Oxford University Press, Oxford, U.K. [Google Scholar]
- Blaskovich M. A. T.; Zuegg J.; Elliott A. G.; Cooper M. A. (2015) Helping chemists discover new antibiotics. ACS Infect. Dis. 1 (7), 285–287. 10.1021/acsinfecdis.5b00044. [DOI] [PubMed] [Google Scholar]
- Müller A. T.; Gabernet G.; Hiss J. A.; Schneider G. (2017) modlAMP: Python for antimicrobial peptides. Bioinformatics 33 (17), 2753–2755. 10.1093/bioinformatics/btx285. [DOI] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 89 (5), 392–400. 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1 (6), 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91 (1–3), 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Best R. B.; Hummer G. (2009) Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 113 (26), 9004–9015. 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. (1987) The missing term in effective pair potentials. J. Phys. Chem. 91 (24), 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Miyamoto S.; Kollman P. A. (1992) Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13 (8), 952–962. 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18 (12), 1463–1472. . [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 (8), 3684. 10.1063/1.448118. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52 (12), 7182. 10.1063/1.328693. [DOI] [Google Scholar]
- Verlet L. (1968) Computer “experiments” on classical fluids. II. Equilibrium correlation functions. Phys. Rev. 165 (1), 201–214. 10.1103/PhysRev.165.201. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103 (19), 8577. 10.1063/1.470117. [DOI] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 (12), 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Gerig J. T. (2015) Further efforts toward a molecular dynamics force field for simulations of peptides in 40% trifluoroethanol–water. J. Phys. Chem. B 119 (16), 5163–5175. 10.1021/acs.jpcb.5b01270. [DOI] [PubMed] [Google Scholar]
- Fioroni M.; Burger K.; Mark A. E.; Roccatano D. (2000) A new 2,2,2-trifluoroethanol model for molecular dynamics simulations. J. Phys. Chem. B 104 (51), 12347–12354. 10.1021/jp002115v. [DOI] [Google Scholar]
- Pillong M.; Hiss J. A.; Schneider P.; Lin Y.-C.; Posselt G.; Pfeiffer B.; Blatter M.; Müller A. T.; Bachler S.; Neuhaus C. S.; Dittrich P. S.; Altmann K.-H.; Wessler S.; Schneider G. (2017) Rational design of membrane-pore-forming peptides. Small 13 (40), 1701316. 10.1002/smll.201701316. [DOI] [PubMed] [Google Scholar]
- Strandberg E.; Tiltak D.; Ieronimo M.; Kanithasen N.; Wadhwani P.; Ulrich A. S. (2007) Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 79 (4), 717–728. 10.1351/pac200779040717. [DOI] [Google Scholar]
- Zhu S.; Aumelas A.; Gao B. (2011) Convergent evolution-guided design of antimicrobial peptides derived from influenza A virus hemagglutinin. J. Med. Chem. 54 (4), 1091–1095. 10.1021/jm1010463. [DOI] [PubMed] [Google Scholar]
- Dennison S. R.; Phoenix D. A. (2011) Influence of C-terminal amidation on the efficacy of modelin-5. Biochemistry 50 (9), 1514–1523. 10.1021/bi101687t. [DOI] [PubMed] [Google Scholar]
- Khara J. S.; Lim F. K.; Wang Y.; Ke X.-Y.; Voo Z. X.; Yang Y. Y.; Lakshminarayanan R.; Ee P. L. R. (2015) Designing α-helical peptides with enhanced synergism and selectivity against Mycobacterium smegmatis: discerning the role of hydrophobicity and helicity. Acta Biomater. 28, 99–108. 10.1016/j.actbio.2015.09.015. [DOI] [PubMed] [Google Scholar]
- Kim H.; Jang J. H.; Kim S. C.; Cho J. H. (2014) De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J. Antimicrob. Chemother. 69 (1), 121–132. 10.1093/jac/dkt322. [DOI] [PubMed] [Google Scholar]
- Kang S. J.; Won H. S.; Choi W. S.; Lee B. J. (2009) De novo generation of antimicrobial LK peptides with a single tryptophan at the critical amphipathic interface. J. Pept. Sci. 15 (9), 583–588. 10.1002/psc.1149. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48 (1), 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Shannon C. E. (1948) A mathematical theory of communication. Bell Syst. Tech. J. 27 (3), 379–423. 10.1002/j.1538-7305.1948.tb01338.x. [DOI] [Google Scholar]
- Lyu P. C.; Sherman J. C.; Chen A.; Kallenbach N. R. (1991) Alpha-helix stabilization by natural and unnatural amino acids with alkyl side chains. Proc. Natl. Acad. Sci. U. S. A. 88 (12), 5317–5320. 10.1073/pnas.88.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtchen A.; Pasupuleti M.; Malmsten M. (2014) Effect of hydrophobic modifications in antimicrobial peptides. Adv. Colloid Interface Sci. 205, 265–274. 10.1016/j.cis.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Dathe M.; Wieprecht T.; Nikolenko H.; Handel L.; Maloy W. L.; MacDonald D. L.; Beyermann M.; Bienert M. (1997) Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 403 (2), 208–212. 10.1016/S0014-5793(97)00055-0. [DOI] [PubMed] [Google Scholar]
- Bhaskaran R.; Ponnuswamy P. K. (1988) Positional flexibilities of amino acid residues in globular proteins. Int. J. Pept. Protein Res. 32 (4), 241–255. 10.1111/j.1399-3011.1988.tb01258.x. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 3 (3), 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Senes A.; Chadi D. C.; Law P. B.; Walters R. F. S.; Nanda V.; DeGrado W. F. (2007) Ez, a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J. Mol. Biol. 366 (2), 436–448. 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Giguère S.; Laviolette F.; Marchand M.; Tremblay D.; Moineau S.; Liang X.; Biron É.; Corbeil J. (2015) Machine learning assisted design of highly active peptides for drug discovery. PLoS Comput. Biol. 11 (4), e1004074. 10.1371/journal.pcbi.1004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimland G.; Eijsink V. G. H.; Nissen-Meyer J. (2002) Mutational analysis of the role of tryptophan residues in an antimicrobial peptide. Biochemistry 41 (30), 9508–9515. 10.1021/bi025856q. [DOI] [PubMed] [Google Scholar]
- Marcotte I.; Wegener K. L.; Lam Y.-H.; Chia B. C. S.; de Planque M. R. R.; Bowie J. H.; Auger M.; Separovic F. (2003) Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem. Phys. Lipids 122 (1–2), 107–120. 10.1016/S0009-3084(02)00182-2. [DOI] [PubMed] [Google Scholar]
- Dennison S. R.; Harris F.; Phoenix D. A. (2005) Are oblique orientated alpha-helices used by antimicrobial peptides for membrane invasion?. Protein Pept. Lett. 12 (1), 27–29. 10.2174/0929866053406039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.