

Abstract
Environmental health studies relate how exposures (eg, chemicals) affect human health and disease; however, in most cases, the molecular and biological mechanisms connecting an exposure with a disease remain unknown. To help fill in these knowledge gaps, we sought to leverage content from the public Comparative Toxicogenomics Database (CTD) to identify potential intermediary steps. In a proof-of-concept study, we systematically compute the genes, molecular mechanisms, and biological events for the environmental health association linking air pollution toxicants with 2 cardiovascular diseases (myocardial infarction and hypertension) as a test case. Our approach integrates 5 types of curated interactions in CTD to build sets of “CGPD-tetramers,” computationally constructed information blocks relating a Chemical- Gene interaction with a Phenotype and Disease. This bioinformatics strategy generates 653 CGPD-tetramers for air pollution-associated myocardial infarction (involving 5 pollutants, 58 genes, and 117 phenotypes) and 701 CGPD-tetramers for air pollution-associated hypertension (involving 3 pollutants, 96 genes, and 142 phenotypes). Collectively, we identify 19 genes and 96 phenotypes shared between these 2 air pollutant-induced outcomes, and suggest important roles for oxidative stress, inflammation, immune responses, cell death, and circulatory system processes. Moreover, CGPD-tetramers can be assembled into extensive chemical-induced disease pathways involving multiple gene products and sequential biological events, and many of these computed intermediary steps are validated in the literature. Our method does not require a priori knowledge of the toxicant, interacting gene, or biological system, and can be used to analyze any environmental chemical-induced disease curated within the public CTD framework. This bioinformatics strategy links and interrelates chemicals, genes, phenotypes, and diseases to fill in knowledge gaps for environmental health studies, as demonstrated for air pollution-associated cardiovascular disease, but can be adapted by researchers for any environmentally influenced disease-of-interest.
Keywords: environmental health, chemical-induced pathways, air pollution, cardiovascular disease, database
An important facet of environmental health explores how chemical stressors affect human disease (Grondin et al., 2016). Although associations between chemical exposures and health outcomes can be documented, there typically remain knowledge gaps about the specific molecular, genetic, and biological mechanisms involved (Birnbaum et al., 2016). Resolving these key intermediary steps should aid in the design of experimental and epidemiological projects as well as the development of molecular tools. Here, using air pollution-induced cardiovascular disease as a test case, we present a bioinformatics method to computationally identify potential steps to help fill in the knowledge gaps.
The toxic effects of air pollutants have been recognized and studied for over 70 years (Costa, 2018) and are estimated to cause more than 4.2 million deaths annually (Cohen et al., 2017). The association between ambient air pollution and cardiovascular mortality rates has been well documented (Dockery et al., 1993; Hadley et al., 2018; Yorifuji et al., 2016; Zhang et al., 2011); however, it is important to resolve the genetic, molecular, and biological mechanisms involved in this complex environmental disease. These in-between steps elucidate the toxicant’s mode-of-action and include activated gene products and phenotypes preceding clinical disease manifestation. In this report, we describe a bioinformatics methodology that condenses and defines this sequence of events (Chemical > Gene > Phenotype > Disease) as a CGPD-tetramer, a reductionist unit of computed information describing a Chemical interaction with a Gene product, which in turn induces a nondisease Phenotype that ultimately can be associated with a Disease. Our strategy integrates high-quality curated content to rapidly generate CGPD-tetramers. We show how these computed tetramers can be used to help inform and identify potential intermediary steps relating ambient air pollutants with myocardial infarction (MI) and hypertension (HT), 2 cardiovascular outcomes associated with air pollution (Rajagopalan et al., 2018).
A CGPD-tetramer includes 4 components (chemical, gene, phenotype, and disease), and extensive, curated information for each of these individual components is available in numerous public repositories; adequately accessing, retrieving, and integrating this composite public data, however, is currently hindered by the necessity of processing different types of vocabularies used by disparate resources (Davis et al., 2019b). To obviate the complex step of querying and mapping heterogeneous data and identifiers across multiple systems, in this report we limit the source information to just 1 public repository, the Comparative Toxicogenomics Database (CTD; http://ctdbase.org/).
CTD is a free resource that provides users with contextualized data for environmental health studies by manually curating chemical-centric interactions from the literature using controlled vocabularies and ontologies to standardize and harmonize heterogeneous data from over 135 000 scientific articles (Davis et al., 2019a). Initially, CTD focused on chemical-gene, chemical-disease, and gene-disease interactions (Davis et al., 2008), but has evolved and extensively grown, developing new curation paradigms to capture chemical-exposure statements (Grondin et al., 2016) and, most recently, novel chemical-phenotype interactions (Davis et al., 2018). The use of controlled vocabularies provides internal consistency for term curation and enables seamless data integration (Davis et al., 2011), facilitating the ability to quickly interrelate chemicals, genes, phenotypes, and diseases to construct CGPD-tetramers. Currently, CTD contains more than 2.6 million manually curated interactions for 16 300 chemicals, 50 900 genes, 5400 phenotypes, and 7200 diseases (http://ctdbase.org/about/dataStatus.go).
Here, we describe a systematic process that leverages CTD content to compute CGPD-tetramers linking ambient air pollutants with unique genes and phenotypes associated with 2 cardiovascular diseases (MI and HT). More importantly, we show how these CGPD-tetramers can be analyzed to help inform the knowledge gaps for environmental health. First, as individual tetramers, component analysis identifies shared genes and phenotypes elicited by different toxicants and implicate oxidative stress, inflammation, immune responses, cell death, and circulatory system processes as important intermediary steps connecting air pollution exposure to cardiovascular disease. Second, assembling blocks of tetramers (via shared genes and phenotypes) generates an extended chemical-induced disease pathway, filling in gaps with detailed molecular, cellular, and system level processes for ozone-associated MI. We validate many of our computed results with reports from the scientific literature for air pollution-associated cardiovascular diseases.
Importantly, our described methodology does not require a priori knowledge of the toxicant, biological system, or adverse outcome, and can be easily adopted by users to study any environmental chemical-induced disease at CTD.
MATERIALS AND METHODS
Data version
Analysis was performed using CTD public data available in July 2019 (revision 15854). CTD is updated with new content on a monthly basis; consequently, counts described here may change over time.
Data collection
Data were extracted from CTD’s public web application in Excel format using the “Download” feature at the bottom of CTD webpages. All CTD curated content is freely available as structured data files for downloading and analysis by users (http://ctdbase.org/downloads/).
Web tools
CTD’s integrated online analytical tools (http://ctdbase.org/tools/) Batch Query, Set Analyzer, and MyVenn were used to analyze chemical, gene, phenotype, and disease datasets. For Batch Query, the direct relationships filter was used to return data for the exact input query terms. For Set Analyzer, gene lists were used as the input type and analyzed for enriched outputs (with the recommended corrected p value threshold .01) for pathways from integrated content from KEGG (Kanehisa et al., 2012) and Reactome (Fabregat et al., 2018). As well, Set Analyzer was used to retrieve common gene-gene interactions (controls set to “merged edges” and “tree layout”) to draw Pathway View maps with integrated data from BioGRID (Oughtred et al., 2019). MyVenn was used for all Venn analyses to identify shared genes and phenotypes.
Air pollutants
The World Health Organization identifies 6 ambient air pollutants with the strongest evidence for global human health effects: particulate matter, ozone, nitrogen dioxide, carbon monoxide, soot (black carbon), and sulfur dioxide (https://www.who.int/airpollution/ambient/pollutants/en/). To these 6 chemicals we added 2 additional CTD chemical terms (Air Pollutants and Vehicle Emissions) that are frequently used in scientific publications when the authors do not identify the specific ambient pollutants. In total, 8 air pollutants were initially analyzed (and their CTD accession identifier): Particulate Matter (MESH:D052638), Ozone (MESH:D010126), Nitrogen Dioxide (MESH:D009585), Carbon Monoxide (MESH:D002248), Soot (MESH:D053260), Sulfur Dioxide (MESH:D013458), Air Pollutants (MESH:D000393), and Vehicle Emissions (MESH:D001335).
Chemical terms were input into CTD’s Batch Query to retrieve curated pollutant-disease associations (direct evidence “molecular mechanism/marker”), and diseases were automatically clustered into MEDIC-Slim Disease Categories (Davis et al., 2012a). Cardiovascular and respiratory tract diseases were the most common adverse outcomes with curated associations for the 8 pollutants (Figure 1). We selected cardiovascular disease as a category to use as a case study to compute potential intermediary genes and biological mechanisms to fill in knowledge gaps between exposure and outcome.
Figure 1.

Ambient air pollutant-associated diseases in Comparative Toxicogenomics Database. The numbers of diseases with curated associations with 8 pollutants are clustered by disease categories (y-axis); only the top 7 disease categories are listed. The category “Pathology” includes pathological processes (eg, chromosome aberration, fibrosis, hemorrhage, and shock), and “Signs & Symptoms” includes clinical symptoms (eg, headache, sneezing, cough, and nausea). Chemicals: AP, Air Pollutants; CO, Carbon Monoxide; NO2, Nitrogen Dioxide; O3, Ozone; PM, Particulate Matter; SO2, Sulfur Dioxide; Soot; VE, Vehicle Emissions.
Cardiovascular diseases
Associated CTD data were collected for 2 cardiovascular diseases with a known association with air pollution (Rajagopalan et al., 2018): Myocardial Infarction (MESH: D009203) and Hypertension (MESH: D006973). Data associated with descendant terms were first removed for the 2 diseases. Because phenotype-disease inferred relationships do not discriminate between chemicals (or genes) that have a “molecular mechanism/marker” or “therapeutic” disease relationship, we combined both sets of data at the outset of analysis.
Phenotypes
At the time of analysis, 4918 unique phenotypes were associated with MI via an inferred relationship (http://ctdbase.org/detail.go?type=disease&acc=MESH%3aD009203&view=phenotype), and 6434 phenotypes were associated with HT (http://ctdbase.org/detail.go?type=disease&acc=MESH%3AD006973&view=phenotype). In CTD, “phenotypes” are operationally distinguished from “diseases” (Davis et al., 2018), wherein a phenotype refers to a nondisease biological event (eg, “cell cycle arrest” is a phenotype, while “liver neoplasms” is a disease; “decreased spermatogenesis” is a phenotype, while “male infertility” is a disease). Two independent controlled vocabularies are used to code this distinction in CTD. To capture disease information, biocurators use terms from the MEDIC disease vocabulary (Davis et al., 2012a), and if the reported outcome does not exist as a term in MEDIC, then, by definition, it is considered a phenotype. For phenotype curation, CTD biocurators use the Gene Ontology (GO) as a source for terms to code chemical-induced nondisease events from the literature (Davis et al., 2018). The GO is a well-known resource used to ascribe gene products with a molecular function, biological process, and cellular component (Ashburner et al., 2000). CTD also imports these gene-GO annotations from NCBI Gene (Brown et al., 2015) and displays them as annotations on all CTD gene pages. Thus, GO terms are used in 2 distinct and independent ways at CTD: as gene-GO annotations (imported from NCBI Gene) and as phenotype terms for the CTD manual curation of direct chemical-phenotype (GO) interactions from the literature. Phenotypes are associated with diseases by inferred relationships via sets of shared chemicals and/or genes (Davis et al., 2016). An inference between a phenotype and a disease is made when both a specific phenotype P1 and a disease D1 independently have a direct interaction with either the same chemical C1 or gene G1. Thus, if chemical C1 has a directly curated interaction with both P1 and independently with D1, then P1 has an inferred relationship to D1 (via the shared C1); similarly, if gene G1 is annotated to P1 (as a GO term by NCBI Gene) and independently has a curated interaction with D1, then P1 can be inferred to D1 (via the shared G1). The set of chemicals shared between P1 and D1 is called the Chemical Inference Network (CIN), whereas the set of genes shared between a phenotype and a disease forms the Gene Inference Network (GIN). All phenotypes with an inferred relationship to a disease are displayed under the “Phenotypes” data-tab on a disease page in CTD. As well, a new file providing the data for all phenotype-disease inferences is freely available to download from CTD (http://ctdbase.org/downloads/#phenotypediseases).
Computing CGPD-tetramers
CGPD-tetramers were derived from curated interactions in CTD, and details about CTD’s manual curation process to capture chemical-gene, chemical-disease, gene-disease, and chemical-phenotype interactions from the scientific literature have been extensively described (Davis et al., 2008, 2011, 2018). We compiled CGPD-tetramers using software written specifically for this task. First, phenotype-disease relationships and their associated CIN and GIN were identified by downloading the “Phenotypes” data-tab for MI and HT, independently, and phenotype-disease relationships were computationally limited to associations established based on the presence of both a CIN and a GIN. Next, CTD curated chemical-gene interactions were downloaded (http://ctdbase.org/downloads/#cg) and used as the source for direct chemical-gene interactions. Finally, to generate CGPD-tetramers, chemicals, and genes of the respective CIN and GIN for a specific phenotype-disease inference were processed against one another in an automated fashion to determine if they also had a curated chemical-gene interaction in CTD; if a directly curated relationship existed, then a tetramer was generated linking the respective chemical, gene, phenotype, and disease. As an example, the phenotype “platelet aggregation” (GO:0070527) was associated with the disease Myocardial Infarction (MESH:D009203) based on a CIN that included the 3 chemical terms Air Pollutants (MESH:D000393), Particulate Matter (MESH:D052638), and Vehicle Emissions (MESH:D001335) and a GIN that included the 2 genes ITGB3 (GENE:3690) and P2RY12 (GENE:64805), providing a total of 6 possible chemical-gene combinations to construct 6 CGPD-tetramers; however, when the 3 chemicals were processed against the 2 genes by looking for direct chemical-gene interactions in CTD, only 3 combinations were validated as having a curated chemical-gene interaction: “Particulate Matter increases the expression of ITGB3 mRNA and protein,” “Vehicle Emissions increases the expression of ITGB3 protein,” and “Vehicle Emissions affects the methylation of P2RY12 gene.” Subsequently, only 3 CGPD-tetramers could be generated: Particulate Matter > ITGB3 > platelet aggregation > Myocardial Infarction; Vehicle Emissions > ITGB3 > platelet aggregation > Myocardial Infarction; and Vehicle Emissions > P2RY12 > platelet aggregation > Myocardial Infarction. Because the third chemical term (Air Pollutants) did not yet have any curated interactions with either of the 2 genes in CTD, no tetramers were generated for those combinations.
RESULTS
The CGPD-tetramer Framework
We define “CGPD-tetramers” as computationally generated information units that interrelate 4 data types curated at CTD: a chemical that molecularly interacts with a gene product, inducing a nondisease phenotype, linked to a disease (Figure 2). To create a CGPD-tetramer, 5 distinct types of manually curated interactions are required as lines of supporting evidence: (1) a direct chemical-gene interaction curated in CTD, (2) a direct chemical-phenotype interaction curated in CTD, (3) a direct chemical-disease interaction curated in CTD, (4) a direct gene-GO annotation imported from NCBI Gene (wherein the GO annotation is the same phenotype term), and (5) a direct gene-disease interaction curated in CTD. A CGPD-tetramer can be generated only if all 5 supporting lines of evidence exist.
Figure 2.
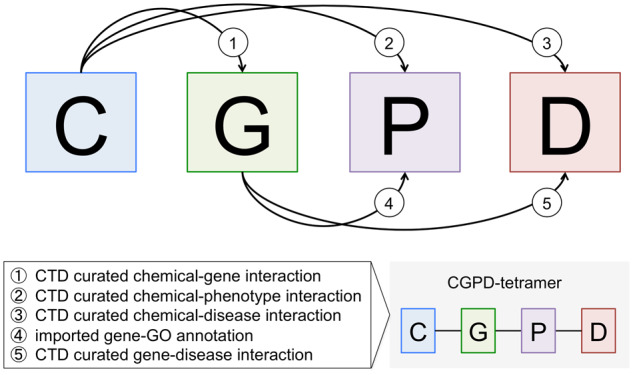
CGPD-tetramers are computationally generated information units that interrelate 4 data types at Comparative Toxicogenomics Database (CTD). To generate a CGPD-tetramer by data integration, 5 lines of supporting evidence are required (box) as directly curated interactions among the 4 data types: C, chemical; G, gene product; P, phenotype; D, disease. If any 1 line of supporting evidence is lacking, the tetramer is not generated.
Computing CGPD-tetramers is simplified by first identifying the phenotypes that have inferred relationships to a disease-of-interest. As aforementioned, a phenotype has an inferred relationship to a disease in CTD via sets of shared chemicals (CIN) or a set of shared genes (GIN). For example, the phenotype “apoptotic process” has an inferred relationship with MI via 198 chemicals (CIN) and 17 genes (GIN) because all 198 chemicals and 17 genes each have an independent, directly curated interaction with both “apoptotic process” and MI in CTD (Figure 3). Thus, all chemicals in any particular CIN have a known, direct interaction with both the listed phenotype and the disease, fulfilling evidence lines Nos 2 and 3 in CGPD-tetramer construction (Figure 2); similarly, all genes in any particular GIN are also directly curated to both the phenotype and disease and fulfill evidence lines Nos 4 and 5.
Figure 3.

Phenotypes associated with myocardial infarction (MI) in Comparative Toxicogenomics Database. A partial screenshot of the “Phenotypes” data-tab for MI lists the phenotypes that have an inferred relationship to this disease via either a Chemical Inference Network or Gene Inference Network. Here, 198 chemicals have direct interactions with both the phenotype “apoptotic process” (evidence line No. 2) and the disease MI (evidence line No. 3); independently, 17 genes also interact with both this phenotype (evidence line No. 4) and disease (evidence line No. 5). Thus, “apoptotic process” can be inferred to MI via 198 chemicals and 17 genes. Note: 5 of the 198 chemicals listed are environmental ambient air pollutants (boxed within the Chemical Inference Network).
Generating CGPD-tetramers for Air Pollution-associated Myocardial Infarction and Hypertension
We generated air pollution-associated CGPD-tetramers for MI and hypertension independently. The inferred phenotypes for MI were filtered and refined in a step-wise method (Figure 4). In the first step, 4918 unique phenotypes are inferred to MI via sets of either shared CIN and/or shared GIN. However, because a CGPD-tetramer requires both a chemical and a gene, we next filtered the dataset to only phenotypes that had both a CIN and a GIN (step 2), resulting in a reduced subset of 892 phenotypes (inferred via 320 chemicals and 94 genes); this subset automatically fulfills evidence lines Nos 2–5. Furthermore, a CGPD-tetramer requires that there not only be a chemical and a gene, but that the chemical has a curated molecular interaction with the gene product (evidence line No. 1, Figure 2). For each inferred phenotype, the corresponding CIN and GIN were programmatically queried to identify any chemical in an associated CIN with a curated interaction in CTD to any gene in the associated GIN (step 3). If a curated interaction exists in CTD between a chemical and a gene for a specific inferred phenotype, then all 5 lines of supporting evidence are met, and a CGPD-tetramer is generated. In total, 14 957 distinct CGPD-tetramers were computed for MI, involving 243 chemicals, 90 genes, and 758 phenotypes (step 4). Of these 243 chemicals, 5 were air pollutant-related chemical terms in CTD: Particulate Matter, Vehicle Emissions, Ozone, Nitrogen Dioxide, and Air Pollutants. Limiting the data to just these pollutants (step 5) results in a final set of 653 CGPD-tetramers, encompassing 5 chemicals, 58 genes, and 117 phenotypes. Of these 653 CGPD-tetramers, 43 of them relate 5 pollutants and 14 genes with the phenotype “apoptotic process” and the cardiovascular disease MI (Figure 5). A file describing all 653 computed CGPD-tetramers is provided (Supplementary Table 1).
Figure 4.

Step-wise process computing CGPD-tetramers for 2 cardiovascular diseases. The disease hierarchy shows 2 cardiovascular diseases: myocardial infarction (MI) (a heart disease) and hypertension (both a heart and vascular disease). In step 1, MI is associated with 4918 phenotypes inferred via either CIN and/or GIN, representing 324 chemicals and 95 genes, respectively. In step 2, the data are filtered for only phenotypes inferred via both a Chemical Inference Network (CIN) and Gene Inference Network (GIN), and then restricted further in step 3 by requiring a chemical in the CIN to have a directly curated Comparative Toxicogenomics Database interaction with a gene in the associated GIN, to yield 758 phenotypes. The data in step 3 are supported by all 5 required lines of evidence and can be used to generate 14 957 CGPD-tetramers (step 4). By limiting the chemicals to ambient air pollutants (step 5), 653 CGPD-tetramers remain, for 5 pollutants, 58 genes, and 117 phenotypes for MI. The same steps are performed for hypertension. Venn analysis discovers 19 genes and 96 phenotypes shared between these 2 cardiovascular diseases associated with air pollution exposure.
Figure 5.

CGPD-tetramers relating air pollutants, intermediary genes, apoptosis, and myocardial infarction (MI). Forty-three individual CGPD-tetramers were computed that relate 5 environmental chemicals (C) with molecular interactions to 14 genes (G) that modulate the phenotype (P) “apoptotic process” inferred to the disease (D) MI. For visualization, the tetramers are condensed into a network schematic. Chemicals: AP, Air Pollutants; NO2, Nitrogen Dioxide; O3, Ozone; PM, Particulate Matter; VE, Vehicle Emissions; genes are depicted using official gene symbols.
The same steps were used to analyze phenotypes for hypertension (Figure 4), resulting in 701 CGPD-tetramers (Supplementary Table 2), composed of 3 environmental air pollutants (Particulate Matter, Carbon Monoxide, and Air Pollutants), 96 genes, and 142 phenotypes.
The CGPD-tetramers for the 2 cardiovascular diseases share 19 genes and 96 phenotypes (Figure 4). Neither Sulfur Dioxide nor Soot had any inferred phenotypes to MI or hypertension; consequently, the 5 levels of required evidence could not be met for these 2 pollutants and they were removed from further analyses and reporting.
CGPD-tetramer Component Analysis
Chemicals
Five environmental air pollutants were involved in the 653 CGPD-tetramers for MI, and 3 air pollutants were used in the 701 tetramers for hypertension (Table 1). For both cardiovascular diseases, Particulate Matter interacted with the most genes and phenotypes to yield the highest number of CGPD-tetramers. This might simply be due to Particulate Matter having 4- to 10-times more curated references in CTD (1463 scientific articles) than the other pollutants: Vehicle Emissions (402 articles), Ozone (303 articles), Air Pollutants (260 articles), Nitrogen Dioxide (198 articles), and Carbon Monoxide (153 articles) at the time of analysis.
Table 1.
Environmental Air Pollutants Used to Construct CGPD-tetramers for Myocardial Infarction and Hypertension
| Myocardial Infarction |
Hypertension |
|||||
|---|---|---|---|---|---|---|
| Pollutant | No. genes | No. phenotypes | No. tetramers | No. genes | No. phenotypes | No. tetramers |
| Particulate matter | 44 | 99 | 315 | 90 | 132 | 582 |
| Air pollutants | 17 | 18 | 46 | 32 | 25 | 76 |
| Vehicle emissions | 47 | 45 | 181 | 0 | 0 | 0 |
| Ozone | 33 | 27 | 91 | 0 | 0 | 0 |
| Nitrogen dioxide | 12 | 9 | 20 | 0 | 0 | 0 |
| Carbon monoxide | 0 | 0 | 0 | 21 | 15 | 43 |
Genes
MI and hypertension were associated with 58 and 96 genes to generate their respective CGPD-tetramers. We first evaluated each gene set independently for enriched pathways and gene interaction modules to provide an indication to their biological mechanisms, as a prelude to understanding their involvement in the computed CGPD-tetramers. First, we performed a pathway analysis, examining the KEGG and Reactome pathways associated with each gene set. The 58 genes for MI were enriched for 82 pathways, and the 96 genes for hypertension were enriched for 160 pathways. The top 10 pathways for each set of genes had extensive overlap, sharing 4 immune system pathways: immune system (REACT:R-HSA-168256), interleukin-4 and 13 signaling (REACT:R-HSA-6785807), cytokine signaling in immune system (REACT:R-HSA-1280215), signaling by interleukins (REACT:R-HSA-449147); and 2 circulatory system pathways: hemostasis (REACT:R-HSA-109582), and fluid shear stress and atherosclerosis (KEGG: hsa05418). These associated pathways suggest that the genes play a key role in immune responses and circulatory system processes. Next, we examined the 2 gene sets for gene/protein network interactions using BioGRID data integrated into CTD. For MI, 30 of the 58 genes (52%) have known gene/protein interactions with each other (Supplementary Figure 1), and for hypertension, 70 of the 96 genes (72%) form a complex, highly interconnected network (Supplementary Figure 2). Thus, for both of these diseases, the majority of genes (52% and 72%) identified from CGPD-tetramers form interaction modules and support development of mechanistic networks to help fill in the knowledge gaps with intermediary steps for air pollution-induced cardiovascular disease. Interestingly, of the 19 genes shared between the CGPD-tetramers for MI and hypertension (Venn analysis, Figure 4), 13 of them (ACE, APOE, BCL2, CAT, GSK3B, ICAM1, MMP2, MMP9, NOS2, NOS3, SOD1, SOD2, and TNF) are also present in each of the gene interaction networks for both diseases, suggesting a potential nexus of molecular mechanisms that can provide a crossover between the 2 air pollution-induced outcomes.
Finally, we directly compared the 2 gene sets to look for functional commonalities. Nineteen genes are shared between the sets for MI and hypertension (Figure 4 and Table 2), and include immune cytokines and receptors (TNF, IL1B, TGFB1, IL6, ICAM1), oxidative stress response genes (CAT, SOD1, SOD2), nitric oxide-generating enzymes (NOS2, NOS3), extracellular matrix proteins (MMP2, MMP9), apoptotic factors (BCL2, GSK3B), blood pressure regulators (ACE, AGT), and an erythropoiesis factor (EPO).
Table 2.
Genes Common in CGPD-tetramers for Both Myocardial Infarction and Hypertension
| Myocardial Infarction |
Hypertension |
|||||
|---|---|---|---|---|---|---|
| Gene | No. Chemicals | No. Phenotypes | No. Tetramers | No. Chemicals | No. Phenotypes | No. Tetramers |
| IL1B | 5 | 26 | 53 | 3 | 24 | 34 |
| TNF | 5 | 24 | 50 | 3 | 23 | 35 |
| BCL2 | 4 | 19 | 33 | 2 | 18 | 20 |
| SOD1 | 4 | 16 | 32 | 2 | 15 | 17 |
| SOD2 | 5 | 14 | 32 | 3 | 14 | 18 |
| TGFB1 | 4 | 17 | 28 | 1 | 15 | 15 |
| IL6 | 4 | 14 | 23 | 3 | 12 | 17 |
| AGT | 2 | 17 | 22 | 2 | 17 | 20 |
| NOS2 | 5 | 5 | 15 | 3 | 5 | 10 |
| CAT | 5 | 8 | 15 | 2 | 6 | 7 |
| MMP9 | 4 | 7 | 14 | 2 | 7 | 8 |
| GSK3B | 2 | 8 | 13 | 2 | 9 | 9 |
| NOS3 | 4 | 7 | 13 | 2 | 6 | 7 |
| GCLC | 4 | 5 | 12 | 2 | 5 | 6 |
| EPO | 3 | 6 | 11 | 2 | 5 | 7 |
| APOE | 3 | 8 | 11 | 1 | 5 | 5 |
| ACE | 2 | 6 | 10 | 1 | 6 | 6 |
| ICAM1 | 3 | 6 | 8 | 2 | 5 | 5 |
| MMP2 | 4 | 4 | 8 | 2 | 4 | 5 |
Phenotypes
We derived 117 phenotypes associated with the 653 CGPD-tetramers for MI and 142 phenotypes for the 701 CGPD-tetramers for hypertension. Interestingly, 96 of the phenotypes are shared between the 2 different cardiovascular diseases, representing 82% (96/117) and 68% (96/142) of the phenotypes for MI and hypertension, respectively (Supplementary Table 3). Many of these phenotypes, however, reflect similar biological concepts and we realized they could be clustered into categories as a way to condense and explore the information at a less granular level: eg, “apoptotic process,” “cell death,” “positive regulation of apoptotic DNA fragmentation,” and “release of cytochrome c from mitochondria,” all reflect cellular death. Upon manual inspection, these 96 common phenotypes could be clustered into 13 categories (Table 3), with some phenotypes mapping to more than 1 group (eg, “positive regulation of neuron apoptotic process” was mapped to both Cell Death and Nervous System). The categories with the greatest number of shared phenotypes as part of their computed CGPD-tetramers include Inflammation-Immune System (150 tetramers for MI, 146 for hypertension), Cell Death (179 tetramers for MI, 104 for hypertension), Circulatory System (77 tetramers for MI, 82 for hypertension), and Oxidative Stress (100 tetramers for MI, 64 for hypertension).
Table 3.
Phenotypes Common in CGPD-tetramers for Both Myocardial Infarction (MI) and Hypertension (HT)
| Phenotype Cluster | Phenotypes | No. Tetramers (MI) | No. Tetramers (HT) |
|---|---|---|---|
| Inflammation-immune system | 26 Phenotypes: acute inflammatory response to antigenic stimulus; B-cell homeostasis; inflammatory response; leukocyte migration; lymphocyte homeostasis; macrophage differentiation; microglial cell activation; monocyte homeostasis; neutrophil homeostasis; platelet activation; positive regulation of cellular extravasation; positive regulation of immunoglobulin secretion; positive regulation of inflammatory response; positive regulation of interferon-gamma production; positive regulation of interleukin-12 production; positive regulation of interleukin-6 production; positive regulation of interleukin-6 secretion; positive regulation of interleukin-8 production; positive regulation of macrophage activation; positive regulation of neutrophil chemotaxis; positive regulation of prostaglandin secretion; positive regulation of T-helper 2 cell cytokine production; positive regulation of tumor necrosis factor production; production of molecular mediator involved in inflammatory response; regulation of inflammatory response; T-cell homeostasis | 150 | 146 |
| Cell death | 10 Phenotypes: apoptotic process; cell death; cellular response to DNA damage stimulus; negative regulation of apoptotic process; positive regulation of apoptotic DNA fragmentation; positive regulation of apoptotic process; positive regulation of cell death; positive regulation of neuron apoptotic process; positive regulation of neuron death; release of cytochrome c from mitochondria | 179 | 104 |
| Circulatory system | 11 Phenotypes: artery smooth muscle contraction; blood vessel development; cardiac muscle contraction; negative regulation of vasoconstriction; platelet activation; positive regulation of vasoconstriction; regulation of blood pressure; regulation of heart contraction; regulation of heart rate; vasoconstriction; vasodilation | 77 | 82 |
| Oxidative stress | 9 Phenotypes: hydrogen peroxide biosynthetic process; nitric oxide biosynthetic process; positive regulation of NAD(P)H oxidase activity; positive regulation of reactive oxygen species metabolic process; positive regulation of superoxide anion generation; reactive oxygen species metabolic process; response to oxidative stress; superoxide anion generation; superoxide dismutase activity | 100 | 64 |
| Cell proliferation | 4 Phenotypes: cell proliferation; mitotic cell cycle arrest; negative regulation of cell proliferation; positive regulation of cell proliferation | 61 | 64 |
| Cell process | 9 Phenotypes: cell migration; collagen fibril organization; establishment of cell polarity; positive regulation of heterotypic cell-cell adhesion; positive regulation of autophagy; positive regulation of mitochondrial membrane potential; positive regulation of protein import into nucleus; regulation of gene expression; regulation of mitochondrial membrane potential | 41 | 39 |
| Nervous system | 6 Phenotypes: cognition; hippocampus development; learning or memory; memory; positive regulation of neuron apoptotic process; positive regulation of neuron death | 38 | 23 |
| Cell signaling | 5 Phenotypes: calcium-mediated signaling; positive regulation of cytosolic calcium ion concentration; positive regulation of NF-kappaB transcription factor activity; positive regulation of NIK/NF-kappaB signaling; positive regulation of p38MAPK cascade | 22 | 32 |
| Cell metabolism | 7 Phenotypes: cholesterol homeostasis; glucose homeostasis; glucose metabolic process; glutathione biosynthetic process; glutathione metabolic process; hemoglobin biosynthetic process; protein dephosphorylation | 22 | 29 |
| Response to stimulus | 4 Phenotypes: response to cholesterol; response to organic substance; response to toxic substance; response to xenobiotic stimulus | 17 | 31 |
| Behavior | 5 Phenotypes: cognition; learning or memory; memory; locomotory exploration behavior; social behavior | 21 | 20 |
| Physiology | 5 Phenotypes: bone development; determination of adult lifespan; muscle contraction; renal system process; spermatogenesis | 21 | 19 |
| Cell development and maintenance | 7 Phenotypes: positive regulation of epithelial to mesenchymal transition; B-cell homeostasis; lymphocyte homeostasis; macrophage differentiation; monocyte homeostasis; neutrophil homeostasis; T-cell homeostasis | 22 | 16 |
Another approach to refine the number of phenotypes derived from CGPD-tetramers (without having to manually review and cluster phenotypes into generic categories) is to consider GO term enrichment of the associated genes, because CTD phenotypes are derived from GO terms, and GO terms have annotated relationships with genes. For example, using the CTD Set Analyzer tool, the 58 genes derived from the CGPD-tetramers for air pollution-associated MI are enriched for 957 GO biological processes (Supplementary Table 4). When these enriched GO terms are compared against the 117 phenotypes derived from CGPD-tetramers, a smaller set of 46 common phenotypes are discovered (Supplementary Figure 3), providing a filtered subset of phenotypes, and, interestingly, this refined set still reflects similar categories derived for the original 117 phenotypes, including: Cell Death (9 phenotypes), Inflammation-Immune System (9 phenotypes), Circulatory System (7 phenotypes), Cell Process (4 phenotypes), and Oxidative Stress (4 phenotypes), amongst others.
Assembling CGPD-tetramers
CGPD-tetramers can be consolidated to construct extended chemical-disease pathways by first aligning and assembling them based on shared chemicals, genes, and phenotypes. For example, 91 CGPD-tetramers connect the chemical ozone with 33 genes and 27 phenotypes to MI (Supplementary File 1). We aligned these 91 ozone-specific tetramers in a matrix to find common overlapping intermediary genes and phenotypes (Figure 6). Most of the phenotypes could be clustered into 6 categories: Oxidative Stress (composed of 10 tetramers using 10 genes and 1 phenotype), Cell Signaling (15 tetramers: 10 genes and 3 phenotypes), Cell Metabolism (8 tetramers: 6 genes, 4 phenotypes), Inflammation-Immune System (21 tetramers: 15 genes and 6 phenotypes), Cell Death (18 tetramers: 12 genes and 3 phenotypes), and Circulatory System (10 tetramers: 8 genes and 6 phenotypes). Aligning CGPD-tetramers identifies genes that can interrelate and connect different phenotype categories. For example, of the 10 genes involved in the Cell Signaling phenotypes, 6 of them (ESR1, HMGB1, IL1B, IL6, TGFB1, and TNF) are also involved in the Inflammation phenotypes, allowing the 2 processes to be molecularly connected by intermediary gene products. Assembling CGPD-tetramers builds an extensive, cohesive, and interrelated chemical-disease pathway (Figure 7), filling in the knowledge gaps with putative intermediary steps at the molecular, cellular, and system levels to connect ozone exposure with MI.
Figure 6.

Aligning CGPD-tetramers. To help inform the knowledge gaps between ozone (O3) exposure and myocardial infarction (MI), the 91 computed CGPD-tetramers are condensed and aligned in a matrix, arranged by shared genes and phenotypes. Here, 33 genes (filled-in boxes) and 27 phenotypes clustered into 6 categories connect this individual pollutant to cardiovascular disease.
Figure 7.

Assembling CGPD-tetramers. Intermediary genes and phenotypes derived from CGPD-tetramers are coalesced and assembled into putative chemical-induced disease pathways to help inform the knowledge gaps connecting ozone (O3) exposure with myocardial infarction (MI) using steps at the molecular, cellular, and system levels. The number of genes (circles) associated with each set of phenotypes is indicated, and phenotype clusters (boxes) are interrelated by shared genes (curved arrows with lists of shared genes), helping to conjoin independent tetramers into a larger integrated network relating ozone exposure to cardiovascular disease. The diagram uses 84 CGPD-tetramers (from Figure 6) linking ozone, 33 genes, 24 phenotypes, and MI.
DISCUSSION
We present a bioinformatics method that leverages curated data from the public database CTD to generate novel sets of information called CGPD-tetramers, relating a chemical interaction with a gene, connected to a phenotype and disease. Five supporting lines of evidence are required to compute tetramers by integrating CTD curated datasets. Our strategy does not require a priori knowledge of the toxicant, biological system, or adverse outcome, and can be used to identify potential molecular and biological intermediary steps that help fill in knowledge gaps connecting chemical exposures with outcomes for environmentally influenced diseases. As a proof-of-concept, we tested the method to explore air pollution-induced cardiovascular disease, but the described strategy can be adopted and applied to any chemical-induced disease in the CTD framework. In total, we generate 653 CGPD-tetramers relating air pollution and MI (5 pollutants, 58 genes, and 117 key events) and 701 CGPD-tetramers for hypertension (3 pollutants, 96 genes, and 142 key events). We identify shared genes and phenotypes between the 2 diseases (highlighting important roles for oxidative stress, inflammation, immune response, cell death, and circulatory system processes), and further show how CGPD-tetramers can be aligned and assembled to yield extended chemical-disease pathways detailing modes-of-action that can be used to support systems toxicology-based applications (Davis et al., 2019b).
Ambient air pollution (especially particulate matter and ozone) has a significant global impact on human health, quality of life, and premature mortality (Lelieveld et al., 2015). Both short- and long-term exposure to air pollutants are associated with increases in cardiovascular death, arteriosclerosis, MI, stroke, and heart failure (Bourdrel et al., 2017). In our analysis, we limited the study to only a small number of air pollutants and only for 2 different types of cardiovascular diseases. MI is an acute cardiovascular event, whereas hypertension represents a chronic condition. Nonetheless, there is clinical evidence to suggest these 2 types of diseases are related (Pedrinelli et al., 2012; Rakugi et al., 1996; Thune et al., 2008). Here, we identify 19 genes (Table 2), 96 phenotypes (Table 3), and a nexus of 13 interacting genes (Supplementary Figs. 1 and 2) that are shared between the 2 diseases, providing both potential genetic and molecular mechanistic roles that additionally relate these 2 outcomes.
The identified genes and phenotypes shared in CGPD-tetramers for MI and hypertension suggest important mechanistic roles for oxidative stress, inflammation, cell death, and circulatory system processes. Of the total 653 CGPD-tetramers linking air pollution with MI, 77% of them group to those 4 categories (Table 3): Oxidative Stress (15%, 100 tetramers), Inflammation-Immune System (23%, 150 tetramers), Cell Death (27%, 179 tetramers), and Circulatory System (12%, 77 tetramers). The 701 CGPD-tetramers for hypertension have a similar distribution (Table 3), with 56% of the tetramers mapping to the same 4 categories: Oxidative Stress (9%), Inflammation-Immune System (21%), Cell Death (15%), and Circulatory System (12%). Finally, in the ozone-specific data subset (Figure 6), 59 of the 91 CGPD-tetramers (65%) also resolve to those same phenotype clusters.
It is important to emphasize that the generation of CGPD-tetramers is inherently dependent on, and limited by, the data quality, completeness, and currency in CTD (Davis et al., 2012b). Thus, it is critical to confirm if these putative intermediary steps computed by CGPD-tetramer generation can be validated in the literature. In our assembled pathway for ozone-associated MI (Figure 6), the first intermediary step “response to oxidative stress” is validated by numerous reports of systemic and pulmonary oxidative stress and reactive oxygen species as key mechanisms for air pollution-associated cardiovascular events (Bourdrel et al., 2017; Fiordelisi et al., 2017; Miller, 2020; Rao et al., 2018) . The subsequent cell signaling phenotypes (“ERK1 and ERK2 cascade,” “p38 MAPK cascade,” and “NF-kappa B factor activity”) are supported by reports describing how inhaled air pollution particles induce oxidative responses to initiate MAPK and NF-kappaB pathways in experimental rodents (Roberts et al., 2003; Shukla et al., 2000), and how MAPK and NF-kappaB signaling are involved with MI (Song et al., 2020; Zhang et al., 2020). As well, air pollution exposure is associated with altered metabolism for cholesterol and triglycerides (Gaio et al., 2019), glutathione (de Oliveira-Fonoff et al., 2017), calcium homeostasis (Holme et al., 2019), and superoxide dismutase (Jiang et al., 2016), substantiating our derived cell metabolism phenotypes from CGPD-tetramers. The subsequent phenotypic events of inflammation (Ji et al., 2018), immune cell deregulation (Barlow et al., 2008; Xu et al., 2013), cell death (Peixoto et al., 2017), blood pressure (Xu et al., 2020), and vascular and circulatory system processes (Bai and van Eeden, 2013; Day et al., 2017) are well documented in the literature in response to air pollution exposure and with roles in MI (Bourdrel et al., 2017; Fiordelisi et al., 2017).
CGPD-tetramers also yield connections that might not be initially appreciated. For instance, “cognition” and “memory” (Table 3), at first, might seem to be nonintuitive phenotypes for MI and hypertension, yet cognitive defects have been reported in cardiovascular patients (Harrison et al., 2014; Lamar et al., 2019) and prenatal ozone exposure inhibits memory and spatial learning in newborn rats (Custodio et al., 2019). Furthermore, the CGPD-tetramer associating ozone with the CASP3 gene product and “positive regulation of neuron apoptotic process” and MI (Figure 6) provides a molecular and biological mechanism connecting nervous system and cardiovascular events, as well as strikingly resembling the results from a study correlating increased CASP3 activity with neuronal apoptosis and cognitive impairments associated with MI (Gilbert et al., 2016). Similarly, the phenotype “spermatogenesis” initially might be considered an outlier (Table 3), yet hypertension has been linked to decreases in sperm concentration in rats (Colli et al., 2019) and ambient air pollution is suggested to influence sperm quality (Bosco et al., 2018; Deng et al., 2016; Lafuente et al., 2016). As a bioinformatics resource, CGPD-tetramers provide investigators with sets of specific genes and biological processes as potential mechanisms to test these reported associations.
Finally, we note that the computed CGPD-tetramers can be used to provide chemical data for adverse outcome pathways (AOPs). AOPs are information networks that interrelate “molecular initiating events” (analogous to CTD chemical-gene interactions), a series of connected “key events” (analogous to CTD phenotypes), and “adverse outcomes” (Edwards et al., 2015; Vinken et al., 2017). Although designed to be chemical-agnostic, AOPs have become a popular framework to represent toxicology information (by linking specific chemicals to them) for human health risk assessment and susceptibility (Ankley and Edwards, 2018; Mortensen et al., 2018; Rycroft et al., 2019). Numerous studies have developed methodologies to generate “computational AOPs” that include chemical components by integrating, at least in part, content from public databases (Bell et al., 2016; Carvaillo et al., 2019; Kosnik et al., 2019; Nymark et al., 2018; Oki et al., 2016). Many of these methods, however, first require the mapping and cross-referencing of data types from a variety of disparate public resources or additional literature mining. Here, CGPD-tetramers contain phenotypes and diseases that can be mapped to AOP “key events” and “adverse outcomes,” respectively, to add content for chemicals and interacting genes as “molecular initiating events” (Davis et al., 2018). Furthermore, we demonstrate how aligning CGPD-tetramers by shared chemicals, genes, and phenotypes can assemble the tetramers to construct and interrelate key phenotypic events to the adverse outcome of MI, which could potentially help inform AOP construction, testing, and refinement. As well, CTD’s use of GO terms as a vocabulary source for phenotypes allow CGPD-tetramers to take advantage of Gene Ontology Causal Activity Modeling (GO-CAM), a project that links multiple independent GO terms into structured biological models for pathway analysis (Thomas et al., 2019); finding commonalities between GO-CAM models and CGPD-tetramers (via shared GO/phenotype terms) will further help connect and organize key phenotypic events for construction of AOPs and chemical-induced mode-of-action pathways.
Our approach using CTD (a single, self-contained, curated database) provides many advantages. First, it obviates the time-consuming requirement to extract, align, and map data concepts from a variety of external repositories to integrate information. Second, it requires no additional text mining of the disease-related literature or a priori knowledge of the toxicant, involved genes, biological mechanisms, or adverse outcome. Third, because all CTD content is seamlessly integrated, users can quickly expand out from any 1 component of a CGPD-tetramer (chemical, gene, phenotype, or disease) to find other potential components or information to consider in the designing of a pathway, such as additional genes associated with the cell signaling phenotype “p38 MAPK cascade” (http://ctdbase.org/detail.go?type=go&acc=GO%3a0038066&view=gene) or other adverse health outcomes associated with ozone exposure (http://ctdbase.org/detail.go?type=chem&acc=D010126&view=disease) or > 10 000 measured exposure marker levels for particulate matter (http://ctdbase.org/detail.go?type=chem&acc=D052638&view=expConsol).
In summary, we describe a novel process that leverages curated content from a single public database to rapidly generate computational CGPD-tetramers linking air pollution toxicants to cardiovascular disease. This method discovers genes, gene/protein interaction modules, and phenotypes to help scientists fill in the knowledge gaps and develop molecular tools to better understand environmental risk. Our strategy can be easily expanded and adopted to generate CGPD-tetramers for any environmental chemical and disease curated in CTD.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
FUNDING
National Institute of Environmental Health Sciences (ES014065, ES023788, ES025128). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary Material
REFERENCES
- Ankley G. T., Edwards S. W. (2018). The adverse outcome pathway: A multifaceted framework supporting 21st century toxicology. Curr. Opin. Toxicol. 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., et al. (2000). Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai N., van Eeden S. F. (2013). Systemic and vascular effects of circulating diesel exhaust particulate matter. Inhal. Toxicol. 25, 725–734. [DOI] [PubMed] [Google Scholar]
- Barlow P. G., Brown D. M., Donaldson K., MacCallum J., Stone V. (2008). Reduced alveolar macrophage migration induced by acute ambient particle (PM10) exposure. Cell Biol. Toxicol. 24, 243–252. [DOI] [PubMed] [Google Scholar]
- Bell S. M., Angrish M. M., Wood C. E., Edwards S. W. (2016). Integrating publicly available data to generate computationally predicted adverse outcome pathways for fatty liver. Toxicol. Sci. 150, 510–520. [DOI] [PubMed] [Google Scholar]
- Birnbaum L. S., Burke T. A., Jones J. J. (2016). Informing 21st-century risk assessments with 21st-centruy science. Environ. Health Perspect. 124, A60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco L., Notari T., Ruvolo G., Roccheri M. C., Martino C., Chiappetta R., Carone D., Lo Bosco G., Carrillo L., Raimondo S., et al. (2018). Sperm DNA fragmentation: An early and reliable marker of air pollution. Environ. Toxicol. Pharmacol. 58, 243–249. [DOI] [PubMed] [Google Scholar]
- Bourdrel T., Bind M. A., Bejot Y., Morel O., Argacha J. F. (2017). Cardiovascular effects of air pollution. Arch. Cardiovasc. Dis. 110, 634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. R., Hem V., Katz K. S., Ovetsky M., Wallin C., Ermolaeva O., Tolstoy I., Tatusova T., Pruitt K. D., Maglott D. R., et al. (2015). Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 43, D36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvaillo J. C., Barouki R., Coumoul X., Audouze K. (2019). Linking bisphenol S to adverse outcome pathways using a combined text mining and systems biology approach. Environ. Health Perspect. 127, 47005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. J., Brauer M., Burnett R., Anderson H. R., Frostad J., Estep K., Balakrishnan K., Brunekreef B., Dandona L., Dandona R., et al. (2017). Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Disease Study 2015. Lancet 389, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colli L. G., Belardin L. B., Echem C., Akamine E. H., Antoniassi M. P., Andretta R. R., Mathias L. S., Rodrigues S. F. D. P., Bertolla R. P., de Carvalho M. H. C., et al. (2019). Systemic arterial hypertension leads to decreased semen quality and alterations in the testicular microcirculation in rats. Sci. Rep. 9, 11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa D. L. (2018). Historical highlights of air pollution toxicology. Toxicol. Sci. 164, 5–8. [DOI] [PubMed] [Google Scholar]
- Custodio V., Rubio C., Paz C. (2019). Prenatal ozone exposure induces memory deficiencies in newborns rats. Front. Mol. Neurosci. 12, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Grondin C. J., Johnson R. J., Sciaky D., McMorran R., Wiegers J., Wiegers T. C., Mattingly C. J. (2019. a). The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 47, D948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Johnson R. J., Lennon-Hopkins K., Sciaky D., Rosenstein M. C., Wiegers T. C., Mattingly C. J. (2012. b). Targeted journal curation as a method to improve data currency at the Comparative Toxicogenomics Database. Database (Oxford) 2012, bas051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Murphy C. G., Rosenstein M. C., Wiegers T. C., Mattingly C. J. (2008). The Comparative Toxicogenomics Database facilitates identification and understanding of chemical-gene-disease associations: Arsenic as a case study. BMC Med. Genomics 1, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Wiegers J., Wiegers T. C., Mattingly C. J. (2019. b). Public data sources to support systems toxicology applications. Curr. Opin. Toxicol. 16, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Wiegers T. C., King B. L., Wiegers J., Grondin C. J., Sciaky D., Johnson R. J., Mattingly C. J. (2016). Generating Gene Ontology-disease inferences to explore mechanisms of human disease at the Comparative Toxicogenomics Database. PLoS One 11, e0155530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Wiegers T. C., Rosenstein M. C., Mattingly C. J. (2012. a). MEDIC: A practical disease vocabulary used at the Comparative Toxicogenomics Database. Database (Oxford) 2012, bar065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Wiegers T. C., Rosenstein M. C., Murphy C. G., Mattingly C. J. (2011). The curation paradigm and application tool used for manual curation of the scientific literature at the Comparative Toxicogenomics Database. Database (Oxford) 2011, bar034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. P., Wiegers T. C., Wiegers J., Johnson R. J., Sciaky D., Grondin C. J., Mattingly C. J. (2018). Chemical-induced phenotypes at CTD help inform the predisease state and construct adverse outcome pathways. Toxicol. Sci. 165, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day D. B., Xiang J., Mo J., Li F., Chung M., Gong J., Weschler C., Ohman-Strickland P. A., Sundell J., Weng W., et al. (2017). Association of ozone exposure with cardiorespiratory pathophysiologic mechanisms in healthy adults. JAMA Intern. Med. 177, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z., Chen F., Zhang M., Lan L., Qiao Z., Cui Y., An J., Wang N., Fan Z., Zhao X., et al. (2016). Association between air pollution and sperm quality: A systematic review and meta-analysis. Environ. Pollut. 208, 663–669. [DOI] [PubMed] [Google Scholar]
- de Oliveira-Fonoff A. M., Mady C., Pessoa F. G., Fonseca K. C. B., Salemi V. M. C., Fernandes F., Saldiva P. H. N., Ramires F. J. A. (2017). The role of air pollution in myocardial remodeling. PLoS One 12, e0176084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockery D. W., Pope C. A. 3rd, Xu X., Spengler J. D., Ware J. H., Fay M. E., Ferris B. G., Speizer F. E. (1993). An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 329, 1753–1759. [DOI] [PubMed] [Google Scholar]
- Edwards S. W., Tan Y. M., Villeneuve D. L., Meek M. E., McQueen C. A. (2015). Adverse outcome pathways—Organizing toxicological information to improve decision making. J. Pharmacol. Exp. Ther. 356, 170–181. [DOI] [PubMed] [Google Scholar]
- Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., et al. (2018). The reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiordelisi A., Piscitelli P., Trimarco B., Coscioni E., Iaccarino G., Sorriento D. (2017). The mechanisms of air pollution and particulate matter in cardiovascular diseases. Heart Fail. Rev. 22, 337–347. [DOI] [PubMed] [Google Scholar]
- Gaio V., Roquette R., Dias C. M., Nunes B. (2019). Ambient air pollution and lipid profile: Systematic review and meta-analysis. Environ. Pollut. 254, 113036. [DOI] [PubMed] [Google Scholar]
- Gilbert K., Godbout R., Rousseau G. (2016). Caspase-3 activity in the rat amygdala measured by spectrofluorometry after myocardial infarction. J. Vis. Exp. 107, e53207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondin C. J., Davis A. P., Wiegers T. C., King B. L., Wiegers J. A., Reif D. M., Hoppin J. A., Mattingly C. J. (2016). Advancing exposure science through chemical data curation and integration in the Comparative Toxicogenomics Database. Environ. Health Perspect. 124, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley M. B., Vedanthan R., Fuster V. (2018). Air pollution and cardiovascular disease: A window of opportunity. Nat. Rev. Cardiol. 15, 193–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S. L., Ding J., Tang E. Y., Siervo M., Robinson L., Jagger C., Stephan B. C. M. (2014). Cardiovascular disease risk models and longitudinal changes in cognition: A systematic review. PLoS One 9, e114431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme J. A., Brinchmann B. C., Le Ferrec E., Lagadic-Gossmann D., Ovrevik J. (2019). Combustion particle-induced changes in calcium homeostasis: A contributing factor to vascular disease? Cardiovasc. Toxicol. 19, 198–209. [DOI] [PubMed] [Google Scholar]
- Ji X., Zhang Y., Li G., Sang N. (2018). Potential role of inflammation in associations between particulate matter and heart failure. Curr. Pharm. Des. 24, 341–358. [DOI] [PubMed] [Google Scholar]
- Jiang S., Bo L., Gong C., Du X., Kan H., Xie Y., Song W., Zhao J. (2016). Traffic-related air pollution is associated with cardio-metabolic biomarkers in general residents. Int. Arch. Occup. Environ. Health 89, 911–921. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosnik M. B., Planchart A., Marvel S. W., Reif D. M., Mattingly C. J. (2019). Integration of curated and high-throughput screening data to elucidate environmental influences on disease pathways. Comput. Toxicol. 12, 100094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente R., Garcia-Blaquez N., Jacquemin B., Checa M. A. (2016). Outdoor air pollution and sperm quality. Fertil. Steril. 106, 880–896. [DOI] [PubMed] [Google Scholar]
- Lamar M., Durazo-Arvizu R. A., Sachdeva S., Pirzada A., Perreira K. M., Rundek T., Gallo L. C., Grober E., DeCarli C., Lipton R. B., et al. (2019). Cardiovascular disease risk factor burden and cognition: Implications of ethnic diversity within the Hispanic Community Health Study/Study of Latinos. PLoS One 14, e0215378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld J., Evans J. S., Fnais M., Giannadaki D., Pozzer A. (2015). The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 525, 367–371. [DOI] [PubMed] [Google Scholar]
- Miller M. R. (2020). Oxidative stress and the cardiovascular effects of air pollution. Free Radic. Biol. Med. 151, 69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen H. M., Chamberlin J., Joubert B., Angrish M., Sipes N., Lee J. S., Euling S. Y. (2018). Leveraging human genetic and adverse outcome pathway (AOP) data to inform susceptibility in human health risk assessment. Mamm. Genome 29, 190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymark P., Rieswijk L., Ehrhart F., Jeliazkova N., Tsiliki G., Sarimveis H., Evelo C. T., Hongisto V., Kohonen P., Willighagen E., et al. (2018). A data fusion pipeline for generating and enriching adverse outcome pathway descriptions. Toxicol. Sci. 162, 264–275. [DOI] [PubMed] [Google Scholar]
- Oki N. O., Nelms M. D., Bell S. M., Mortensen H. M., Edwards S. W. (2016). Accelerating adverse outcome pathway development using publicly available data sources. Curr. Environ. Health Rep. 3, 53–63. [DOI] [PubMed] [Google Scholar]
- Oughtred R., Stark C., Breitkreutz B. J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R., et al. (2019). The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrinelli R., Ballo P., Fiorentini C., Denti S., Galderisi M., Ganau A., Germano G., Innelli P., Paini A., Perlini S., et al. (2012). Hypertension and acute myocardial infarction: An overview. J. Cardiovasc. Med. (Hagerstown) 13, 194–202. [DOI] [PubMed] [Google Scholar]
- Peixoto M. S., de Oliveira Galvao M. F., de Medeiros S. R. B. (2017). Cell death pathways of particulate matter toxicity. Chemosphere 188, 32–48. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S., Al-Kindi S. G., Brook R. D. (2018). Air pollution and cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 72, 2054–2070. [DOI] [PubMed] [Google Scholar]
- Rakugi H., Yu H., Kamitani A., Nakamura Y., Ohishi M., Kamide K., Nakata Y., Takami S., Higaki J., Ogihara T. (1996). Links between hypertension and myocardial infarction. Am. Heart J. 132, 213–221. [PubMed] [Google Scholar]
- Rao X., Zhong J., Brook R. D., Rajagopalan S. (2018). Effect of particulate matter air pollution on cardiovascular oxidative stress pathways. Antioxid. Redox Signal. 28, 797–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E. S., Richards J. H., Jaskot R., Dreher K. L. (2003). Oxidative stress mediates air pollution particle-induced acute lung injury and molecular pathology. Inhal. Toxicol. 15, 1327–1346. [DOI] [PubMed] [Google Scholar]
- Rycroft T. E., Foran C. M., Thrash A., Cegan J. C., Zollinger R., Linkov I., Perkins E. J., Garcia-Reyero N. (2019). AOPERA: A proposed methodology and inventory of effective tools to link chemicals to adverse outcome pathways. Altex 37, 64–74. [DOI] [PubMed] [Google Scholar]
- Shukla A., Timblin C., BeruBe K., Gordon T., McKinney W., Driscoll K., Vacek P., Mossman B. T. (2000). Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am. J. Respir. Cell Mol. Biol. 23, 182–187. [DOI] [PubMed] [Google Scholar]
- Song K.-Y., Zhang X.-Z., Li F., Ji Q.-R. (2020). Silencing of ATP2B1-AS1 contributes to protection against myocardial infarction in mouse via blocking NFKBIA-mediated NF-κB signalling pathway. J. Cell. Mol. Med. 24, 4466–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P. D., Hill D. P., Mi H., Osumi-Sutherland D., Van Auken K., Carbon S., Balhoff J. P., Albou L.-P., Good B., Gaudet P., et al. (2019). Gene Ontology Causal Activity Modeling (GO-CAM) moves beyond GO annotations to structured descriptions of biological functions and systems. Nat. Genet. 51, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thune J. J., Signorovitch J., Kober L., Velazquez E. J., McMurray J. J., Califf R. M., Maggioni A. P., Rouleau J. L., Howlett J., Zelenkofske S., et al. (2008). Effect of antecedent hypertension and follow-up blood pressure on outcomes after high-risk myocardial infarction. Hypertension 51, 48–54. [DOI] [PubMed] [Google Scholar]
- Vinken M., Knapen D., Vergauwen L., Hengstler J. G., Angrish M., Whelan M. (2017). Adverse outcome pathways: A concise introduction for toxicologists. Arch. Toxicol. 91, 3397–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N., Lv X., Yu C., Guo Y., Zhang K., Wang Q. (2020). The association between short-term exposure to extremely high level of ambient fine particulate matter and blood pressure: A panel study in Beijing, China. Environ. Sci. Pollut. Res. Int. 27, 28113–28122. [DOI] [PubMed] [Google Scholar]
- Xu X., Jiang S. Y., Wang T.-Y., Bai Y., Zhong M., Wang A., Lippmann M., Chen L.-C., Rajagopalan S., Sun Q. (2013). Inflammatory response to fine particulate air pollution exposure: Neutrophil versus monocyte. PLoS One 8, e71414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T., Kashima S., Doi H. (2016). Fine-particulate air pollution from diesel emission control and mortality rates in Tokyo: A quasi-experimental study. Epidemiology 27, 769–778. [DOI] [PubMed] [Google Scholar]
- Zhang F., Li L., Krafft T., Lv J., Wang W., Pei D. (2011). Study on the association between ambient air pollution and daily cardiovascular and respiratory mortality in an urban district of Beijing. Int. J. Environ. Res. Public Health 8, 2109–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-G., Wei Y., Jiang J., Wang L., Liang H.-Y., Lei C.-B. (2020). Effect of TGF-beta1 on myocardial cell apoptosis in rats with acute myocardial infarction via MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 24, 1350–1356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.