

Abstract
Marek's disease (MD) is a contagious avian viral disease that is responsible for large economic losses to farmers. The disease is caused by Marek's disease virus (species Gallid alphaherpesvirus 2), which causes neurological lesions, immune suppression, and tumor proliferation of lymphoid cells that invade a large number of organs and tissues. Despite widespread vaccination, Marek's disease virus (MDV), has shown a continuous increase in its virulence and has acquired the ability to overcome immune responses induced by vaccines. In the present study, the oncogenic serotype MDV-1 was detected by real-time PCR in DNA samples extracted from organs developing tumor infiltrations. Identification of the pathotype based on a 132-bp tandem repeat and sequencing and phylogenetic analysis of the Meq gene and its encoded protein allowed classification of the isolated viruses as "very virulent", with two new and unique mutations in the Meq gene resulting in amino acid substitutions. Sequencing of pp38, vIl-8, UL1 and UL44 genes did not reveal any new mutations that were characteristic of the Tunisian isolates or correlated with virulence. These results raised concerns about the ability of HVT and CVI988 vaccines, which are currently used in Tunisia and other countries, to protect chickens against highly virulent virus strains.
Introduction
Marek's disease (MD) is a highly contagious immunosuppressive disease that is characterized by paralysis and lymphoma development of T-cells in viscera and muscles [1]. The etiological agent is Marek's disease virus (MDV), or gallid herpesvirus 2 (GaHV-2), a member of the genus Alphaherpesvirus, family Herpesviridae, that has an enormous impact on the poultry industry [2]. The molecular basis for increased MDV virulence is currently unknown, but mutations in some regions of the viral genome, including the Meq (Marek's disease virus EcoRI fragment Q), phosphoprotein 38 (pp38), viral interleukin-8 (vIL-8), UL1, and UL44 genes have been implicated as virulence factors [3–7].
The MDV genome consists of a double-stranded linear DNA molecule of about 180 kbp. It consists of several regions, namely, long unique (UL) and unique short (US) regions flanked by long terminal repeats (TRLs) and short terminal repeats (TRSs), long internal repeats (IRLs) and short internal repeats (IRSs). The genome structure and the gene content of each region are similar in all GaHV-2 serotypes, but there are key differences [8]. Oncogenic serotype 1 is characterized by the presence of the Meq oncogene and other unique genes, including pp38, vIL-8, and vTR, in the repeat regions, particularly the TRL region. The Meq gene is considered the major factor responsible for tumorigenesis in chickens, since its deletion leads to a breakdown of T-cell transformation [9]. In addition to the Meq gene, which is directly responsible for transformation, the major lytic antigen pp38 has been reported to be associated with enhanced virulence and is highly expressed during lytic infection and lymphoma formation [10, 11]. There are other genes, such as vIL-8, whose deletion or mutation may decrease tumor development and lead to attenuation of virulence [5, 12]. Another gene that is suspected to contribute to the virulence of some hypervirulent MDV strains is the UL1 gene, which codes for glycoprotein L (gL) [6]. This protein forms a hetero-oligomeric complex with glycoprotein H (gH-L) that plays an important role in entry of the virus into host cells and cell-to-cell infection [13] An MDV-specific cytotoxic immune response is induced by the MDV gC protein and, to a lesser extent, the gL protein [14]. The gC protein, which is encoded by the UL44 gene, appears to be involved in virus-host interactions and is important for attenuation of virulence [7].
Due to mutations in the viral genome, the severity of disease varies among GaHV-2 isolates, leading to ever-changing pathotypes that overwhelm vaccine protection, and efforts have been made to identify new emerging pathotypes in order to ensure vaccine protection [15]. Sporadic epidemics of MD have been reported worldwide, even in vaccinated flocks [16], indicating that the virulence of MDV has increased in recent decades, and some of the more recent isolates are more pathogenic to chickens than older isolates [17, 18]. Thus, the increased use of vaccines has led the virus to evolve toward higher virulence, which can overcome the protection conferred by currently available vaccines [19]. It has been suggested that the widespread use of the CVI988 vaccine strain may have led to the emergence of new, more virulent pathotypes [20], and there are serious concerns that such pathotypes are circulating in Tunisia, especially due to the excessive use of vaccines. This poses a threat to the poultry industry and gives a higher priority to research, especially in the absence of relevant studies, particularly in Tunisia. Therefore, the main objective of this study was to determine the pathotypes of circulating GaHV-2 strains on Tunisian farms based on analysis of the Meq, pp38, UL1, UL44, and v-IL8 genes of two MDV field isolates. This will provide a basis for the development of more effective vaccines.
Materials and methods
Sample collection
Two field isolates were isolated in 2016 from samples of diseased broiler chickens reared on two different farms located in the same region. The birds had developed MD symptoms, including paralysis of the legs and lethargy. At necropsy, macroscopic examination of lesions revealed tumor infiltrations in the form of nodules 2 cm in diameter, associated with several smaller nodules, in the spleen, heart and liver as well as an enlarged spleen and an enlargement and a loss of the pearly white appearance of the sciatic nerves. The liver, kidneys and heart were collected and preserved at − 80 °C until used. These birds had been vaccinated with monovalent CVI-988 vaccine when they were one day old. Specific information about these birds is shown in Table 1.
Table 1.
Summary of the data samples
| Sample | Bird number | Type | Age (weeks) | Onset of symptoms (week) | Mortality (%) | Origin |
|---|---|---|---|---|---|---|
| TN 1013/16 | 90 | Broiler | 18 | 7th | 11.1 | Cherfech-Ariana |
| TN 1014/16 | 40 | Broiler | 6 | 4th | 30 | Borjyoussef-Ariana |
Virus purification, isolation, and identification
Organs were homogenized in a blender in the presence of Dulbecco's modified Eagle medium (DMEM) containing 5% antibiotics. The mixture was clarified by centrifugation at 1500 rpm for 15 min and passed through a 0.22-µm filter. A volume of 3 ml of each viral suspension was placed on the top of a layer of sucrose and ultracentrifuged at 40,000 rpm for 2 h. The resulting virus particle layer was recovered, suspended in DMEM, and stored at -80 °C. Primary cultures of chicken embryo fibroblasts (CEFs) were prepared according to standard protocols [21].
A volume of 100 μl of purified virus suspension was used to inoculate the cells, which were then incubated at 37 °C for 10 days. The cells were harvested after the appearance of a cytopathic effect (CPE) and lysed by two cycles of freezing and thawing.
DNA extraction and detection of the viral genome by PCR
Extraction of viral DNA from the internal organs was performed using an EasyPure Genomic DNA Kit (TransGen Biotech) according to the manufacturer's instructions. The highly conserved MDV-1-A antigen region of the glycoprotein C gene was targeted using specific primers described by Zhu et al. [22]. Real-time amplification reactions were carried out using a Swift Spectrum 48 Thermal Cycler (ESCO) and using KAPA SYBR FAST qPCR Master Mix using the following reaction conditions: incubation for 3 min at 95 °C to activate the enzyme, followed by 40 cycles of 3 s at 95 °C and 1 min at 60 °C. Extracted DNA from the CVI 988 Rispens vaccine and a negative control were included in each experiment. The extracted DNA was also tested for avian leukosis virus subgroups J and K, using real-time PCR to confirm specificity [23, 24].
Virus pathotyping by conventional PCR
DNA extracted from the tissues was amplified using primer pairs described by Tian et al. [4], Becker et al. [25], and Hassanin et al. [26]. for the Meq, vIL-8, pp38, UL1, and UL44 genes and the 132-bp tandem repeat regions, generating fragments of 1094, 830, 1006, 576, 1506 and 434 bp, respectively. The reaction mixtures were optimized using a KAPA Taq PCR kit with 2 μl (0.2 μM) of each of the two primers for each gene, 5 μl of 10 × KAPA Taq buffer, 1 μl of dNTP mix (0.2 mM), 0.2 μl of Taq polymerase (0.02 U/μl), 2 μl of DNA extract, and nuclease-free water in a final volume of 50 μl. Amplification was carried out in a Bio-Rad T100 thermal cycler, using the following reaction steps: denaturation for 4 min at 95 °C, followed by 35 cycles consisting of a second denaturation for 1 min at 94 °C, hybridization of the primer pairs for Meq, vIL-8, pp38, 132-bp tandem repeat, UL1, and UL44 for 1 min at 59, 56, 60, 52, 55 and 55 °C, respectively, and elongation for 1 min/kb at 72 °C, followed by a final elongation for 10 min at 72 °C. The amplified products were separated by electrophoresis on a 2% agarose gel containing 0.5 mg of ethidium bromide (BET, Sigma) per ml, visualized using a Gel Doc 2000 system (Bio-Rad), and used for sequencing.
Sequencing and phylogenetic analysis
To analyse the Meq, v-IL8, pp38, and UL1 and UL44 genes and the 132-bp tandem repeat of MDV, PCR products were purified using a Gel and PCR Extraction Kit (BioBASIC) and sequenced using an ABI PRISM 3500 Genetic Analyzer (Applied Biosystems). A multiple alignment of the sequences of the Tunisian isolates with reference sequences downloaded from the GenBank database was performed using the programs Bioedit (version 7.2.5.0) and ClustalW. Phylogenetic analysis was done by the maximum-likelihood method in MEGA6 (Version 6) and evaluated statistically by analyzing 100 bootstrap replications. The genome sequences of the GaHV-2 isolates were submitted to the GenBank database and assigned the accession numbers KY113150 and MK041219 for Meq, KY113151 and MK058702 for vIL8, KY113152 and MK058701 for the 132-bp tandem repeat, MN128713 and MN128714 for pp38, MN018231 and MN030639 for UL1, and MN480312 and MN480313 for UL44 of TN1013/16 and TN1014/16, respectively.
Results
Virological analysis
Isolation and identification of viruses
After two passages of organ suspensions in chicken embryo fibroblasts, a cytopathic effect was observed, with refractive rounded cells clustering together, as is typical for Marek's disease virus. During the third passage, a progressive development of CPE in the form of rounded cells was observed. In the fourth passage, clear CPE was observed as early as day 4 post-inoculation in the form of aggregated rounded cells with characteristic cytoplasmic extensions. No detached cells were observed during the four passages. Virus titration of both isolates, performed in a Vero cell culture in a 96-well microtiter plate with flat-bottomed wells, resulted in TCID50 titers of 106.30/ml, and 105.4/ml for TN1013/16 and TN1014/16, respectively.
Macroscopic lesions in embryos
All chicken embryos that were infected with 0.2 ml of GaHV-2 at a concentration of 105 TCID50/ml died on day 9, and necropsy revealed macroscopic lesions showing atrophy, liver pallor, and leg malformations. In addition, small white vesicles were observed on the allantoic membrane.
Molecular analysis
Detection of the viral genome
The results of SYBR Green real-time PCR amplification, carried out on organ suspensions, revealed a single type of amplicon for both samples with a specific melting temperature of 82.4 °C, confirming the presence of the GaHV-2 genome.
Sequence alignment and phylogeny of the Meq gene
Complete nucleotide sequences of Meq oncogene as well as their encoded protein sequences were analyzed. The Meq gene sequences of the isolates TN1013/16 and TN1014/16 were 100% identical. The sequences of Meq gene of various strains having different degrees of attenuation (att), including the currently used vaccine strain (CVI988), and moderately virulent (mv), virulent (v), very virulent (vv) and very virulent plus (vv +) strains from different countries, were selected and included in this analysis. Alignment of nucleotide sequences of all these strains showed that the Meq oncogene sequences of the Tunisian isolates shared the highest percentage of sequence identity with vv and vv + strains from Italy (100%) and China (99% and 99.6%, respectively). The lowest sequence similarity was observed with strains from the USA and Australia, with 98.8% to 99.4% and 84.4% to 84.6% identity, respectively.
Moreover, the Meq oncogene sequences of both TN1013/16 and TN1014/16 shared 98.8–99%, 84.4–99.4%, 84.5–99.3%, and 84.3–94.6% identity with the vv + , vv, v, and attenuated vaccine strains, respectively. An alignment of the predicted amino acid sequences indicated that the proteins of both Tunisian isolates showed the lowest percentage of identity (83%) to the current vaccine strain CVI988 (DQ534538) and were 100% identical to that of the Italian strain GaHV-2/Italy/Ck/674/16 (MK139667) and 99.8% identical to those of the highly virulent Chinese strains GXY2 (EF546430), YLO (DQ174459), and GX070060 (EU427303).
An exhaustive comparison of the nucleotide sequences of Meq oncogenes of the Tunisian isolates with those of the reference strains in the GenBank database, showed the presence of the substitutions T > C and G > A at positions 328 and 811, respectively, which are specific to the Tunisian and the Italian isolates.
The protein encoded by the Meq gene was found to be 339 amino acids long in most virulent strains, except for the Australian strains, 398 amino acids long in the attenuated and v and vv Australian strains, and 399 amino acids long in the vaccine strain CVI988. The TN1013/16 and TN1014/16 isolates both contained arginine instead of cysteine at position 110 and glycine at position 271.
A phylogenetic analysis based on the nucleotide sequences of the Meq gene, using the maximum-likelihood algorithm, is presented in Fig. 1. The phylogenetic tree showed that the GaHV-2 isolates are divided into two heterogeneous clusters. Cluster 1 is composed of two branches, the first of which is composed of strains with different levels of virulence and from different geographic locations and includes attenuated strains such as strain 3004 from Russia and strain CVI988 from the Netherlands, the mv-like CU-2 from the USA, and the v-like 04CRE and vv-like 02LAR from Australia. The second branch is composed of vv and vv + American and Colombian strains.
Fig. 1.

Phylogenetic tree of GaHV-2 based on the Meq gene. The tree was constructed using maximum-likelihood algorithm implemented in MEGA 6.0.6 with 1000 bootstrap replicates. Tunisian isolates are indicated by red diamonds
The second cluster is composed of two branches, the first of which includes the Tunisian and Italian strains, and the second of which includes Egyptian and vv + Chinese strains.
Pathotyping based on the sequence of Meq protein
Molecular determination of the pathotype of GaHV-2 strains is typically based on the sequence of the Meq protein, in particular, the number of proline repeat (PPPP) motifs. The v, vv and vv + strains are characterized by the presence of two to five repeats [27]. The v strains have five repeats (04CRE [EF523773], MPF57 [EF523774]); the vv strains may also have five (RB1B [AY243332], 02LAR [EF523772], Woodlands1 [EF523775]) or four (Md5 [AF243438] and GaHV-2/Italy/Ck/674/16 [MK139667]), and the vv + strains may have three (GXY2 [EF546430], YLO40920 [DQ174459], GX070060 [EU427303], 0297 [AF493553]) or even two PPPP motifs (686 [AY362715], 686A [AY362725], GaHV-2/Italy/Ck/855/17 [MK139678]).
The TN1013/16 and TN1014/16 isolates have four PPPP motifs, like the majority of the vv strains, such as Md5. This suggests that the Tunisian isolates are probably vv strains.
A low proline percentage has been shown to correlate with high virulence [27]. We therefore calculated the proline percentage of the Tunisian isolates and compared them to the reference strains of different pathotypes (Table 2). The Meq protein of isolates TN1013/16 and TN1014/16 was composed of only 21.18% proline, which represents a relatively small percentage compared to that of mv and v strains, and even with the majority of vv strains. This reinforces the hypothesis that these isolates are hypervirulent vv or vv + .
Table 2.
Number of PPPP signatures and proline percentage in different GaHV-2 pathotypes
| Isolate | Country | Accession number | Size of Meq (aa) | Proline % | Number of PPPP repeats | Pathotype |
|---|---|---|---|---|---|---|
| CVI988 | Netherlands | DQ534538 | 399 | 23.3 | 8 | AttMDV |
| JM/102 W | United States | DQ534539 | 399 | 23.1 | 6 | vMDV |
| MPF57 | Australia | EF523771 | 398 | 22.9 | 5 | vMDV |
| RB1B | United States | AY243332 | 339 | 21.5 | 5 | vvMDV |
| Md5 | United States | AF243438 | 339 | 21.3 | 4 | vvMDV |
| TN1013/16 | Tunisia | MK041219 | 339 | 21.18 | 4 | vvMDV |
| TN1014/16 | Tunisia | KY113150 | 339 | 21.18 | 4 | vvMDV |
| 648A | United States | AY362725 | 339 | 20.9 | 2 | vv + MDV |
| GaHV-2/Italy/Ck/855/17 | Italy | MK139678 | 298 | 19.4 | 2 | – |
| GaHV-2/Italy/Ck/674/16 | Italy | MK139667 | 339 | 21.18 | 4 | – |
The length of the Meq proteins of the TN1013/16 and TN1014/16 isolates was compared to those of reference strains of known pathotypes (Table 2). All of the hypervirulent strains that were analyzed were found to have a Meq protein of 339 aa. This was the case for the TN1013/16 and TN1014/16 isolates, again suggesting that these isolates are pathogenic.
Pathotyping based on the 132-bp tandem repeat region
The 132-bp tandem repeat region, present in the BamHI-H segment of the viral genome, may be present one or more copies in MDV strains. The number of repetitions of one sequence is an indicator of the pathogenicity of the virus [25]. The electrophoretic profiles of strains from this study showed amplification of a 434-bp fragment from both TN1013/16 and TN1014/16, which can be explained by the presence of two copies of the 132-bp tandem repeat sequence, thus confirming their pathogenicity and suggesting their high virulence. On the other hand, the attenuated vaccine strain (CVI988 Rispens) showed an electrophoretic profile with three bands of 434, 830 and 1094 bp, indicating the presence of two, five and seven copies, respectively, of the 132-bp tandem repeat in the genome of the vaccine strain (Fig. 2).
Fig. 2.
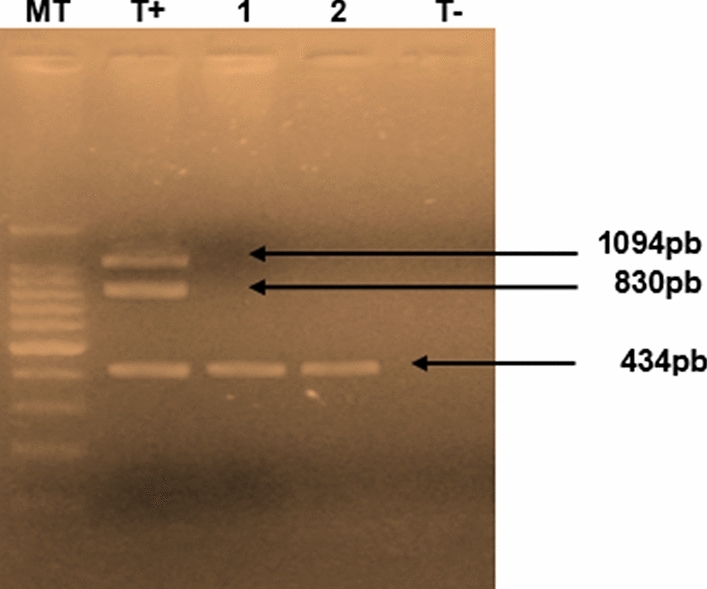
Amplification profile of the 132-bp tandem repeat on a 1.5% agarose gel. MT, 100-bp size marker; T + , vaccine (CVI988 Rispens); 1, sample TN1013/16; 2, sample TN1014/16; T-, negative control
Sequence comparisons and phylogeny of the VIL-8, pp38, UL1 and UL44 genes
Sequencing of the amplified products of each field isolate yielded sequences of 682, 1006, 585 and 1506 bp, corresponding to the complete size of the vIl-8, pp38, UL1 and UL44 genes, respectively. These gene sequences were 100% identical in TN1013/16 and TN1014/16.
Multiple alignments of amino acid sequences showed variability at positions 4 and 31 of the vIL-8 protein. The v and vv strains BY (HQ638179), LS (HQ638183), SD2012 (KC511812), MS01 (HQ638184) and YA (HQ638196) have a serine residue at position 4, while the Tunisian and other strains, including the v strain GA, vv and vv + strains 584a (DQ534532) and 648a (AF489277), have a leucine residue at this position. The Tunisian isolates and the vv Chinese and American strains J1 (HQ638182), Md5 (AF489275), 595 (DQ534533), RB1B (EF523390) and vv + strains 584a (DQ534532) and 648a (JQ809692) have aspartic acid residue at position 31. However, the v strains LMS (JQ314003), LS (HQ638183), Ind/TN12/02 (KP644422) and Ind/TN11/01(KP644421) and the vv strain Ind/KA12/02 (KP644422) have a glycine at the same position. Thus, the four amino acids at positions 4 and 31 do not appear to determine the degree of virulence of the strain.
Analysis of the pp38 gene showed that the Tunisian isolates as well as the majority of the other strains, regardless of their degree of virulence, have a glycine at position 109. Strains with a glutamate at this position have different degrees of virulence. A nucleotide sequence alignment of the pp38 genes of the Tunisian isolates and the reference strains showed 100% identity to the Chinese v strain SD2012 (KC511813) and mv strain CU-2 (EU499381) from the USA.
Glycoprotein L exhibited a high degree of sequence stability, and its deduced amino acid sequence was 97.9% to 100% identical to those of reference strains from other countries. Our strains did not show the deletion of 12 nucleotides in the cleavage site at positions 54–65, that were reported previously for Japanese (Gifu 2 (LC208808) and American TK (AY129998) and 06-L (AY130000) strains.
A phylogenetic analysis based on the UL1 gene also showed that the isolates were divided into two distinct phylogenetic groups. The first group included the Tunisian isolates TN1013/16 and TN1014/16 as well as all other strains with different degree of virulence from different countries, while the second branch included vv and vv + strains from the United States (TK and L) and Japan (Gifu2) with a deletion of 12 nucleotides.
The UL44 gene was found to be 100% identical to those of the Chinese vv strain LMS and the American strain Md5. The UL44 gene displayed high sequence conservation, regardless of virulence and was the most conserved among sequenced genes in this study.
A phylogenetic tree based on the nucleotide sequences of the vIL-8, pp38, UL1 and UL44 genes showed that TN1013/16 and TN1014/16 both belong to a heterogenous group, with different levels of virulence.
Discussion
In recent years, numerous isolations of vv and vv + MDV strains from vaccinated chickens have been reported, and very common forms of proliferative lymphatic diseases have been described [20]. The virulence of MDV isolates has increased in recent decades, and some recently isolated vv and vv + strains have been shown to be more pathogenic for chickens than the older ones [28]. Recombination events among MDV strains could be among the mechanisms by which virulence increases [29]. It is therefore justified to be concerned about new emerging strains that can break vaccine protection.
Some GaHV-2-specific genes, including Meq, pp38, UL1, UL44 and vIL-8, contain sequence differences that are associated with oncogenicity, viral pathogenicity and virulence [26, 30]. In this study, to better understand the evolution of Tunisian isolates, we studied the pathotypes of MDV strains circulating on farms based on the Meq, vIL-8, pp38, UL1 and UL44 genes.
The Meq gene has been studied extensively and appears to correlate with virulence [3]. Indeed, it has been reported that a change of serine to alanine at position 71 is a characteristic of hypervirulent strains of MDV [3]. This has been observed in the Chinese vv isolate SD2012-1 (KC511815) and vv + isolate JZ2014 (KP144355), the American isolates N (AY362718), W (AY362723), 584A (DQ534532), L (AY362717) and X (AY362724), and the Tunisian isolates TN1013/16 (MK041219) and TN1014/16 (KY113150). However, some vv strains, such as 02 LAR (EF523772) and Woodlands1 (EF523775), have a serine at this position, casting doubt on whether the presence of this mutation is a reliable criterion for classifying MDV pathotypes.
The substitution of glutamate by leucine at position 77 has been reported by Shamblin et al. [3]. to be a feature of hypervirulent strains. However, isolate JZ2014 (KP144355), the Colombian isolates UDEACO04 (KU058701) and SD2012-1 (KC511815), and the Tunisian isolatesTN1013/16 (MK041219) and TN1014/16 (KY113150), have glutamic acid at position 77 but are classified as vv and vv + strains. This suggests that the presence of glutamic acid at position 77 is not necessarily a characteristic of low-virulence MDV strains. On the other hand, the presence of the amino acids R119, Q153 and A176, has been shown to be a common feature of the American vv isolates 549 (AY362714) and 595 (AY362715) and the American vv + isolates N, L, X, 584A, and 684A (AY362725), in contrast to a report by Tian et al. [4], who suggested that these mutations are unique in vv + strains. However, the Tunisian isolates share R119, and P176 as well as P153.
MDV strains with high virulence have been shown to contain mutations at the second position of the proline-rich region (PRR): PPPP > P (Q / A / R) PP [3]. Attenuated strains have more PPPP motifs, whereas most virulent strains have more interrupted motifs. Analysis of the Meq protein amino acid sequences of theTN1013/16 and TN1014/16 isolates revealed a point mutation in the proline-rich region at position 217, interrupting the PPPP motif. A variable number of PPPP motifs in the central PRR of the Meq protein was observed in low-virulence strains, and in fact, eight repeats were detected in the vaccine strain CVI988, and nine and 10 in the Italian strains GaHV-2/Italy/Ck/847/15 and GaHV-2/Italy/Ck/847/17, respectively [31]. In contrast, hypervirulent strains have low number of these motifs, and the smallest number was observed in the vv + strain 648A, with only two motifs. The hypervirulent vv and vv + strains contained interruptions in the proline repeats at the second position (P > Q153, P > A176 and P > A276), and the largest number of such interruptions was observed in the most virulent strains [32]. Both TN1013/16 and TN1014/16 contained only the interruption at position 217 that is present in all virulent strains except RB1B.
On the other hand, Wajid et al. [33]. have suggested a correlation between the proline content and the virulence of emerging strains. Analysis of reference strains showed that this parameter is inversely proportional to virulence. In this case, this percentage is around 21.18% in the Tunisian isolates, reinforcing the hypothesis that they are very virulent.
Mutations at positions 110 and 271 appear to be characteristic of Tunisian and Italian isolates [31]. Such mutations and their overall sequence similarity might suggest that migratory birds such as ducks and white footed geese play a role in transporting the virus from one country to another [34]. The genetic divergence of MDV strains in the Meq gene is more obvious than in the vIL-8, pp38, UL1 and UL44 genes, making the Meq gene most appropriate for phylogenetic analysis.
In addition, the vIL-8 gene, located in the long repeat (RL) region, was initially identified as a spliced variant of the Meq gene [35]. It is highly conserved in all strains but with some variability at sites 4 and 31 of the vIL-8 protein. However, these mutations are not characteristic of virulent strains.
As reported by Shamblin et al. [3], we did find some mutations in the UL1 and UL44 genes that strictly correlate with the virulence level. These mutations map to the putative signal cleavage site of the UL1 genes, and are found in four out of 11 vv + MDVs, but also in one vvMDV (643P), indicating that it does not correlate with enhanced virulence.
Although several early genes have been shown to be essential for viral replication, and thus for initiating the transformation pathway and subsequent development of clinical signs of MD, the Meq gene is considered to be the primary oncogene of MDV, with other genes serving as auxiliary factors [10]. Similarly, although the Meq gene is mainly expressed during the latent state, studies have indicated that the Meq gene can also be expressed early in infection [10], suggesting that variations in the Meq gene sequence can have a significant influence on virulence. However, previous gene sequencing studies have not revealed mutations in other genes that are consistently correlated with virulence [3].
Our findings showed that there is significant polymorphism in the MDV Meq gene, which is a key gene in the induction of lymphoid tumors. The Tunisian isolates had two point mutations in the Meq gene that might be associated with virulence. In addition, the overall proline content and pattern of PPPP repeats showed strong correlation with the virulent pathotype. Although the virulence of a specific isolate is unlikely to be determined solely by mutations in a single gene, the results of our work suggest that Meq gene sequencing and protein analysis can provide a useful indication of the virulence of these isolates. However, confirmation of the pathotype requires in vivo experiments.
The CVI988 vaccine is widely used around the world. In China, it has been noted that immune failure tends to occur in only a few chickens. A number of vv strains have been isolated from different parts of China [36], raising the question whether the currently available vaccines are able to protect chickens against these highly virulent strains.
The weak protective effect of MDV vaccines has led to MD outbreaks [16], and the emergence of MDV strains with increasing virulence is therefore an important issue for the poultry industry.
Conclusion
Our results have made it possible, for the first time, to be aware of the presence of highly virulent strains circulating on Tunisian farms. Variations in the Meq protein sequence, the composition and the number of PPPP motifs present in the PRR region, and the size of the Meq protein can serve as indicators for determining the pathotype of MDV isolates. Simple sequencing of the Meq gene can be used to identify any changes in the virus pathotype and consequently to prevent a potential ??epidemic?? following the emergence of new more virulent pathotype.
In addition, we found two mutations in the Tunisian isolates that had not been observed in any other strain with sequences in the GenBank database until 2019, when Mescolini et al. [31] found the same mutations. In addition, the presence of two copies of the 132-bp tandem repeat segment has made it possible to characterize these strains and confirm their pathogenicity. This will be useful for developing rapid diagnostic methods for detection and assessment of the pathogenicity of circulating MDV strains.
Acknowledgements
This study was supported by a grant from the Ministry of Higher Education and Scientific Research of Tunisia (no. 18PJEC07-02).
Author contributions
JL contributed to the study design and interpretation of the results with HM and wrote the manuscript. HM and FA contributed to isolation, detection and amplification of gene sequences. KK, IC and AA contributed to various aspects of sample collection. JN and JL performed the phylogenetic analyses; AG revised and approved the manuscript.
Data availability
Data and materials from the current study are available from the corresponding author on reasonable request.
Compliance with ethical standards
Conflict of interest
The authors declare that they are no competing interests.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gimeno IM, Cortes AL, Reddy SM, de Juan L, Abad B, Käser T, Limsatanun A. Highly virulent Marek's disease virus strains affect T lymphocyte function and viability of splenocytes in commercial meat type chickens. Avian Pathol. 2019 doi: 10.1080/03079457.2019.1643451. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy DA, Cairns C, Jones MJ, Bell AS, Salathé RM, Baigent SJ, Nair VK, Dunn PA, Read AF. Industry-wide surveillance of Marek's disease virus on commercial poultry farms. Avian Dis. 2017 doi: 10.1637/11525-110216-Reg.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shamblin CE, Greene N, Arumugaswami V, Dienglewicz RL, Parcells MS. Comparative analysis of Marek’s disease virus (MDV) glycoprotein-, lytic antigen pp38-and transformation antigen Meq-encoding genes: association of meq mutations with MDVs of high virulence. Vet Microbiol. 2004 doi: 10.1016/j.vetmic.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Tian M, Zhao Y, Lin Y, Zou N, Liu C, Liu P, Cao S, Wen X, Huang Y. Comparative analysis of oncogenic genes revealed unique evolutionary features of field Marek's disease virus prevalent in recent years in China. Virol J. 2011 doi: 10.1186/1743-422X-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui X, Lee LF, Hunt HD, Reed WM, Lupiani B, Reddy SM. A Marek's disease virus vIL-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 2005 doi: 10.1637/7277-091004. [DOI] [PubMed] [Google Scholar]
- 6.Shaikh SA, Katneni UK, Dong H, Gaddamanugu S, Tavlarides-Hontz P, Jarosinski KW, Osterrieder N, Parcells MSA. Deletion in the glycoprotein L (gL) gene of US Marek's disease virus (MDV) field strains is insufficient to confer increased pathogenicity to the bacterial artificial chromosome (BAC)-based strain, RB-1B. Avian Dis. 2013 doi: 10.1637/10450-112012-Reg.1. [DOI] [PubMed] [Google Scholar]
- 7.Wilson MR, Southwick RA, Pulaski JT, Tieber VL, Hong Y, Coussens PM. Molecular analysis of the glycoprotein C-negative phenotype of attenuated Marek's disease virus. Virology. 1994 doi: 10.1006/viro.1994.1137. [DOI] [PubMed] [Google Scholar]
- 8.Lee SI, Takagi M, Ohashi K, Sugimoto C, Onuma M. Difference in the Meqgene between oncogenic and attenuated strains of Marek's disease virus serotype 1. J Vet Med Sci. 2000 doi: 10.1292/jvms.62.287. [DOI] [PubMed] [Google Scholar]
- 9.Lupiani B, Lee LF, Cui X, Gimeno I, Anderson A, Morgan RW, Reddy SM. Marek's disease virus-encoded Meqgene is involved in transformation of lymphocytes but is dispensable for replication. Proc Natl Acad Sci USA. 2004 doi: 10.1073/pnas.0404508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen XB, Sondermeijer PJ, Velicer LF. Identification of a unique Marek's disease virus gene which encodes a 38-kilodalton phosphoprotein and is expressed in both lytically infected cells and latently infected lymphoblastoid tumor cells. J Virol. 1992;66:85–94. doi: 10.1128/JVI.66.1.85-94.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy SM, Lupiani B, Gimeno IM, Silva RF, Lee LF, Witter RL. Rescue of a pathogenic Marek's disease virus with overlapping cosmid DNAs: use of a pp38 mutant to validate the technology for the study of gene function. Proc Natl Acad Sci USA. 2002 doi: 10.1073/pnas.092152699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel AT, Selvaraj RK, Kamil JP, Osterrieder N, Kaufer BB. Marek's disease viral interleukin-8 promotes lymphoma formation through targeted recruitment of B cells and CD4+ CD25+ T cells. J Virol. 2012 doi: 10.1128/JVI.00556-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu P, Reed WM, Lee LF. Glycoproteins H and L of Marek's disease virus form a hetero-oligomer essential for translocation and cell surface expression. Arch Virol. 2001;146:983–992. doi: 10.1007/s007050170130. [DOI] [PubMed] [Google Scholar]
- 14.Markowski-Grimsrud CJ, Schat KA. Cytotoxic T lymphocyte responses to Marek's disease herpesvirus-encoded glycoproteins. Vet Immunol Immunopathol. 2002;90:133–144. doi: 10.1016/s0165-2427(02)00229-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu C, Yan F, Liu A, Cheng Y, Li Z, Sun G, Lv H, Wang X. Recombinant Gallid herpesvirus 2 with interrupted Meqgenes confers safe and efficacious protection against virulent field strains. Vaccine. 2017 doi: 10.1016/j.vaccine.2017.07.048. [DOI] [PubMed] [Google Scholar]
- 16.Gong Z, Zhang L, Wang J, Chen L, Shan H, Wang Z, Ma H. Isolation and analysis of a very virulent Marek's disease virus strain in China. Virol J. 2013 doi: 10.1186/1743-422X-10-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Padhi A, Parcells MS. Positive selection drives rapid evolution of the meq oncogene of Marek's disease virus. PLoS ONE. 2016 doi: 10.1371/journal.pone.0162180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun GR, Zhang YP, Lv HC, Zhou LY, Cui HY, Gao YL, Qi XL, Wang YQ, Li K, Gao L, Pan Q, Wang XM, Liu CJ. A Chinese variant Marek's disease virus strain with divergence between virulence and vaccine resistance. Viruses. 2017 doi: 10.3390/v9040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ralapanawe S, Walkden-Brown SW, Renz KG, Islam AF. Protection provided by Rispens CVI988 vaccine against Marek's disease virus isolates of different pathotypes and early prediction of vaccine take and MD outcome. Avian Pathol. 2016 doi: 10.1080/03079457.2015.1110850. [DOI] [PubMed] [Google Scholar]
- 20.Gimeno IM. Marek's disease vaccines: a solution for today but a worry for tomorrow? Vaccine. 2008 doi: 10.1016/j.vaccine.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez R, Brown DT. Growth and maintenance of chick embryo fibroblasts (CEF) Curr Protoc Microbiol. 2010 doi: 10.1002/9780471729259.mca04is17. [DOI] [PubMed] [Google Scholar]
- 22.Zhu GS, Ojima T, Hironaka T, Ihara T, Mizukoshi N, Kato A, Ueda S, Hirai K. Differentiation of oncogenic and nononcogenic strains of Marek’s disease virus type 1 by using polymerase chain reaction DNA amplification. Avian Dis. 1992;36:637–645. doi: 10.2307/1591759. [DOI] [PubMed] [Google Scholar]
- 23.Qin L, Gao Y, Ni W, Sun M, Wang Y, Yin C, Qi X, Gao H, Wang X. Development and application of real-time PCR for detection of subgroup J avian leukosis virus. J Clin Microbiol. 2013 doi: 10.1128/JCM.02030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Zhao Z, Chen Y, Zhang J, Yan L, Zheng X, Liao M, Cao W. Development and application of a SYBR green real-time PCR for detection of the emerging avian leukosis virus subgroup K. Poult Sci. 2018 doi: 10.3382/ps/pey086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Becker Y, Tabor E, Asher Y, Davidson I, Malkinson M, Witter RL. PCR detection of amplified 132 bp repeats in Marek's disease virus type 1 (MDV-1) DNA can serve as an indicator for critical genomic rearrangement leading to the attenuation of virus virulence. Virus Genes. 1993 doi: 10.1007/BF01702588. [DOI] [PubMed] [Google Scholar]
- 26.Hassanin O, Abdallah F, El-Araby IE. Molecular characterization and phylogenetic analysis of Marek's disease virus from clinical cases of Marek's disease in Egypt. Avian Dis. 2013 doi: 10.1637/10337-082912-Reg.1. [DOI] [PubMed] [Google Scholar]
- 27.Renz KG, Cooke J, Clarke N, Cheetham BF, Hussain Z, Fakhrul Islam AF, Tannock GA, Walkden-Brown SW. Pathotyping of Australian isolates of Marek's disease virus and association of pathogenicity with meq gene polymorphism. Avian Pathol. 2012 doi: 10.1080/03079457.2012.656077. [DOI] [PubMed] [Google Scholar]
- 28.Witter RL. Increased virulence of Marek's disease virus field isolates. Avian Dis. 1997;41:149–163. doi: 10.2307/1592455. [DOI] [PubMed] [Google Scholar]
- 29.He L, Li J, Zhang Y, Luo J, Cao Y, Xue C. Phylogenetic and molecular epidemiological studies reveal evidence of recombination among Marek's disease viruses. Virology. 2018 doi: 10.1016/j.virol.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 30.Nair VK, Kung HJ. Marek's disease virus oncogenicity: molecular mechanisms. Biol Anim Infect. 2004 doi: 10.1016/B978-012088379-0/50008-6. [DOI] [Google Scholar]
- 31.Mescolini G, Lupini C, Felice V, Guerrini A, Silveira F, Cecchinato M, Catelli E. Molecular characterization of the meq gene of Marek's disease viruses detected in unvaccinated backyard chickens reveals the circulation of low- and high-virulence strains. Poult Sci. 2019 doi: 10.3382/ps/pez095. [DOI] [PubMed] [Google Scholar]
- 32.López-Osorio S, Piedrahita D, Espinal-Restrepo MA, Ramírez-Nieto GC, Nair V, Williams SM, Baigent S, Ventura-Polite C, Aranzazu-Taborda DA, Chaparro-Gutiérrez JJ. Molecular characterization of Marek's disease virus in a poultry layer farm from Colombia. Poult Sci. 2017 doi: 10.3382/ps/pew464. [DOI] [PubMed] [Google Scholar]
- 33.Wajid SJ, Katz ME, Renz KG, Walkden-Brown SW. Prevalence of Marek's disease virus in different chicken populations in Iraq and indicative virulence based on sequence variation in the EcoRI-Q (meq) gene. Avian Dis. 2013 doi: 10.1637/10342-083112-Reg.1. [DOI] [PubMed] [Google Scholar]
- 34.Boodhoo N, Gurung A, Sharif S, Behboudi S. Marek's disease in chickens: a review with focus on immunology. Vet Res. 2016;47:119. doi: 10.1186/s13567-016-0404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng F, Bradley G, Tanaka A, Lancz G, Nonoyama M. Isolation and characterization of cDNAs from BamHI-H gene family RNAs associated with the tumorigenicity of Marek's disease virus. J Virol. 1992;66:7389–7396. doi: 10.1128/JVI.66.12.7389-7396.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teng LQ, Wei P, Song ZB, He JJ, Cui ZZ. Molecular epidemiological investigation of Marek’s disease virus from Guangxi, China. Arch Virol. 2011 doi: 10.1007/s00705-010-0840-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and materials from the current study are available from the corresponding author on reasonable request.