

Abstract
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer and remains a main cause of death in children despite recent improvements in cure rates. In the past decade, development of massively parallel sequencing has enabled large scale genome profiling studies of ALL, which not only led to identification of new subtypes in both B-cell precursor ALL (BCP-ALL) and T-cell ALL (T-ALL), but has also identified potential new therapeutic approaches to target vulnerabilities of many subtypes. Several of these approaches have been validated in preclinical models and are now being formally evaluated in prospective clinical trials. In this review, we provide an overview of the recent advances in our knowledge of genomic bases of BCP-ALL, T-ALL, and relapsed ALL, and discuss their clinical implications.
Keywords: Acute lymphoblastic leukemia, ALL, B-ALL, T-ALL, DUX4, ETV6-RUNX1, Hyperdiploid, Hypodiploid, KMT2A, TCF3, IKZF1, MEF2D, PAX5, Ph-like, ZNF384, ETP, TLX1/3, TAL1, relapse
Introduction
Acute lymphoblastic leukemia (ALL) is a neoplasm of B- or T-lineage lymphoid progenitors, and comprises multiple distinct subtypes characterized by constellations of genetic alterations, including aneuploidy, chromosomal rearrangements, DNA copy number alterations, and sequence mutations [1–6]. Among them, chromosomal translocations are generally considered to be as initiating genetic events in leukemogenesis. The prevalence and prognosis of these alterations vary according to age, ethnicity, and inherited cancer susceptibility, and thus, accurate diagnosis and detection of these lesions are important for appropriate risk classification and precision medicine (Figure 1). Recent advances in massively parallel sequencing have revolutionized our understanding of genomic landscape of ALL by enabling comprehensive characterization of leukemia subtypes (Figure 2) [7–10]. Herein, we will review the genomic landscape of B-cell precursor ALL (BCP-ALL), T-lineage ALL (T-ALL), and relapsed ALL with particular emphasis on newly described entities and targets during the past decade.
Figure 1.

t-SNE plot showing major B-cell precursor acute lymphoblastic leukemia (BCP-ALL) subtypes based on gene expression profiling of 1988 cases [7].
Figure 2.
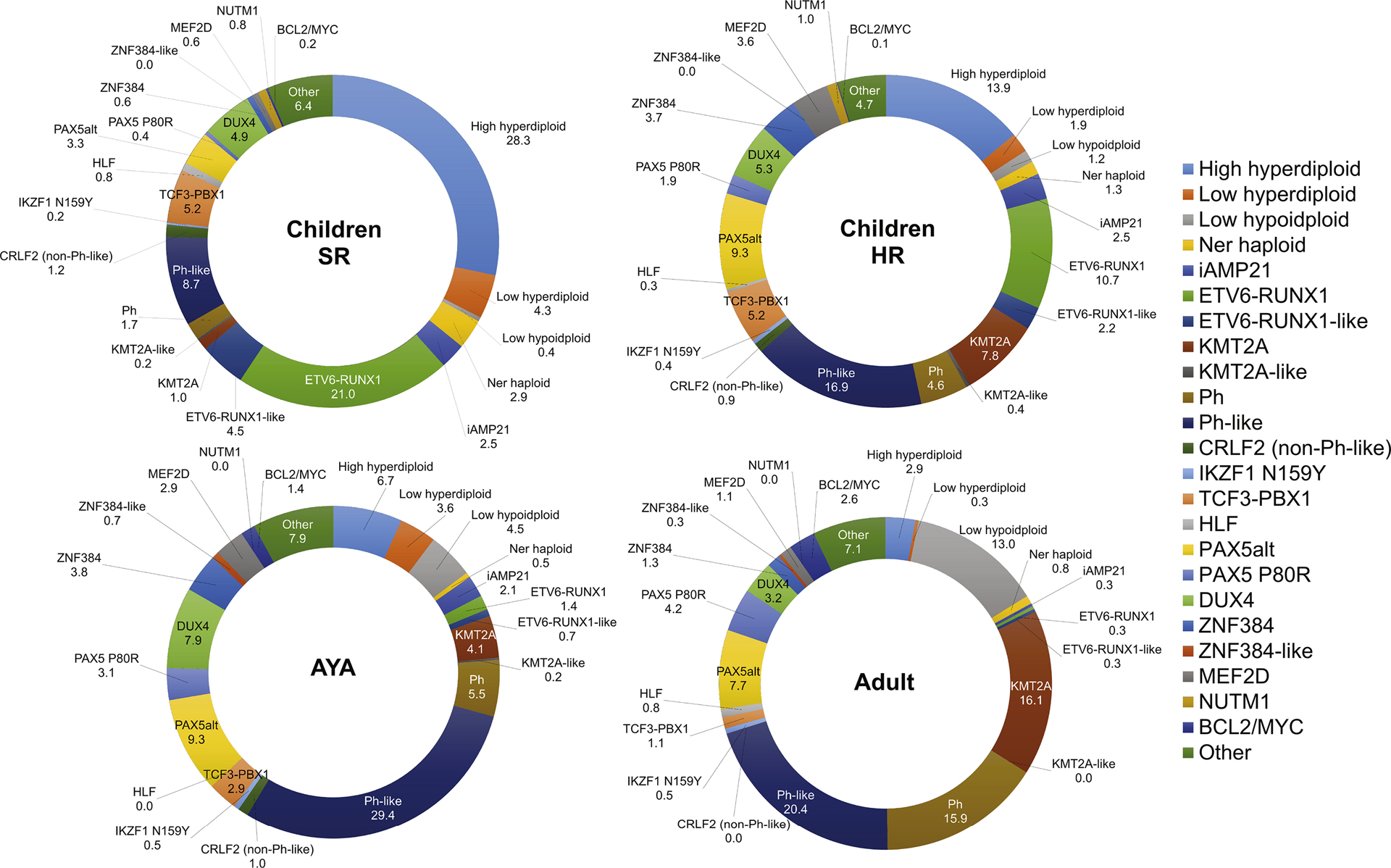
Prevalence of each major subtype in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) across age or risk. SR, standard risk; HR, high risk [7].
B-cell Precursor Acute Lymphoblastic Leukemia
Recurring chromosomal alterations
High hyperdiploidy (51–67 chromosomes) is one of the most common subtypes of ALL in children, comprising ~30% of pediatric BCP-ALL and is associated with a favorable prognosis (Table 1) [7,11]. Genetically, high hyperdiploidy is characterized by a nonrandom gain of chromosomes, typically +X, +4, +6, +10, +14, +17, +18, and +21 [11]. In particular, combined gain of chromosome 4, 10, and 17 is associated with favorable prognosis [12]. Alterations involving the Ras pathway (KRAS, NRAS, FTL3, PTPN11) and epigenetic modifiers (CREBBP, WHSC1) are frequent genetic events [11]. Importantly CREBBP mutations in the histone acetyl transferase (HAT) domain are generally selected for and preserved during disease evolution, and with Ras pathway mutations are common in aneuploid B-ALL cases, including those with hyperdiploidy and hypodiploidy [13].
Table 1.
Characteristics of each BCP-ALL subtype.
| Molecular subtype | Category | Median age (yrs) | Peak prevalence | Genomic alterations | Characteristics | Ref |
|---|---|---|---|---|---|---|
| High hyperdiploid (51–67 chromosomes) | Aneuploid | 4 | Children (25%) | Ras pathway, epigenetic modifiers |
|
[11] |
| Low hypodiploid (32–39 chromosomes) | Aneuploid | 47 | Adults (10–15%) | IKZF2 deletion, biallelic TP53 alterations |
|
[14] |
| Near haploid (24–31 chromosomes) | Aneuploid | 5.4 | < 3% in all ages | Ras pathway (NF1), IKZF3 deletions |
|
[14] |
| iAMP21 | Copy number gain | 10 | < 3% in children and AYA | Gain of three or more extra copies of a region of chromosome 21 including RUNX1 |
|
[21] |
| ETV6-RUNX1 | TF rearrangement | 4 | Children (25%) | deletion of the non-rearranged ETV6 allele, PAX5 deletions, WHSC1 mutations |
|
[26,28] |
| ETV6-RUNXHike ALL | TF rearrangement | 3 | Children (3%) | Alterations (fusions/deletions) in ETV6, IKZF1, TCF3 |
|
[7,25,31] |
| TCF3-PBX1 | TF rearrangement | 8 | Children (8%) |
|
[7,33] | |
| TCF3-HLF | TF rearrangement | 15 | < 1% in all ages | PAX5 deletions, Ras pathway |
|
[7,33] |
| KMT2A-rearranged | TF rearrangement | 40 | Infants (80%) and adults (15%) | Ras pathway (subclonal), PI3K pathway |
|
[37] |
| BCR-ABL1 (Ph+) | Kinase driven | 40–45 | Adults (40–50%) | IKZF1 alterations, CDKN2AIB deletions |
|
[41,51,10] |
| Ph-like | Kinase driven | 21 | AYA (25–30%) | Multiple kinase alterations, IKZF1 alterations, CDKN2AIB deletions |
|
[41,51] |
| DUX4-rearranged | TF rearrangement | 14.3 | AYA (8%) | ERG deletions (polyclonal), IKZF1 deletions, Ras pathway |
|
[60–62] |
| MEF2D-rearranged | TF rearrangement | 14 | AYA (7%) | Ras pathway |
|
[64,66] |
| ZNF384-rearranged | TF rearrangement | 15 | AYA (5%) | Ras pathway, epigenetic modifiers |
|
[61,69,70] |
| PAX5alt | Other TF driven | 10 | Children (10%) | PAX5 rearrangements |
|
[7] |
| PAX5 P80R | Other TF driven | 22 | Adults (4%) | Ras pathway, JAK-STAT pathway |
|
[7] |
| NUTM1-rearranged | TF rearrangement | 3 | Children (l%) |
|
[7] | |
| IKZF1 N159Y | Other TF driven | < 1% in all ages | Gain of whole chromosome 21 |
|
[7] | |
| BCL2/MYC-rearranged | Other TF driven | 48 | AYA and adults (3%) |
|
[7] |
Hypodiploid ALL is further subdivided into two principal subtypes with unfavorable prognosis according to the severity of aneuploidy: near haploidy (24–31 chromosomes) and low hypodiploidy (32–39 chromosomes) [14,15]. Accurate identification of hypodiploid ALL is important in view of the poor prognosis, and inherited genetic basis of low hypodiploid ALL in children. Duplication of the aneuploid genome, or masked hypodiploidy is common and may be mistaken for high hyperdiploidy [3]. This can be distinguished in that masked hypodiploidy typically has diploid and tetraploid chromosomes, whereas hyperdiploidy has a mixture of triploid and some tetraploid (e.g. 21, X), that is, the specific gained chromosomes differ. Although still associated with unfavorable prognosis, minimal residual disease (MRD) risk-stratified therapy has improved the outcome of hypodiploid ALL [16]. Near haploid ALL presents at a younger age and leukemic cells exhibit frequent gene alterations activating the Ras pathway (particularly NF1) and inactivating mutations/deletions of IKZF3 (AIOLOS) [14]. Importantly, unlike all other chromosomes that may be aneuploid in hypodiploid ALL, loss of chromosome 21 is not observed even in near haploidy, nor in other forms of ALL [14]. Low hypodiploid ALL is rare but increases with age. The pattern of aneuploidy within low hypodiploidy is not random, and as with near haploidy, chromosome 21 is universally retained [14]. Importantly, almost all low hypodiploid ALL has biallelic alterations of TP53 and 43% of pediatric (but not adult) cases harbor germline TP53 alterations indicating that low hypodiploid ALL is a manifestation of Li-Fraumeni syndrome [17]. Frequent secondary alterations include IKZF2 (HELIOS), RBI, and CDKN2A/CDKN2B [14]. Hypodiploid ALL cells have sensitivity to PI3K and BCL2 inhibitors, which might be a promising target [14,18].
Intrachromosomal amplification of chromosome 21 (iAMP21) is more common in older children and is characterized by gain of three or more extra copies of a region of chromosome 21 including RUNX1 generated by breakage-fusion-bridge cycles and chromothripsis [19,20]. The germline Robertsonian translocation rob(15;21) or a germline ring chromosome 21 are associated with a markedly elevated risk of iAMP21 [21]. Patients with iAMP21 usually lack other key cytogenetic alterations [3]. Clinically, this subtype showed a poor outcome with high rate of relapse when they treated as standard risk, however, intensive therapy can greatly improve the outcome [22,23].
ETV6-RUNX1 and ETV6-RUNX1-like ALL
The t(12:21)(p13:q22) translocation gives rise to ETV6-RUNX1 fusion which is the most common fusion in BCP-ALL (20%−25% in children) with a favorable prognosis [7,24]. In general, this translocation is cryptic on cytogenetic analysis, and leukemic cells have a distinct immunophenotype (CD27 positive and CD44 low/negative) [25]. The ETV6-RUNX1 fusion may be identified in umbilical cord blood, and thus, is considered to arise in utero as a leukemia-initiating alteration [24]. However, ETV6-RUNX1 itself is insufficient to induce overt leukemia and requires the prolonged latency with additional genetic events including deletion of the non-rearranged ETV6 allele, focal deletion of PAX5 and WHSC1 [4,24,26–28]. This is consistent with heterogeneity in the sub-clonal composition of ETV6-RUNX1 ALL [24,29,30].
Recent studies defined a new subtype, ETV6-RI JNXHike ALL, showing similar gene expression profiles and immunophenotype to ETV6-RUNX1 ALL despite the lack of ETV6-RUNX1 fusion [7,8,25,31]. As with ETV6-RUNX1 ALL, ETV6-RJJNX1-like ALL shows the highest prevalence in children and relatively favorable outcome [6,25,31]. This subtype includes several alternate rearrangements in ETV6 (e.g. ETV6-ELMO1), IKZF1 (e.g. IKZF1-ETV6), TCF3 (e.g. TCF3-FLII1), and FUS-ERG as well as copy number alterations in ETV6, IKZF1, and ARPP21, suggesting that alteration of multiple ETS and other transcription factors are converging on the same mechanism of transformation (although not ERG, which is distinct in the DJX4-rearranged ALL) [7,25,31].
TCF3-PBX1/TCF3-HLF
The translocation t(1;19)(q23;p13) generating TCF3-PBX1 fusion is present in 5%−6% of pediatric BCP-ALL and TCF3-PBX1 ALL associates with a pre-B immunophenotype expressing cytoplasmic immunoglobulin heavy chain [3,4]. This subtype was previously considered as high risk due to higher central nervous system involvement and relapse, however with present intensive treatment, TCF3-PBX1 ALL is classified as favorable or intermediate risk [3]. Conditional activation of TCF3-PBX1 in B cell progenitors showed enhancement of self-renewal and led to leukemia with PAX5 deletion and activation of JAK-STAT and Ras pathways after a pre-leukemia phase [32]. Although alterations of JAK-STAT and Ras pathways are not common in TCF3-PBX1 ALL, targeting pre-BCR signaling by inhibition of the JAK/STAT pathway might be a good option [7,32,33]. Importantly, TCF3-PBX1 ALL exhibits sensitivity to dasatinib and ponatinib, but not imatinib, which occurs as a result of inhibition of pre-BCR signaling by SRC kinases. Due to compensatory upregulation of ROR1 expression, combination with ROR1 inhibition could enhance the sensitivity of dasatinib [34].
A variant of the t(1;19) translocation, t(17;19)(q22;p13), generates TCF3-HLF fusion, which defines a rare subtype of ALL associated with an extremely poor prognosis [3,7]. Comparison between TCF3-PBX1 and TCF3-HLF ALL revealed distinct gene expression profiles and mutational landscape [33]. TCF3-HTF ALL exhibited stem cell and myeloid features with enrichment of PAX5 deletions and alterations of Ras pathway genes [7,33]. TCF-HLF fusion works as a pioneer transcription factor to recruit EP300 for driving MYC and shows vulnerability to EP300 inhibition [35]. Primary cells with TCF3-HLF show sensitivity to the BCL2 inhibitor venetoclax (ABT-199), representing a potential target therapeutic approach [33].
KMT2A-rearranged ALL
KMT2A (MLL) on chromosome 11q23 is rearranged to more than 80 different partner genes, and these rearrangements describe a distinct subtype of leukemia with both lymphoid and myeloid features and poor outcome [3,36]. This subtype is typically of the pro-B phenotype, lacking CD10 expression, with co-expression of myeloid markers. Approximately 80% of KMT2A-rearranged ALL is observed in infants, in whom KMT2A rearrangement is acquired in utero [4,36]. There is also a second peak in prevalence in adults and more than 75% of cases are fused to AFF1 [36]. Less frequently exposure to topoisomerase II inhibitors induces therapy-related leukemia with KMT2A rearrangements [36]. Intriguingly KMT2A breakpoints in most infant ALL and therapy-related leukemia are similar, suggesting common mechanism of rearrangement [36]. In infant ALL, the most commonly perturbed pathways include PI3K and Ras pathways [37,38]. KMT2A rearrangement results in assembly of a large multi-protein complex that results in aberrant transcription and epigenetic dysregulation (e.g. high level of H3K79 methylation) through recruiting excessive DOT1L, H3K79 methyltransferase, which interacts with multiple KMT2A rearrangement partners [39]. Thus, targeted therapy against this complex is developing, which includes inhibition of DOT1L, bromodomain, Menin, and the polycomb repressive complex [36,39,40].
BCR-ABL1 (Ph+) ALL and Ph-like ALL
The Philadelphia chromosome (Ph), derivative chromosome 22, derives from the reciprocal t(9;22)(q34;q11) translocation, and encodes BCR-ABL1 [7,41]. Although BCR-ABL1 ALL is associated with poor prognosis, the addition of tyrosine kinase inhibitors (TKIs) to the conventional chemotherapy has greatly improved outcome [4,42,43]. However, the major secondary cooperative alterations, IKZF1 deletions, are still predictive of an unfavorable outcome irrespective of TKI exposure [42]. Moreover, mutations in the kinase domain of ABL1 (most frequently T315I) induces TKI resistance, which is more common in TKI monotherapy or in adults treated with less intensive chemotherapy, and less common in children treated with intensive chemotherapy [44]. In addition to ponatinib, targeting Y-catenin, a key component of BCR-ABL1 downstream, is promising irrespective of TKI resistance [4,45]. The deleterious effect of IKZF1 mutations is in part due to loss of IKZF1 repression of stemness and cell-cell adhesion, phenotypes that may be reversed by rexinoids (via agonism of rexinoid X receptor alpha, which induces expression of wild type IKZF1) and focal adhesion kinase inhibitors (which inhibit downstream integrin signaling pathways) [46]. Before consensus guidelines for MRD assessment in BCR-ABL1 ALL have been provided [47], several approaches have been tested for MRD monitoring (genome or transcriptome BCR-ABL1 and Ig/TCR rearrangements) [48]. Importantly, some patients showed discrepancy of MRD results in Ig/TCR and BCR-ABL1 transcript, which was caused by the presence of BCR-ABL1 fusion outside of the blast population [48]. This BCR-ABL1 positive clonal hematopoiesis is suggestive of a CML-like disease exhibiting lymphoid blast crisis.
Ph-like or BCR-ABL1-like ALL exhibits a gene expression profile similar to BCR-ABL1 ALL despite the lack of the BCR-ABL1 fusion [49,50]. The prevalence and outcome of Ph-like ALL are similar to those of BCL-ABL1 ALL, increasing in incidence with age and associated with elevated MRD levels and/or higher rates of treatment failure, although there is a difference in the prevalence of Ph-like ALL in AYA (higher than BCR-ABL1 ALL) [41,51]. Similar to BCR-ABL1 ALL, IKZF1 alterations are characteristic, which result in acquisition of stem cell-like features and poor responsiveness to TKI. Although Ph-like ALL is genetically heterogeneous, this subtype can be fall into four main groups (Table 2): (1) alterations driving JAK-STAT signaling, including rearrangements and mutations/deletions of CRLF2, JAK2, EPOR, TYK2, IL7R, SH2B3, JAK1, JAK3, TYK2, IL2RB, (2) fusions involving ABL-class genes (ABL1, ABL2, CSF1R, LYN, PDGFRA, PDGFRB); (3) mutations activating Ras signaling (NRAS, KRAS, PTPN11); and (4) less common fusions (FLT3, FGFR1, NTRK3) [4,6,41,51]. Among them, CRLF2 alterations are found in almost half of Ph-like ALL in AYAs and adults. CRLF2 is located in the pseudoautosomal region of the sex chromosomes (PAR1) at Xp22.3/Yp11.3, and its alterations were incorporated into the criteria for “IKZF1plus”, a subtype with higher risk of relapse defined by co-occurrence of the IKZF1 deletion with deletion of CDKN2A, CDKN2B, PAX5, and/or PAR1 region in the absence of ERG deletion [52]. Importantly, most alterations in Ph-like ALL can, theoretically be targeted by FDA-approved TKIs: JAK-STAT signaling (JAK inhibition); ABL-class fusions (ABL inhibitor); FLT3 and NTRK3 fusions (FLT3 and NTRK3 inhibitor) [6,41,53–55]. Combination of kinase inhibitors against multiple signaling shows synergistic effect in PDX model of CRLF2/JAK mutant (JAK and PI3K/mTOR inhibitors) and ABL/PDGFR mutant (dasatinib and PI3K/mTOR inhibitor) [56]. Some of these (ruxolitinib, imatinib, dasatinib, ponatinib) are being tested in frontline studies [57–59].
Table 2.
Four main groups in Ph-like ALL
| Category | Kinase gene | Representative alterations | Target therapy |
|---|---|---|---|
| JAK-STAT signaling | CRLF2 | mutations (F232C), fusions (CSF2RA, IGH, P2RYS) | JAK inhibitor |
| EPOR | fusions (IGH, IGK, LAIR1, THADA) | JAK inhibitor | |
| TYK2 | fusions (MYB, SMARCA4, ZNF340) | TYK2 inhibitor | |
| TSLP | fusions (IQGAP2) | JAK inhibitor | |
| SH2B3 | Deletion/mutations | JAK inhibitor | |
| IL7RA | mutations, indels | JAK inhibitor | |
| JAK1 | mutations (V658F) | JAK inhibitor | |
| JAK2 | mutations (R683G), fusions (ATF7IP, BCR, EBF1, ETV6, PAX5, SNX29, SSBP2, ZNF340) | JAK inhibitor | |
| JAK3 | mutations | JAK inhibitor | |
| IL2RB | fusions (MYH9) | JAK inhibitor | |
| ABL signaling | ABL1 | fusions (ETV6, FOXP1, NUP214, RANBP2) | Imatinib/dasatinib |
| ABL2 | fusions (PAG1, RCSD1, ZC3HAV1) | Imatinib/dasatinib | |
| CSF1R | fusions (MEF2D, SSBP2, TBL1XR1) | Imatinib/dasatinib | |
| LYN | fusions (GATAD2A, NCOR1) | Imatinib/dasatinib | |
| PDGFRA | fusions (FIP1L1) | Imatinib/dasatinib | |
| PDGFRB | fusions (ATF7IP, ETV6, SNX29, SSBP2, ZMYNDS) | Imatinib/dasatinib | |
| Ras signaling | NRAS | mutations | |
| KRAS | mutations | ||
| PTPN11 | mutations | ||
| NF1 | Mutations/deletions | ||
| BRAF | mutations | ||
| CBL | fusions (KANK1) | ||
| Other signaling | FLT3 | FLT3-ITB, fusions (AMYM2) | FLT3 inhibitor |
| NTRK3 | fusions (ETVS) | NTRK3 inhibitor | |
| FGFR1 | fusions (BCR) | Ponatinib | |
| PTK2B | fusions (KDM6A, STAG2, TMEM2) | FAK inhibitor | |
| DGKH | fusions (ZFAND3) | ||
| BLNK | fusions (DNTT) |
DUX4-rearranged ALL
Rearrangement and overexpression of the homeobox transcription factor gene DUX4 constitutes a distinct subgroup, DUX4-rearranged ALL [7,31,60,61]. This subtype is also driven by deregulation of the ETS family transcription factor ERG, and comprises up to 5%–10% of BCP-ALL with a slight peak in AYAs. It has a distinct immunophenotype (CD2 and CD371 positive) and favorable outcome [7,31,60–62]. Deregulation of DUX4 is induced by rearrangement to strong enhancer elements, most commonly the immunoglobulin heavy chain (IGH) enhancer, which results in expression of a C-terminal truncated DUX4 protein that is not normally expressed in B cells [60,61]. This truncated isoform of DUX4 then binds to an intragenic region of ERG resulting in transcriptional deregulation and expression of multiple aberrant coding and non-coding ERG isoforms, and deletion of ERG in up to 70% of DUX4-rearranged cases [60]. One isoform is ERGalt, a C-terminal fragment that retains the DNA-binding and transactivating domain of ERG, exerts dominant negative effect and is transforming [60]. The deletions of ERG are commonly polyclonal, and may encompass the region of ERG that encodes the N-terminus of ERGalt. These findings support a model in which an initiating rearrangement of DUX4 results in gross transcriptional deregulation of ERG and primes the locus for RAG-mediated deletion; and loss of ERG activity, either through deletion and/or expression of ERGalt, cooperates with DUX4 deregulation in leukemogenesis [60,63]. DUX4-rearrangement is associated with favorable outcome in children and adults, even with IKZF1 deletion [60,61]. As clonal ERG deletions are not present in all DUX4-rearranged cases, the use of ERG deletion as a surrogate for this subtype, as is used in the definition of IKZFplus is suboptimal and should be avoided. Accurate identification of this favorable subtype of ALL requires identification of DUX4 rearrangement (either directly or through identification of elevated DUX4 expression) [63]. In this regard, detection of strong CD371 cell surface expression by flow cytometry might serve as a promising surrogate marker for this subtype [62].
MEF2D-rearranged ALL
Rearrangement of MEF2D is associated with older age of onset and relatively inferior outcome due to early relapse [64–67]. MEF2D-rearranged ALL is characterized by an aberrant immunophenotype (low or absent expression of CD10, high expression of CD38 and cytoplasmic μ chain), mature B-ALL-like morphology, and distinct expression profiles [64,66,67]. The N-terminal of MEF2D is fused to several partner genes, retaining its DNA binding domain [64,66,67]. High expression of MEF2D fusion protein is induced by evasion from miRNA-mediated degradation [68], and results in transcriptional activation of MEF2D targets [66]. Dysregulated MEF2D targets includes overexpression of HDAC9, which confers therapeutic sensitivity to HDAC inhibitors such as panobinostat [66].
ZNF384-rearranged ALL
ZNF384-rearranged ALL is a unique subtype that can be diagnosed as BCP-ALL or B/myeloid mixed phenotype acute leukemia (MPAL) [69]. In BCP-ALL, peak age of onset and prognosis varies by fusion partners: EP300-ZNF384 (median age 11, excellent outcome); TCF3-ZNF384 (median age 5, frequent late relapse) [7,70,71]. In contrast, ZNF384-rearranged ALL shows uniformly distinct immunophenotype (weak CD10 and aberrant CD13 and/or CD33 expression) and gene expression profiles [69,70]. The secondary genomic alterations and gene expression profiles of ZNF384-rearranged BCP-ALL and MPAL cases are essentially indistinguishable, and both have lineage plasticity at diagnosis and relapse (lymphoid disease to myeloid disease and vice versa) [6,69]. Transplantation of sorted subpopulations of cells from ZNF384-rearranged cell line showed propagation of the immunophenotypic diversity [69]. Moreover, twin case of ZNF384-rearranged ALL indicated a fetal hematopoietic progenitor as the cell of origin of this case [72]. These are clinically important results which confer a compelling genomic rationale that ZNF384-rearranged cases should be treated in a uniform strategy rather than using lymphoid or lymphoid leukemia-directed therapy according to predominant lineage [6]. In this regard, FLT3 overexpression is characteristic and can be targeted with the multi kinase inhibitor sunitinib [6].
PAX5-driven ALL (PAX5alt/PAX5 P80R)
The paired box DNA-binding transcription factor PAX5 is an essential regulator of the early stages of B cell development, and PAX5alterations are important in the pathogenesis of BCP-ALL as initiating or cooperation lesions. These include (1) disease initiating alterations (PAX5 P80R, rearrangements/focal intragenic amplifications in P4X5-altered ALL [PAX5alt]); (2) secondary lesions (e.g. PAX5 focal deletions in 30% of ETV6-RUNX1 ALL, and PAX5mutations in multiple subtypes), and (3) germline alterations that predispose to ALL [5,7,8,26,74]. In mouse models, Pax5 heterozygosity cooperates with constitutive activation of the JAK-STAT pathway in the development of BCP-ALL, supporting its role as a haploinsufficient tumor suppressor [73]. PAX5alt represents a group of cases with similar leukemic cell gene expression profiles, but diversity in the nature of underlying PAX5 alterations. These include (1) cases with diverse (>20) PAX5 rearrangements that typically preserve the N-terminal DNA-binding domain of PAX5, but with loss of the C-terminal transactivation domain; (2) cases with focal intragenic amplification of the PAX5 DNA-binding paired domain (PAX5amp), and (3) cases with sequence mutations. Within this group, specific lesions are associated with variation in gene expression profile: for example, cases with PAX5ETV6 rearrangement, or compound heterozygosity for p.Arg38 and p.Arg140 mutations in the DNA-binding paired domain have distinct gene expression profiles. PAX5alt is most common in children and the AYA population [7].
The PAX5 P80R subtype is characterized by the presence of the PAX5 P80R mutation with inactivation of the wild-type PAX5 allele by deletion, loss-of-function mutation or copy-neutral loss of heterozygosity [7,75]. Notably, heterozygous Pax5P80R/+ knock-in mice develop transplantable BCP-ALL, with genetic inactivation of the wildtype Pax5 allele [7]. Thus, biallelic PAX5 alterations are a hallmark of this subtype, and sequence mutations of lymphoid transcription factors such as PAX5 P80R and IKZF1 N159Y (see below) may be initiating events in leukemogenesis. The prevalence of PAX5 P80R increases with age and is associated with intermediate to favorable prognosis [7,74]. Additional important cooperating lesions include structural rearrangements of chromosomal arms 9p and 20q, which associate with the presence of dic(9:20) [74,75]. Moreover, mutations in the Ras and JAK-STAT pathway members are particularly enriched, highlighting the potential for targeted therapies [7,74].
Other subtypes of BCP-ALL.
BCP-ALL with NUTM1 rearrangements is a rare distinct subtype observed exclusively in children [7,8]. NUTM1 is a chromatin modifier, recruiting EP300 to increase local histone acetylation [76]. While the common partner, BRD9-NUTM1 is reported in BCP-ALL, BRD4-NUTM1 is a hallmark of NUT midline carcinoma (NMC) and acts to repress differentiation in NMC by widespread repression of histone acetylation, indicating therapeutic approach with bromodomain and HDAC inhibitors [6,76].
IKZF1 alterations, like PAX5, are also common across the spectrum of B-ALL (particularly in BCR-ABL1-positive, Ph-like and DUX4 rearranged cases) but a specific mutation, IKZF1 pAsn159Tyr defines a subtype with gene expression profile cases [7,8]. In this subtype, the nonmutated wild-type allele of IKZF1 is retained, and most cases have concurrent gain of chromosome 21 [7]. This mutation induces misregulation of IKZF1 transcriptional activation, in part through distinctive nuclear mislocalization and enhanced intercellular adhesion [77].
T-cell Acute Lymphoblastic Leukemia
Genetic heterogeneity in T-ALL
T-ALL is derived from thymic T-cell progenitors. T-ALL accounts for approximately 15% and 25% of pediatric and adult ALLs, respectively, and is twice as prevalent in males as in females [78–81]. Disruption of the normal differentiation, proliferation and survival during thymocyte development as a result of accumulation of genetic alterations results in the development of T-ALL [78,82]. NOTCH1 activating mutations and loss of CDKN2A locus are a hallmark secondary events in T-ALL, found in over 70% of cases [78,80,81]. The majority of T-ALL cases harbor leukemia-initiating rearrangements or mutations that result in the aberrant expression of transcription factors and oncogenes, including basic helix-loop-helix factors (TAL1, TAL2, LYL1), LMO genes (LMO1, LMO2), homeobox genes (TLX1 (HOXH), TLX3 (HOX11L2), NKX2.1, NKX2.2, NKX2.5, HOXA), MYB, and SPI1 [9,10,78,80,81]. These aberrant expression and dysregulated pathways confer unique gene expression signatures and classify T-ALL into several subtypes which reflect the point of differentiation arrest during T-cell development (Figure 3) [82]. T-ALL may also be classified by DNA methylation profiles which are associated with expression signatures, immunophenotypic profiles, and T-cell development stage [83]. In addition to fusion genes, somatic mutations and copy number alterations induce dysregulation in several pathways and genes, including JAK-STAT (IL7R, JAK1, JAK3, DNM2), Ras (NRAS, KRAS, and NF1), PI3K-AKT (PTEN, AKT1, PIK3CA PIK3CD), epigenetic regulators (PHF6, SUZ12, EZH2, KDM6A), transcription factors and regulators (ETV6, GATA3, RUNX1, LEF1, WT1, BCL11B), and translation regulators (CNOT3, RPL5, RPL10) [9,10,78,80,81].
Figure 3.
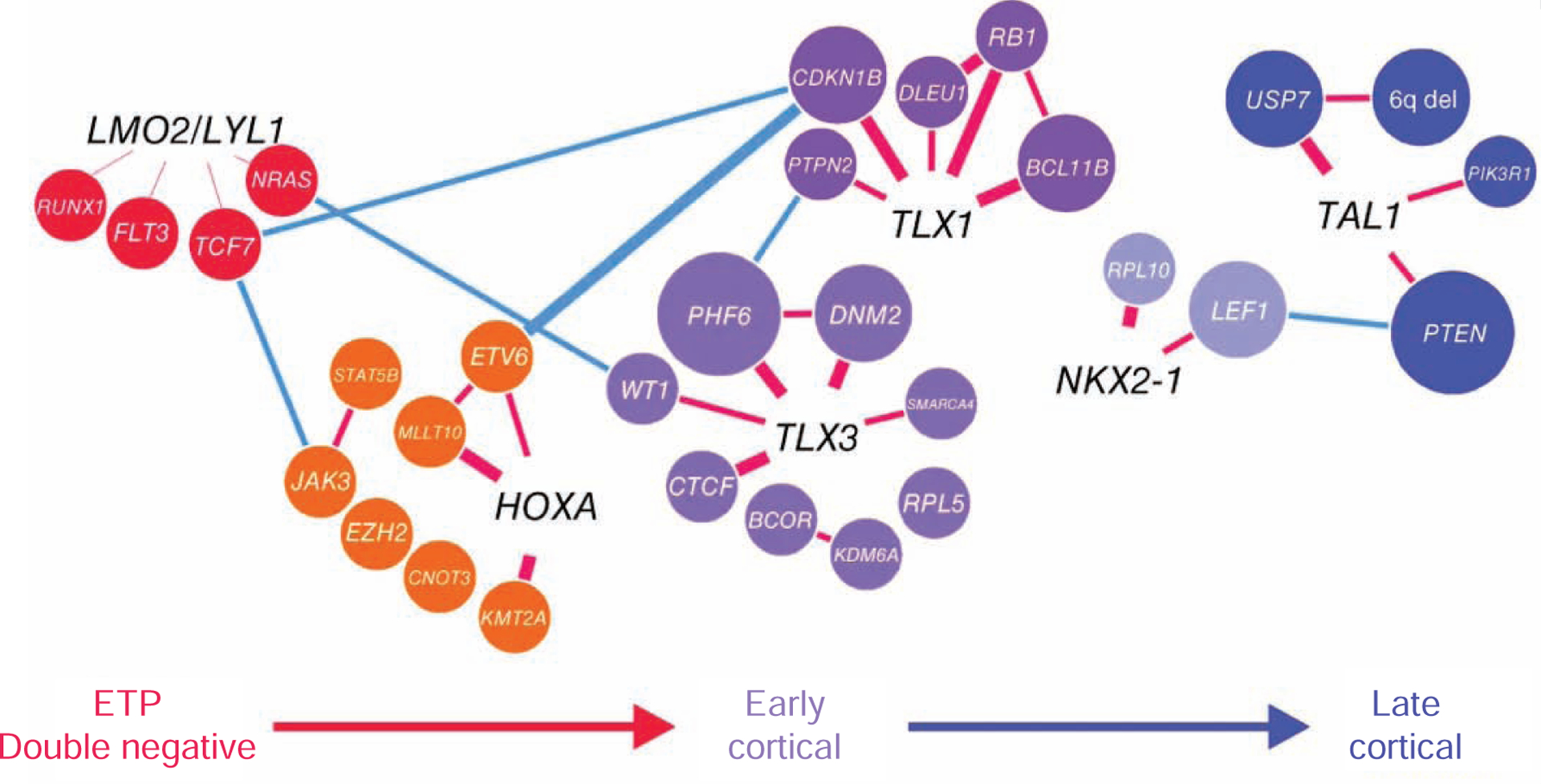
Associations between genetic alterations, each T-cell acute lymphoblastic leukemia (T-ALL) subgroups, and T-cell differentiation stages [9].
Early T-cell precursor (ETP) ALL
ETP ALL is a distinct subtype of immature leukemia, that despite its name, exhibits genetic and transcriptomic features more similar to a hematopoietic stem cell than a T cell precursor, and should be considered a subgroup of immature, lineage ambiguous leukemias that are variably classified by immunophenotype, rather than genetic/biologic features. Other cases in this spectrum include undifferentiated leukemia and T/myeloid mixed phenotype acute leukemia (T/M MPAL). ETP ALL is defined by an immunophenotype similar to the earliest stages of T-cell development (cytoplasmic CD3+, CD7+; CD8−, CD1a−, CD5weak) with aberrant expression of myeloid and/or stem-cell markers [84,85]. Previously, ETP-ALL has had an inferior outcome, although this is mitigated by contemporary risk-adapted therapy [86]. ETP-ALL is genetically characterized by somatic mutations in multiple signaling pathways (Ras, JAK-STAT, and epigenetic regulators) and transcription factors important for T-cell development [9,10,87]. Furthermore, ETP-ALL shows high-DNA methylation profiles at binding sites of polycomb repressive complex targets or components [83]. Importantly, ETP-ALL shares genomic and epigenomic features with T/M MPAL, in terms of frequent biallelic WT1 alterations, and alterations in other several transcription factors (ETV6, RUNX1, CEBPA) and signaling pathways (JAK-STAT, FLT3, Ras) [69,87]. These data suggest that they are similar entities in the spectrum of immature leukemias and both might have sensitivity to FLT3 and/or JAK inhibition.
T-ALL in early stages of cortical thymocyte maturation
T-ALL that associates with aberrant activation of homeobox genes shows CD1a+, CD4+, and CD8+ immunophenotype, reflecting a differentiation arrest in early stages of cortical thymocyte maturation, while some HOXA dysregulated cases show immature phenotype [80,84,88]. Clinically, this subtype of T-ALL represented favorable prognosis [10]. Genetically this subtype has the highest prevalence of both NOTCH1 and CDKN2A alterations [9,10,78]. Dysregulated expression of homeobox genes is mostly induced by chromosomal translocations and inversions that relocate homeobox genes under the control of strong enhancers in the TCR and BCL11B regulatory regions [9]. TLX1 and TLX3 rearranged T-ALL is representative and has a similar gene expression signatures, DNA methylation profiles, and overlapping downstream targets [9,10,83]. Notably, NUP214-ABL1 is found concurrently with TLX1 and TLX3 rearrangements and shows promising sensitivity to several TKIs [78]. Furthermore, due to frequent JAK-STAT pathway gene mutations in this subgroup, ruxolitinib, a JAK-STAT inhibitor, has been administered to patients with these alterations [58].
TAL1-driven T-ALL
T-ALL with aberrant expression of the TAL1 oncogene has a late cortical thymocyte immunophenotype (CD4+, CD8+, CD3+) and accounts for approximately 40% of T-ALL [9,10]. This subtype of T-ALL is further classified into two expression subtypes [10,88]. In normal T-cell development, TAL1 is transcriptionally silenced by double-positive stages [82]. Aberrant expression of TAL1 is induced by chromosomal translocations, the sub-microscopic interstitial deletion (STIL-TAL1), disruption of insulated neighborhoods [89], and heterozygous somatic indels in a noncoding intergenic element upstream of the TAL1 transcription start site [90]. Furthermore, TAL1 forms a positive auto-regulatory loop and complex with GATA3, RUNX1, and MYB [91]. Although TAL1 is such a major oncogene in T-ALL, only 30% of transgenic mice develop T-ALL after a latent period, and thus, additional abnormalities are required for leukemogenesis [79]. In addition, PI3K-AKT pathway gene mutations are frequently detected in TAL1-driven T-ALL [9,10], which associates with glucocorticoid resistance and can be reversed by the inhibition of this pathway [9,78].
NOTCH1 activating mutations in T-ALL
NOTCH1 acts as a ligand-dependent transcription factor and the NOTCH1 signaling pathway is essential for the commitment of T-cell lineage specification and for further T-cell development [82]. Aberrant activation of NOTCH1 pathway is mostly induced by somatic mutations that disrupt the negative regulatory region leading to ligand-independent activation, or impairment of the proteasomal degradation of intracellular domain of NOTCH1 which includes truncation of the PEST domain, NOTCH1 mutations in 3’ untranslated region, and FBXW7mutations [78,92–94]. These two types of NOTCH1 activating mutations co-occur in more than 20% of T-ALL, which has synergistic effects [92]. However, the oncogenic activity of NOTCH1 is dose dependent and most mutations in human T-ALL do not generate sufficient signals to initiate leukemia development [95]. Moreover, most T-ALL harbor multiple subclonal NOTCH1 activating mutations, and thus, these mutations are acquired as a late secondary event, which has been confirmed by clonal analysis using single-cell sequencing [9,93,96,97]. One of the main direct targets of NOTCH1 is MYC which is regulated by T-cell-specific NOTCH1-controlled distal enhancer called “NMe” [94,98]. Furthermore, NOTCH1 and MYC control many overlapping target genes to promote cell growth [9,94].
Due to the importance of NOTCH1 signaling in T-ALL, several preclinical studies inhibiting NOTCH1 signaling with Y-secretase inhibitors (GSIs) have been initiated. However, lack of cytotoxic antitumor responses and dose-limiting gastrointestinal toxicities still limit their direct translation into patient benefit, although the combination of GSIs and glucocorticoids is promising [78,99].
Relapsed ALL
Although cure rate of primary ALL has greatly improved with risk-adjusted therapy, relapsed ALL is still a leading cause of death for children mainly due to therapy resistance. This resistance is frequently induced by acquired mutations but also influenced by the bone marrow microenvironment. Recent genomic analyses of paired primary and relapsed ALL samples have revealed that these secondary mutations have acquired during the disease progression with Darwinian selection, highly branched clonal architectures, especially in early relapse (9–36 months) [29,93,100–102]. Furthermore, chemotherapy of ALL has been postulated to induce bona fide drug resistance mutations including NT5C2, PRPS1, NR3C1, and TP53 [100]. However, relapse-fated subclones present at diagnosis commonly exhibit drug resistance prior to the administration of any therapy [103].
One of the representative relapse specific somatic alterations is CREBBP alterations which occur in up to 20% of relapsed B-ALL and impair glucocorticoid sensitivity [13]. Most early relapse is associated with 6-MP resistance, as a result of NT5C2 gain-of function mutations, PRPS1 mutations, and loss of MSH6. NT5C2 mutations confer resistance to purine analogs at the cost of impaired tumor cell growth and reduced leukemia-initiating cell activity [104]. NT5C2 inhibitors might be promising, however, several problems lie ahead such as development of mutant specific inhibitor [105]. Importantly, NT5C2 and PRPS1 mutations are not detectable at primary samples even in a minor clone [100,104,106]. Other recurrent somatic alterations in relapsed ALL include mutations in SETD2, KDM6, and KMT2D (MLL2) [29,100,102]. Tracking of these mutations as MRD may offer the opportunity to identify the relapse-fated clone early in disease evolution and modulate therapy accordingly to circumvent relapse.
Summary
Recent development of sequence technology and integrated large-scale analyses have greatly improved our knowledge of the genetic basis of ALL in terms of identification of new subtypes and dysregulated pathways associated with therapeutic targets. Many clinically important alterations are not evident using conventional cytogenetic and molecular approaches, and optimal ALL diagnostics requires next-generation sequencing, with RNA-seq capturing most clinically relevant information [58]. These alterations have several major implications for clinical management including appropriate risk stratification, monitoring of treatment response, and most importantly development of precision medicine. Indeed, the use of TKIs has brought a great impact on the outcome of newly diagnosed BCR-ABL1 ALL, however, most targeted therapies have been tested as single agents in relapsed or refractory patients. In this point, several ongoing studies using targeted therapies in frontline treatment are underway. However, although the majority of ALL subtypes are found to be driven by alterations of transcription factor, they are not intuitively directly druggable. Newly developed antibody and cellular immunotherapy are efficacious in a proportion of patients, however many these approaches fail in a substantial proportion of patients. It is thus now critical to take these genomic discoveries forward to characterize mechanisms of leukemogenesis, and explore novel therapeutic approaches to find subtype specific vulnerabilities for the improvement of treatment strategy in future.
Practice Points.
Comprehensive genetic analysis has provided major implications for clinical management.
Several genomically-informed targeted therapies have been incorporated into frontline studies for the evaluation of the efficacy.
Exploring genetic basis to find subtype specific vulnerabilities is important for new therapeutic approaches.
Acknowledgements
The authors are supported by the St Jude Comprehensive Cancer Center Support Grant NCI P30 CA021765 and an NCI Outstanding Investigator Award R35 CA197695, the Henry Schueler 41&9 Foundation, and a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (to C.G.M.), and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
Declaration of competing interest
Shunsuke Kimura declares no conflict of interest. Charles Mullighan has received research funding from Abbvie, Pfizer and Loxo Oncology, and consulting fees from Illumina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Mullighan CG. Genomic Characterization of Childhood Acute Lymphoblastic Leukemia. Semin Hematol 2013;50:314–24. doi: 10.1053/j.seminhematol.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iacobucci I, Mullighan CG. Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol 2017;35JC0.2016.70.783. doi: 10.1200/jco.2016.70.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Harrison CJ, Moorman AV, Schwab C, Iacobucci I, Mullighan C. Childhood Acute Lymphoblastic Leukemia 2017:61–98. doi: 10.1007/978-3-319-39708-5_4. [DOI] [Google Scholar]
- [4].Roberts KG, Mullighan CG. The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Csh Perspect Med 2019:a034835. doi: 10.1101/cshperspect.a034835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pui C-H, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nature Reviews Clinical Oncology 2019;16:227–40. doi: 10.1038/s41571-018-0136-6. [DOI] [PubMed] [Google Scholar]
- [6].Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin Haematol 2019;32:101095. doi: 10.1016/j.beha.2019.101095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019;51:296 307. doi: 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li J-F, Dai Y-T, Lilljebjorn H, Shen S-H, Cui B-W, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc National Acad Sci 2018;115:201814397. doi: 10.1073/pnas.1814397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49:1211 1218. doi: 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Seki M, Kimura S, Isobe T, Yoshida K, Ueno H, Nakajima-Takagi Y, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 2017;49:1274 1281. doi: 10.1038/ng.3900. [DOI] [PubMed] [Google Scholar]
- [11].Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 2015;47:672–6. doi: 10.1038/ng.3301. [DOI] [PubMed] [Google Scholar]
- [12].Sutcliffe MJ, Shuster JJ, Sather HN, Camitta BM, Pullen J, Schultz KR, et al. High concordance from independent studies by the Children’s Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children’s Oncology Group (COG) initiative. Leukemia 2005;19:734–40. doi: 10.1038/sj.leu.2403673. [DOI] [PubMed] [Google Scholar]
- [13].Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013;45:242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Comeaux EQ, Mullighan CG. TP53 Mutations in Hypodiploid Acute Lymphoblastic Leukemia. Csh Perspect Med 2017;7:a026286. doi: 10.1101/cshperspect.a026286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mullighan CG, Jeha S, Pei D, Payne-Turner D, Coustan-Smith E, Roberts KG, et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 2015;126:2896–9. doi: 10.1182/blood-2015-09-671131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 2018;36:JCO.2017.75.521. doi: 10.1200/jco.2017.75.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Diaz-Flores E, Comeaux EQ, Kim KL, Melnik EM, Beckman K, Davis KL, et al. BCL-2 is a Therapeutic Target For Hypodiploid B-Lineage Acute Lymphoblastic Leukemia. Cancer Res 2019;79:2339 2351. doi: 10.1158/0008-5472.can-18-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Harrison CJ, Haas O, Harbott J, Biondi A, Stanulla M, Trka J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfurt-Munster study group. Brit J Haematol 2010;151:132–42. doi: 10.1111/j.1365-2141.2010.08314.x. [DOI] [PubMed] [Google Scholar]
- [20].Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014;508:98–102. doi: 10.1038/nature13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Harrison CJ, Schwab C. Constitutional abnormalities of chromosome 21 predispose to iAMP21-acute lymphoblastic leukaemia. Eur J Med Genet 2016;59:162–5. doi: 10.1016/j.ejmg.2016.01.006. [DOI] [PubMed] [Google Scholar]
- [22].Heerema NA, Carroll AJ, Devidas M, Loh ML, Borowitz MJ, Gastier-Foster JM, et al. Intrachromosomal Amplification of Chromosome 21 Is Associated With Inferior Outcomes in Children With Acute Lymphoblastic Leukemia Treated in Contemporary Standard-Risk Children’s Oncology Group Studies: A Report From the Children’s Oncology Group. J Clin Oncol 2013;31:3397–402. doi: 10.1200/jco.2013.49.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moorman AV, Robinson H, Schwab C, Richards SM, Hancock J, Mitchell CD, et al. Risk-Directed Treatment Intensification Significantly Reduces the Risk of Relapse Among Children and Adolescents With Acute Lymphoblastic Leukemia and Intrachromosomal Amplification of Chromosome 21: A Comparison of the MRC ALL97/99 and UKALL2003 Trials. J Clin Oncol 2013;31:3389–96. doi: 10.1200/jco.2013.48.9377. [DOI] [PubMed] [Google Scholar]
- [24].Sundaresh A, Williams O. Mechanism of ETV6-RUNX1 Leukemia. Adv Exp Med Biol 2017;962:201–16. doi: 10.1007/978-981-10-3233-2_13. [DOI] [PubMed] [Google Scholar]
- [25].Zaliova M, Kotrova M, Bresolin S, Stuchly J, Stary J, Hrusak O, et al. ETV6/RUNX1 nlike acute lymphoblastic leukemia: A novel BDcell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosomes Cancer 2017;56:608–16. doi: 10.1002/gcc.22464. [DOI] [PubMed] [Google Scholar]
- [26].Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007;446:758 764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- [27].Kuiper RP, Schoenmakers EFPM, Reijmersdal SV van, Hehir-Kwa JY, Kessel AG van, Leeuwen FN van, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 2007;21:1258–66.doi: 10.1038/sj.leu.2404691. [DOI] [PubMed] [Google Scholar]
- [28].Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014;46:116–25.doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mullighan CG, Mullighan CG, Phillips LA, Su X, Phillips LA, Su X, et al. Genomic Analysis of the Clonal Origins of Relapsed Acute Lymphoblastic Leukemia. Science 2008;322:1377 1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Anderson K, Lutz C, Delft FW van, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2010;469:356–61. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- [31].Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, Palffy S von, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 2016;7:11790. doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duque-Afonso J, Feng J, Scherer F, Lin C-H, Wong SHK, Wang Z, et al. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J Clin Investigation 2015;125:3667–80. doi: 10.1172/jci81158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz H-J, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 2015;47:1020 1029. doi: 10.1038/ng.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell 2012;22:656–67. doi: 10.1016/j.ccr.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huang Y, Mouttet B, Warnatz H-J, Risch T, Rietmann F, Frommelt F, et al. The Leukemogenic TCF3-HLF Complex Rewires Enhancers Driving Cellular Identity and Self-Renewal Conferring EP300 Vulnerability. Cancer Cell 2019. doi: 10.1016/j.ccell.2019.10.004. [DOI] [PubMed] [Google Scholar]
- [36].Winters AC, Bernt KM. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Frontiers Pediatrics 2017;5:4. doi: 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 2015;47:330–7. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Valentine MC, Linabery AM, Chasnoff S, Hughes AEO, Mallaney C, Sanchez N, et al. Excess congenital non-synonymous variation in leukemia-associated genes in MLL- infant leukemia: a Children’s Oncology Group report. Leukemia 2014;28:1235–41. doi: 10.1038/leu.2013.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen C-W, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med 2015;21:335–43. doi: 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019;36:660–673.e11. doi: 10.1016/j.ccell.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. New Engl J Med 2014;371:1005–15. doi: 10.1056/nejmoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Slayton WB, Schultz KR, Kairalla JA, Devidas M, Mi X, Pulsipher MA, et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J Clin Oncol 2018;36:2306 2314. doi: 10.1200/jco.2017.76.7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shen S, Chen X, Cai J, Yu J, Gao J, Hu S, et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Jama Oncol 2020;6. doi: 10.1001/jamaoncol.2019.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chang BH, Willis SG, Stork L, Hunger SP, Carroll WL, Camitta BM, et al. Imatinib resistant BCR-ABL1 mutations at relapse in children with Ph+ALL: a Children’s Oncology Group (COG) study. Brit J Haematol 2012;157:507–10. doi: 10.1111/j.1365-2141.2012.09039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Luong-Gardiol N, Siddiqui I, Pizzitola I, Jeevan-Raj B, Charmoy M, Huang Y, et al. Y-Catenin-Dependent Signals Maintain BCR-ABL1+ B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2019;35:649–663.e10. doi: 10.1016/j.ccell.2019.03.005. [DOI] [PubMed] [Google Scholar]
- [46].Churchman ML, Evans K, Richmond J, Robbins A, Jones L, Shapiro IM, et al. Synergism of FAK and tyrosine kinase inhibition in Ph+ B-ALL. Jci Insight 2016;1:e86082. doi: 10.1172/jci.insight.86082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pfeifer H, Cazzaniga G, Velden VHJ van der, Cayuela JM, Schafer B, Spinelli O, et al. Standardisation and consensus guidelines for minimal residual disease assessment in Philadelphia-positive acute lymphoblastic leukemia (PhD+DALL) by real-time quantitative reverse transcriptase PCR of e1a2 BCR-ABL1. Leukemia 2019;33:1910–22. doi: 10.1038/s41375-019-0413-0. [DOI] [PubMed] [Google Scholar]
- [48].Hovorkova L, Zaliova M, Venn NC, Bleckmann K, Trkova M, Potuckova E, et al. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 2017;129:2771–81. doi: 10.1182/blood-2016-11-749978. [DOI] [PubMed] [Google Scholar]
- [49].Boer MLD, Slegtenhorst M van, Menezes RXD, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009;10:125–34. doi: 10.1016/s1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. New Engl J Med 2009;360:470–80. doi: 10.1056/nejmoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen I-M, et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J Clin Oncol 2017;35:394–401. doi: 10.1200/jco.2016.69.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 2018;36:1240 1249. doi: 10.1200/jco.2017.74.3617. [DOI] [PubMed] [Google Scholar]
- [53].Roberts KG, Janke LJ, Zhao Y, Seth A, Ma J, Finkelstein D, et al. ETV6-NTRK3 induces aggressive acute lymphoblastic leukemia highly sensitive to selective TRK inhibition. Blood 2018;132:861 865. doi: 10.1182/blood-2018-05-849554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nardi V, Ku N, Frigault MJ, Dubuc AM, Tsai HK, Amrein PC, et al. Clinical response to larotrectinib in adult Philadelphia chromosome-like ALL with cryptic ETV6-NTRK3 rearrangement. Blood Adv 2020;4:106–11.doi: 10.1182/bloodadvances.2019000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schewe DM, Lenk L, Vogiatzi F, Winterberg D, Rademacher AV, Buchmann S, et al. Larotrectinib in TRK fusion-positive pediatric B-cell acute lymphoblastic leukemia. Blood Adv 2019;3:3499–502. doi: 10.1182/bloodadvances.2019000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen I-M, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2016;129:177–87. doi: 10.1182/blood-2016-05-707653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017;130:2064–72. doi: 10.1182/blood-2017-06-743252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Inaba H, Azzato EM, Mullighan CG. Integration of Next-Generation Sequencing to Treat Acute Lymphoblastic Leukemia with Targetable Lesions: The St. Jude Children’s Research Hospital Approach. Frontiers Pediatrics 2017;5:258. doi: 10.3389/fped.2017.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of Tyrosine Kinase Inhibitors in Ph-like Acute Lymphoblastic Leukemia harboring ABL-class Rearrangements. Blood 2019;134:1351–5. doi: 10.1182/blood.2019001244. [DOI] [PubMed] [Google Scholar]
- [60].Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 2016;48:1481 1489. doi: 10.1038/ng.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet 2016;48:569 574. doi: 10.1038/ng.3535. [DOI] [PubMed] [Google Scholar]
- [62].Schinnerl D, Mejstrikova E, Schumich A, Zaliova M, Fortschegger K, Nebral K, et al. CD371 cell surface expression: A unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019;104:haematol.2018.214353. doi: 10.3324/haematol.2018.214353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zaliova M, Potuckova E, Hovorkova L, Musilova A, Winkowska L, Fiser K, et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on method’s sensitivity. Haematologica 2019;104:haematol.2018.204487. doi: 10.3324/haematol.2018.204487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, et al. MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 2016;34:3451–9. doi: 10.1200/jco.2016.66.5547. [DOI] [PubMed] [Google Scholar]
- [65].Liu Y-F, Wang B-Y, Zhang W-N, Huang J-Y, Li B-S, Zhang M, et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. Ebiomedicine 2016;8:173–83. doi: 10.1016/j.ebiom.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 2016;7:13331. doi: 10.1038/ncomms13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, et al. Clinical and molecular characteristics of MEF2D fusion-positive precursor B-cell acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2018;104:128–37. doi: 10.3324/haematol.2017.186320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hirano D, Hayakawa F, Yasuda T, Tange N, Yamamoto H, Kojima Y, et al. Chromosomal translocation-mediated evasion from miRNA induces strong MEF2D fusion protein expression, causing inhibition of PAX5 transcriptional activity. Oncogene 2018;38:2263–74. doi: 10.1038/s41388-018-0573-9. [DOI] [PubMed] [Google Scholar]
- [69].Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018;562:84. doi: 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017;102:118–29. doi: 10.3324/haematol.2016.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hirabayashi S, Butler E, Ohki K, Kiyokawa N, Bergmann AK, Boer JM, et al. Acute Lymphoblastic Leukemia with Zinc-Finger Protein 384 (ZNF384)-Related Rearrangements: A Retrospective Analysis from the Ponte Di Legno Childhood ALL Working Group. Blood 2019;Supplement_1:652. doi: 10.1182/blood-2019-123236. [DOI] [Google Scholar]
- [72].Bueno C, Tejedor JR, Bashford-Rogers R, Gonzalez-Silva L, Valdes-Mas R, Agraz-Doblas A, et al. Natural history and cell of origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood 2019;134:900–5. doi: 10.1182/blood.2019000893. [DOI] [PubMed] [Google Scholar]
- [73].Dang J, Wei L, Ridder J de, Su X, Rust AG, Roberts KG, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood 2015;125:3609–17. doi: 10.1182/blood-2015-02-626127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Passet M, Boissel N, Sigaux F, Saillard C, Bargetzi M, Ba I, et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2018;133:280–4. doi: 10.1182/blood-2018-10-882142. [DOI] [PubMed] [Google Scholar]
- [75].Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kampf S, et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019;33:1895–909. doi: 10.1038/s41375-019-0430-z. [DOI] [PubMed] [Google Scholar]
- [76].French C NUT midline carcinoma. Nat Rev Cancer 2014;14:149–50. doi: 10.1038/nrc3659. [DOI] [PubMed] [Google Scholar]
- [77].Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015;28:343–56. doi: 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gianni F, Belver L, Ferrando A. The Genetics and Mechanisms of T-Cell Acute Lymphoblastic Leukemia. Csh Perspect Med 2019:a035246.doi: 10.1101/cshperspect.a035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tan TK, Zhang C, Sanda T. Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia. Int J Hematol 2018;109:1059. doi: 10.1007/s12185-018-2518-z. [DOI] [PubMed] [Google Scholar]
- [80].Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 2016;16:494 507. doi: 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- [81].Girardi T, Vicente C, Cools J, Keersmaecker KD. The genetics and molecular biology of T-ALL. Blood 2017;129. doi: 10.1182/blood-2016-10-706465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 2014;14:529 545. doi: 10.1038/nri3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kimura S, Seki M, Kawai T, Goto H, Yoshida K, Isobe T, et al. DNA methylation-based classification reveals difference between pediatric T-cell acute lymphoblastic leukemia and normal thymocytes. Leukemia 2019:1–6. doi: 10.1038/s41375-019-0626-2. [DOI] [PubMed] [Google Scholar]
- [84].Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- [85].Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 2009;10:147 156. doi: 10.1016/s1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Patrick K, Wade R, Goulden N, Mitchell C, Moorman AV, Rowntree C, et al. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Brit J Haematol 2014;166:421 424. doi: 10.1111/bjh.12882. [DOI] [PubMed] [Google Scholar]
- [87].Devidas M, Zhang J, Ding L, Wu G, Winter S, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481:157 163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Soulier J, Clappier E, Cayuela JM, Regnault A, Garda-Peydro M, Dombret H, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005;106:274 286. doi: 10.1182/blood-2004-10-3900. [DOI] [PubMed] [Google Scholar]
- [89].Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016;351:1454 1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346:1373 1377.doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012;22:209 221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269 271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- [93].Kimura S, Seki M, Yoshida K, Shiraishi Y, Akiyama M, Koh K, et al. NOTCH1 pathway activating mutations and clonal evolution in pediatric T-cell acute lymphoblastic leukemia. Cancer Sci 2019;110:784 794. doi: 10.1111/cas.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017;129:1124–33. doi: 10.1182/blood-2016-09-692582. [DOI] [PubMed] [Google Scholar]
- [95].Chiang MY, Xu L, Shestova O, Histen G, L’heureux S, Romany C, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest 2008;118:3181 3194. doi: 10.1172/jci35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bie JD, Demeyer S, Alberti-Servera L, Geerdens E, Segers H, Broux M, et al. Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 2018;32. doi: 10.1038/s41375-018-0127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Mansour MR, Duke V, Foroni L, Patel B, Allen CG, Ancliff PJ, et al. Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin Cancer Res 2007;13:6964 6969. doi: 10.1158/1078-0432.ccr-07-1474. [DOI] [PubMed] [Google Scholar]
- [98].Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014;20:1130 1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, Stanchina E de, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 2008;15:50–8. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2019. doi: 10.1182/blood.2019002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ferrando AA, Lopez-Otm C. Clonal evolution in leukemia. Nat Med 2017;23:1135 1145. doi: 10.1038/nm.4410. [DOI] [PubMed] [Google Scholar]
- [102].Oshima K, Khiabanian H, Silva-Almeida AC da, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc National Acad Sci 2016;113:11306–11. doi: 10.1073/pnas.1608420113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Dobson SM, Garcia-Prat L, Vanner RJ, Wintersinger J, Waanders E, Gu Z, et al. Relapse fated latent diagnosis subclones in acute B lineage leukaemia are drug tolerant and possess distinct metabolic programs. Cancer Discov 2020:CD-19–1059. doi: 10.1158/2159-8290.cd-19-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tzoneva G, Oshima K, Dieck CL, Ambesi-Impiombato A, Sanchez-Martin M, Madubata CJ, et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018;553:511 514. doi: 10.1038/nature25186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dieck CL, Ferrando AA. Genetics and mechanisms of NT5C2-driven chemotherapy resistance in relapsed ALL. Blood 2019;133:blood-2019-01-852392. doi: 10.1182/blood-2019-01-852392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Waanders E, Gu Z, Dobson SM, Antic Z, Crawford JC, Ma X, et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov 2020. doi: 10.1158/0008-5472.bcd-19-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008;453:110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]