

Abstract
Microglia, a type of CNS immune cell, have been shown to contribute to ethanol-activated neuronal death of the stress regulatory proopiomelanocortin (POMC) neuron-producing β-endorphin peptides in the hypothalamus in a postnatal rat model of fetal alcohol spectrum disorders. We determined whether the microglial extracellular vesicle exosome is involved in the ethanol-induced neuronal death of the β-endorphin neuron. Extracellular vesicles were prepared from hypothalamic tissues collected from postnatal rats (both males and females) fed daily with 2.5 mg/kg ethanol or control milk formula for 5 d or from hypothalamic microglia cells obtained from postnatal rats, grown in cultures for several days, and then challenged with ethanol or vehicle for 24 h. Nanoparticle tracking analysis and transmission electron microscopy indicated that these vesicles had the size range and shape of exosomes. Ethanol treatments increased the number and the β-endorphin neuronal killing activity of microglial exosomes both in vivo and in vitro. Proteomics analyses of exosomes of cultured microglial cells identified a large number of proteins, including various complements, which were elevated following ethanol treatment. Proteomics data involving complements were reconfirmed using quantitative protein assays. Ethanol treatments also increased deposition of the complement protein C1q in β-endorphin neuronal cells in both in vitro and in vivo systems. Recombinant C1q protein increased while C1q blockers reduced ethanol-induced C3a/b, C4, and membrane attack complex/C5b9 formations; ROS production; and ultimately cellular death of β-endorphin neurons. These data suggest that the complement system involving C1q-C3-C4-membrane attack complex and ROS regulates exosome-mediated, ethanol-induced β-endorphin neuronal death.
SIGNIFICANCE STATEMENT Neurotoxic action of alcohol during the developmental period is recognized for its involvement in fetal alcohol spectrum disorders, but the lack of clear understanding of the mechanism of alcohol action has delayed the progress in therapeutic intervention of this disease. Proopiomelanocortin neurons known to regulate stress, energy homeostasis, and immune functions are reported to be killed by developmental alcohol exposure because of activation of microglial immune cells in the brain. While microglia are known to use extracellular vesicles to communicate with neurons for maintaining homeostasis, we show here that ethanol exposure during the developmental period hijacks this system to spread apoptotic factors, including complement protein C1q, to induce the membrane attack complex and reactive super-oxygen species for proopiomelanocortin neuronal killing.
Keywords: apoptosis, complements, exosomes, fetal alcohol syndrome, microglia, proopiomelanocortin
Introduction
Fetal alcohol spectrum disorders (FASDs) indicate a range of maladies caused by chronic alcohol exposure during pregnancy. It is documented that there are ∼2%–5% of children born in the United States with FASD, and its prevalence is higher among low socioeconomic populations (May et al., 2018). The individuals with FASD show a broad spectrum of defects comprising definite physical, cognitive, psychomotor, behavioral, learning disabilities, and stress hyperactivity, and sometimes they may require long-term support (Sarkar et al., 2019; Wozniak et al., 2019). Previous research shows that prenatal ethanol exposures in preclinical models, during second- and third-trimester–equivalent periods of brain development, decrease brain weight (Maier et al., 1999a,b) and reduce the number of hippocampal neurons (Miller, 1995), cortical neurons (Jacobs and Miller, 2001), cerebral granule and Purkinje neurons (Maier et al., 1999a), and hypothalamic neurons (De et al., 1994). Within the hypothalamus, proopiomelanocortin (POMC) neurons that are known to regulate the hypothalamic, pituitary, and adrenal axis, energy homeostasis, and immune functions are reported to be susceptible to the adverse effects of fetal alcohol exposure (Sarkar et al., 2007; Hellemans et al., 2008). A large number of POMC neurons have been shown to undergo apoptotic cell death during prenatal alcohol exposure (Chen et al., 2006). The mechanism by which POMC neurons experience apoptosis following alcohol exposure is not completely understood. The involvement of microglia in ethanol's neurotoxic action in the fetal and adult brain has been determined by several researchers (Narita et al., 2007; Toyama et al., 2008; Crews and Nixon, 2009; Fernandez-Lizarbe et al., 2009; Boyadjieva and Sarkar, 2010; Shrivastava et al., 2017). These studies reported that activated microglia release neurotoxic cytokines or chemokines that might be responsible for neuronal damage. In addition, it has been shown that inflammatory cytokines are released from microglia in small extracellular vesicles (Bianco et al., 2005). Two of the most studied extracellular vesicles are exosomes (released to the extracellular space through exocytosis) and microvesicles (generated by budding and shed into the extracellular environment) (El Andaloussi et al., 2013; Iraci et al., 2016). Exosomes are small-membrane vesicles (ranging from 30 to 150 nm) comprised of a lipid bilayer, transmembrane proteins, and cytosolic components derived from their host cells. Once formed, exosomes can travel to nearby or distant cells and be taken up through a number of mechanisms, including membrane fusion, receptor-mediated endocytosis, and phagocytosis (Mulcahy et al., 2014). Once exosomes enter the target cell, their cargo is released into the cytoplasm and can alter cellular homeostasis. This form of signaling allows cells to communicate with each other via exosomes. Exosomes are known to transfer different biomolecules between cells, including proteins, lipids, and RNAs (van Niel et al., 2006). Moreover, the exosomes have been shown to carry mRNAs and microRNAs and are thereby capable of transferring genetic information between cells. In the CNS, exosomes can be released from all cell types, including microglia, oligodendrocytes, and neurons. It was proposed that the exosomes may contribute to the physiology of the nervous system and to the neuron-glia communication (Turola et al., 2012; Prada et al., 2013). Although neurons and microglia are known to be rich producers of exosomes, the role of exosomes in neuron-glial responses to ethanol-induced pathologic stress is unknown (Janas et al., 2016; Morton et al., 2018; Tseng et al., 2019). For this reason, we determined whether postnatal alcohol exposure (PAE) induces apoptosis of β-endorphin neurons through the release of proapoptotic exosomes from microglia. Therefore, we evaluated the role microglial activation played in the activity and cargo of hypothalamic exosomes. Since proteomics analysis of exosomal extracts from ethanol-activated microglia identified a large number of complement proteins, including C1q, we also determined whether and how C1q contributed to the exosome-mediated killing of β-endorphin neurons.
Materials and Methods
Animals
Adult Sprague Dawley rats were obtained from Charles River Laboratories. All animals were kept under standard lighting conditions (12 h lights on; 12 h lights off) and provided rodent chow and water ad libitum. These rats were bred to generate neonatal animals, which were used in this study. Animal care and treatment were performed in accordance with institutional guidelines, and protocols were approved by the Rutgers Institutional Animal Care and Facilities Committee and complied with National Institutes of Health policy.
In vivo studies
Postnatal rat pups (PND2; both sexes) were fed by gavage a milk formula containing 11.34% ethanol (v/v; 0.1-0.2 ml/animal; during a period of 1 min), yielding a total daily ethanol dose of 2.5 g/kg (AF), or isocaloric control (PF), or they were left in the litter with their mother (AD) as described by us previously (Shrivastava et al., 2017; Chastain et al., 2019). Gavage feeding was conducted at 10:00 A.M. and 12:00 P.M. from PND2 to PND6. After feeding, these pups were immediately returned to the litter. Additionally, some animals were treated subcutaneously with minocycline (45 µg/kg; 1 h before the first feeding) or C1NH, (SRP3318, 100 U/kg; 1 h before the first feeding). All of these drugs were purchased from Sigma Millipore. C1NH was reconstituted in water to a concentration of 100 U/kg of body weight and was used for subcutaneous injection (50-60 µl according to the pup's body weight was administered by using a BD insulin syringe with a 31 G needle, which was placed between the skin layers and the muscle to create a virtual space by lifting the skin). Two hours after the last feeding on PND6, some of the pups were transcardially perfused with 4% PFA, postfixed overnight, cryoprotected in 30% sucrose, and cut into 30 µm coronal sections for immunohistochemistry. Additional pups were killed, and the mediobasal hypothalamus (MBH; the mediobasal portion of the hypothalamus extended ∼1 mm rostral to the optic chiasma and just caudal to the mammillary bodies, lateral to the hypothalamic sulci, and dorsal to 2 mm deep) was collected for exosome preparation for measurement of protein measurement.
Primary microglial culture
In vitro studies were conducted using microglial cells. These cultures were prepared using the PND1 rat pups' hypothalami (both sexes) using the method described by us previously (Boyadjieva and Sarkar, 2013). Cells were plated in T25 flasks (1 × 106 cells/flask). Cultures were fed every 4 d with DMEM/MEM/Hams F12 in a 4:5:1 ratio with 10% FBS and an antibiotic solution (100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B). To confirm the purity of isolated microglia cells, the culture was stained with IBA-1, a microglial marker, or the astrocyte marker GFAP and visualized under microscope. The isolated microglia cultures were 99% IBA1-positive cells considered as pure microglial culture. Before treatment, microglial cells were fed with DMEM/MEM/Hams F12 containing serum supplement (30 nm selenium, 20 nm progesterone, 1 μm iron-free human transferrin, 100 μm putrescine, and 5 μg/ml insulin). Microglial cells were treated with various doses of ethanol (25-100 mm) or vehicle for 24 h. In some experiments, microglial cells were treated with 10 μm GW4869 with or without 50 mm ethanol or vehicle (control) for 24 h. The conditioning medium was harvested for exosome isolation. All chemicals were purchased from Sigma Millipore.
Primary POMC/β-endorphin neuronal culture
Enriched β-endorphin neuronal cultures were prepared by differentiation of neural stem cells in vitro by the methods described by us previously (Sarkar et al., 2008). In brief, pregnant rats of the Sprague Dawley strain at 16-18 d of gestation were killed, and the fetuses were removed by aseptic surgical procedure. Brains from the fetuses were immediately removed, and hypothalami were dissected out and placed in ice-cold Hanks balanced salt solution containing an antibiotic solution, 0.1% BSA, and 200 μm ascorbic acid (all from Sigma Millipore). The hypothalamic cells were dissociated using mechanical procedures (De et al., 1994), washed, and then incubated at 37°C for 5 min using the same medium. Neurons were separated from glial cells by filtering mixed hypothalamic cells through a 48 μm nylon mesh. Hypothalamic cells were then sedimented at 400 × g for 10 min, pellets were resuspended in HEPES-buffered DMEM (HDMEM, 4.5 g/L glucose), and cells were cultured into polyornithine (100 μg/ml)-coated 25 cm2 tissue culture flasks (3 million cells/flask) in HDMEM-containing 10% FBS and 1% penicillin/streptomycin. On day 2, the medium was replaced with HDMEM-containing 10% FBS, 33.6 μg/ml uridine, and 13.2 μg/ml 5-fluorodeoxyuridine (Sigma Millipore) to stop the overgrowth of astroglial cells. On day 3, the culture medium was replaced with HDMEM-containing serum supplement with antibiotics. Cells were maintained for the next 2 d with this medium. By this time, these cultures were ∼85%-90% neurons, as determined by MAP-2 positivity. Enriched hypothalamic neurons were maintained in HDMEM containing 10% FBS for 3 weeks. Cells were trypsinized and cultured weekly. By the beginning of the third week, many neurospheres started to develop. These spheres were separated and dissociated into single cells using trypsin/EDTA (Sigma Millipore) solution and cultured in poly-L-ornithine–coated 24-well plates (20,000 cells/well) in stem cell medium (DMEM-F-12, LIF, 0.1 mg/ml; L-glutamine, 10 mm; rat bFGF), 20 ng/ml; MEM amino acid solution, 0.5%; all of the chemicals were from Sigma Millipore, except bFGF, which was obtained from R&D Systems). Cells were cultured for a period of 2 weeks. These cells grew and developed secondary neurospheres. These neurospheres were then differentiated by treating them for 7 d with pituitary adenylate cyclase activating peptide (10 μm; SynPep) and dibutyryl cAMP (μm; Sigma Millipore) and then maintaining them in HDMEM-containing serum supplement and antibiotics without the drugs for 1 week. These differentiated cells were all stained for POMC-derived peptide β-endorphin.
Exosome preparation from microglial culture media
Microglia (1 × 106 cells/flask) were treated with ethanol (50 mm) or with vehicle. After 24 h of incubation, the cell culture media was harvested, centrifuged at 2000 × g for 30 min to remove cells and debris, and the supernatant containing the cell-free culture media was then transferred to a new tube and also strained by a 0.2 µm filter. Cell-free culture media were then centrifuged again at 300 × g and 3000 × g for 10 min and then at 10,000 × g for 30 min. The exosome isolation reagent (500 µl) from Invitrogen (catalog #4478359) was then added to 1 ml of cell-free culture media. The cell-free culture media and reagent mixture were thoroughly mixed by vortexing or pipetting up and down until it became a homogeneous solution. The mixture was then incubated at 4°C overnight. After incubation, the mixture was centrifuged at 10,000 × g for 1 h at 4°C. The supernatant was then aspirated and discarded from the tube. The exosomes were present in the pellet at the bottom of the tube. The pellet was resuspended in 100 µl of 1× PBS. The resuspended pellet was used for downstream analysis.
Exosome preparation from MBH tissue
Rat MBH, as previously described with modifications (Shrivastava et al., 2017), were dissected and placed in cold HBSS media (Sigma Millipore). Then MBH were mechanically dissociated in 5 ml syringes using 18-gauge needles followed by 21-gauge needles. The homogenate was filtered through 40 µm cell strainers and then centrifuged at 3000 × g for 10 min at 4°C to remove cellular debris. Each supernatant was then passed through a 0.2 µm filter. Next, the samples were centrifuged serially at 4°C: 300 × g for 10 min, 2000 × g for 10 min, and at 10,000 × g for 30 min. An equal amount of Exosome Isolation Reagent (Thermo Fisher Scientific) was added to each supernatant and incubated overnight at 4°C. On the next day, the mixtures were centrifuged at 10,000 × g for 60 min at 4°C and the exosome pellets were collected. The pellets were suspended in 50 µl PBS; and two equal aliquots were prepared: one for Western blot and one for CD13 aminopeptidase activity assay, transmission electron microscopy (TEM), and nanoparticle tracking analysis. For Western blot, resuspended exosomes were prepared in Exosome Resuspension Buffer (Total Exosome RNA and Protein Isolation Kit, Thermo Fisher Scientific). Nanoparticle tracking analysis was performed on intact exosomes using a NanoSight Range (NanoSight) equipped with a 405 nm laser and an automatic syringe pump system.
Western blot analysis of various proteins
The exosomes (20 μg total protein) from different treatment groups were separated by 4%-20% SDS-PAGE and transferred overnight to immobilon-P PVDF membranes. Membranes were incubated with primary antibody for CD63 (1:200; MX-49.129.5; sc-5275, Santa Cruz Biotechnology), CD13 (1:200; D6V1W, Cell Signaling Technology), cytochrome c (1;200; D18C7, Cell Signaling Technology), GM130 (1:100, SC55590, Santa Cruz Biotechnology), calnexin (1:200; C5C9, Cell Signaling Technology), HSP70 (1:1000; 5A5; ab2787, Abcam), or CD9 (1;2000; EPR2949, ab92726, Abcam) for 18 h at 4°C in blocking buffer. Membranes were then washed and incubated with peroxidase-conjugated secondary antibody (1: 5000) for 1 h. Afterward membranes were washed and then incubated with ECL Western blot chemiluminescence reagent (Pierce). Membranes were exposed to X-ray films and developed using X-Ray developer. The protein band intensities were determined by Image Studio Lite (LI-COR Biotechnology). We added an equal amount of protein lysate from each group. We did not use other exosome marker protein levels as loading control because it has been shown that the levels of CD63, HSP70, and CD9 are altered in different experimental conditions (Chettimada et al., 2018; Crenshaw et al., 2019). We presented the level of protein value as a ratio of corresponding band intensities of control.
Nanosight analysis of exosomes
The Nanosight LM10 was used to analyze the size distribution and concentration of exosomes. The exosome samples were diluted at 1/200 with PBS and were injected in the sample chamber with sterile syringes (BD Discardit II) until the liquid reached the tip of the nozzle. This unique technology utilizes the properties of both light scattering and Brownian motion to obtain the size distribution and concentration measurement of particles in liquid suspension. A laser beam is passed through the sample chamber, and the particles in suspension in the path of this beam scatter light in such a manner that they can easily be visualized via a 20× magnification microscope onto which a camera is mounted. The camera operates at 30 frames per second, capturing a video file of the particles moving under Brownian motion. The software tracks many particles individually and, using the Stokes-Einstein equation, calculates their hydrodynamic diameters. All measurements were performed at room temperature.
TEM
Exosome samples were prepared for TEM inspection as described previously. Briefly, exosomes were mounted on copper grids (FCF400-Cu-50 Formvar/Carbon 400 Mesh, Cu from Electron Microscopy Sciences) and then fixed by 1% glutaraldehyde in cold DPBS for 5 min to stabilize the immunoreaction. It was then washed in sterile distilled water and contrasted by uranyl-oxalate solution at pH 7 for 5 min. Finally, it was embedded by methyl cellulose-UA for 10 min on ice. A JEOL 1010 TEM was used to image exosome samples at a voltage of 80 kV.
Measurement of exosome activity
Aminopeptidase activity assay: Exosomes were mixed with 50 mm of leucine-p-nitroaniline (Sigma Millipore) in a 96-well plate (20 μg/ml total exosomal protein in 150 ml of PBS/well). The release of p-nitroaniline at 60 min intervals was used to follow aminopeptidase activity by measuring the absorbance at 405 nm.
Total MMP2 assay
A rat MMP-2 ELISA kit was used to detect total MMP2 concentration in exosomes released from microglia treated with different doses of ethanol. This ELISA kit detects MMP-2 and includes zymogen and active enzymes.
In vitro labeling of exosomal membrane components
We used BODIPY TR ceramide (Invitrogen) to stain exosomal membranes; 1 µl of the dye stock solution was added to the 100 µl exosomes containing solution to obtain a final dye concentration of 10 μm. The mixture was then incubated at 37°C for 20 min. Exosome spin columns (MW 3000), (Thermo Fisher Scientific) were used to remove the excess unincorporated dye from the labeled exosomes.
Immunohistochemistry for β-endorphin
Serial coronal sections of perfused brains were made using a Leica cryostat at 30 µm in thickness from stereotaxic plates 19 to plates 23 (bregma −2.3 to −4.3 mm) spanning the arcuate nucleus. Perfused sections were mounted on Superfrost Plus glass slides (VWR) containing one AD, one PF, and one AF brain section. The sections were washed in PBS twice followed by antigen retrieval in citrate buffer, pH 6.2, at 100°C for 10 min. After two washes in PBS-T (0.05% Triton-X), the sections were incubated in blocking buffer (2.5% normal horse serum in PBS-T) at room temperature for 30 min. The sections were subsequently incubated overnight at 4°C with the rabbit anti-β-endorphin (1:1000; Peninsula Laboratories). After the primary antibody incubation, samples were washed in PBST, and then sections were incubated with an AlexaFluor-488 donkey anti-rabbit secondary antibody (1:500; Thermo Fisher Scientific). Sections were mounted with DAPI (Vector Laboratories) and sealed with nail polish. To evaluate the immunohistochemical staining intensity, animals in each experimental group were photographed using a Nikon-TE 2000 inverted microscope (Nikon Instruments). Cell counting was quantified using ImageJ software (National Institutes of Health).
Tracing uptake of the labeled exosomes by β-endorphin neurons
A 1-well chamber slide system (Thermo Fisher Scientific) was used for this study, and ∼5 × 104 β-endorphin cells were grown in each well. Fluorescently labeled exosomes (5 × 107 particles/well) were then added to the recipient cells and incubated at 37°C for 30 min to 3 h. The cells were then fixed with 4% PFA and stained with β-endorphin antibody using immunohistochemistry procedures.
Proteomic analysis
SDS-PAGE of exosomal protein gel loading
First, 50 mm DTT was added to exosome samples (10 μg) from different microglia culture groups and heated at 95°C for 5 min. Samples were then cooled to room temperature and centrifuged for 5 min at 25,000 × g. Then 35 μl of sample was loaded to gel (Nupage Novex, 10% Tris glycine gel, 1.5 mm thick) and run into gel at ∼1 cm at 100 V. The gel was stained with 0.1% Coomassie Brilliant Blue R250 in 7% acetic acid and 35% methanol, and destained with destain buffer (7% acetic acid, 30% ethanol). The band was cut out, and we performed in-gel digestion: Each gel band was subjected to reduction with 10 mm DTT for 30 min at 6°C, alkylation with 20 mm iodoacetamide for 45 min at room temperature in the dark, digestion with trypsin (sequencing grade, Thermo Fisher Scientific, catalog #90058), and incubated overnight at 37°C. Peptides were extracted twice with 5% formic acid and 60% acetonitrile and dried under vacuum.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Samples were then analyzed by LC-MS using Nano LC-MS/MS (Dionex Ultimate 3000 RLSC nano System) interfaced with QExactive HF. Samples were loaded onto a self-packed 100 µm × 2 cm trap (Magic C18AQ, 5 µm 200 Å, Michrom Bioresources) and washed with loading Buffer A (0.1% trifluoroacetic acid) for 5 min with a flow rate of 10 µl/min. The trap was brought in-line with the analytical column (Magic C18AQ, 3 µm 200 Å, 75 µm × 50 cm) and peptides were fractionated at 300 nl/min using a segmented linear gradient: 4%–15% B in 30 min (where A: 0.2% formic acid, and B: 0.16% formic acid, 80% acetonitrile), 15%–25% B in 40 min, 25%–50% B in 44 min, and 50%–90% B in 11 min. Solution B then returned at 4% for 5 min for the next run.
Mass spectrometry data were acquired using a data-dependent acquisition procedure with a cyclic series of a full scan acquired in Orbitrap with resolution of 120,000 followed by MS/MS (HCD relative collision energy 27%) of the 20 most intense ions and a dynamic exclusion duration of 20 s. The peak list of the LC-MSMS was generated by Thermo Proteome Discoverer (version 2.1) into MASCOT Generic Format and searched against the rat ensembl database (version 6), plus a database composed of common laboratory contaminants using an in-house version of X!Tandem (GPM Furry) (Craig et al., 2004). Search parameters are as follows: fragment mass error: 20 ppm, parent mass error: ±7 ppm; fixed modification: carbamidomethylation on cysteine; flexible modifications: oxidation on methionine; and protease specificity: trypsin (C-terminal of R/K unless followed by P), with 1 miss-cut at preliminary search and 5 miss-cut during refinement. Only spectra with log ≤2 were included in the final report. To identify differences in protein abundance between different groups (control, ethanol-treated groups), protein spectral count (PSC) data were normalized by dividing the raw PSC for an individual protein (GAPDH or actin) by the total PSCs generated from within that sample. Zero values were replaced with 1 to avoid fallacies related to division by zero. Log2 ratios were calculated for individual proteins across all groups: log2 ratio = log2 (normalized E/normalized C PSC). To determine differences between PSCs from different groups, Student's t tests were performed using mean PSCs over all groups. We focused our subsequent analyses on proteins with a p value of <0.05.
Bioinformatics analysis
The proteomic dataset, which included NCBI GeneInfo identifiers, log2 ratios, and p values for each identified protein, was input into Ingenuity Pathway Analysis (IPA) using the core analysis platform (Ingenuity Systems). The core analysis matched the proteins in our dataset with those in the Ingenuity Knowledge Base. The analysis also created molecular networks, divided the data into biological functions that were overrepresented in the dataset, and determined overrepresented canonical pathways. The unmapped proteins were excluded from further analysis. The core analysis was performed by using default settings. The default settings considered the direct and indirect relationships between the molecules supported by experimentally observed data and the networks that did not exceed 35 molecules. A cutoff log2 ratio of >1.0 and ≤1.0 (twofold change in abundance) and a p value cutoff of 0.05 were applied. The priority lists were generated for enriched molecular networks, biological functions, canonical pathways, differentially abundant proteins, and predicted upstream regulators. Each network was scored based on the number of mapped proteins in the network, its size, the number of mapped proteins in the dataset, and the number of proteins in the Ingenuity Knowledge Base that could be included in the network. Score values were calculated from hypergeometric distribution and the right-tailed Fisher's exact test. IPA also predicted potential upstream regulators of the proteins in our dataset, which were designated activated or inhibited based on a Z score, a statistical measurement of differential expression based on the log2 ratio, the p value, and the statistical effect size.
Apoptosis studies
During experimentation, β-endorphin cells were cultured (5 × 104/well) in a 6-well plate in a neuronal medium for 2 d. The cells were then exposed to medium containing exosomes (∼5 × 107 particles/well) isolated from microglia and activated with or without ethanol or vehicle for a period of 24 h. The cells were then lysed with nucleosome lysis buffer and used for nucleosome assay using the ELISA kit (Sigma Millipore, catalog #11774425001) for determination of β-endorphin neuronal apoptosis.
Complement levels in exosomes
A Rat Complement C3 ELISA kit (ab157731, Abcam) was used to quantify C3 in exosomes. Rat Complement C1q, C2, and C4 (catalog #EKU09022, EKU08453, and EKU03393) ELISA kits (Biomatik) were used to quantify C1q, C2, and C4 levels in exosomes. The manufacturer protocol was followed to perform the ELISA.
Complement blocking activity of anti-C1q monoclonal antibody
Monoclonal anti-C1q antibody [JL-1] (ab71940; 1:25 dilution) was added to the β-endorphin cells (5 × 104/well in 6-well plate) 30 min before the activation with exosomes (5 × 107 particles/well) from different culture groups (C and E). The nucleosome assay was done to detect apoptosis after 24 h of incubation with both the antibody and exosomes.
C1q deposition on β-endorphin neuron
Monoclonal anti-C1q antibody [JL-1] (ab71940) was used to detect the level of C1q deposition in exosome-treated β-endorphin neurons in cultures. β-endorphin cells were incubated with exosomes from control and microglial cell cultures for 2 h. The cell culture slides were then fixed in 4% PFA for 30 min and then underwent double-labeled immunocytochemistry. Fluorescent images were captured with a Cool SNAP-pro CCD camera coupled to a Nikon-TE 2000 inverted microscope. Images were processed with Adobe Photoshop 7.0.
Flow-cytometric analysis of downstream signaling proteins of C1q in β-endorphin neurons, treated with ethanol-activated microglial exosomes or recombinant C1q
β-Endorphin cells were cultured (5 × 104/well) in a 6-well plate in neuronal medium for 2 d. The cells were then exposed to medium containing exosomes (∼5 × 107 particles/well) isolated from microglia, activated with or without ethanol or vehicle or C1Q protein (500 ng) for a period of 2 h. Fluorescence intensity was measured using a BD Accuri C6 Flow Cytometer (Beckman Coulter). Five thousand events were read for each sample, and data were analyzed using the C6 Accuri software. Fluorochrome-labeled antibodies (Santa Cruz Biotechnology, C5b-9 antibody, 2A; AlexaFluor-488 C3a/b antibody, 11H9; AlexaFluor-488 CD55 antibody,143-3, PE item sc-21769 PE and C4 antibody, 16 D2; FITC, item sc-58930 FITC) were used during the analysis. Flow cytometric gates were set using unstained cells with the forward scatter and side scatter plot, and labeled cells were read on the FL-1A (488 nm) and FL-2A (545/40 nm) channel. The median fluorescent intensity values of positively labeled cells were expressed as ± SEM for each sample. The data were normalized in this way to account for variation in fluorescent intensities between batches.
Detection of ROS in β-endorphin neurons
The cellular ROS production was measured by determining the rate of H2O2 production using the CM-H2DCFDA fluorescent probe. The mitochondrial ROS production was measured by determining mitochondrial superoxide production using the MitoSOX red fluorescent probe. β-endorphin cells (5 × 103/well) were grown in 96-well clear-bottom black plates for 2 d. On day 3, the cells were treated with medium containing exosomes (1 × 106 particles/well) isolated from microglia, activated with or without ethanol (50 mm), vehicle, or C1q protein (50 ng) for a period of 2 h and with H2O2 (40 µm) for 30 min. In some cultures, ROS activity was blocked using 10 µM N-acetyl-L-cysteine (NAC). Cells grown in multiwell plates were washed twice with PBS to remove the medium, and then 0.5 ml of measurement buffer containing 2 mm H2DCFDA was added. Immediately after addition, fluorescence was measured at 485 nm excitation (Ex bandwidth: 20 nm) and 520 nm emission (Em bandwidth: 10 nm) wavelengths by using a BioTek plate reader. For mitochondrial ROS, cells were washed twice with PBS and subsequently incubated for 10 min (to allow the probe to enter the cell and start the reaction within the mitochondria) at 37°C in 0.5 ml of measurement buffer containing 5 mm MitoSOX Red (Abcam). After the incubation, the cells were washed twice with PBS. The fluorescence was monitored in the measurement buffer with a microplate reader (Bio-Tek) set to 510 nm excitation (Ex bandwidth: 10 nm) and 595 nm emission (Em bandwidth: 35 nm) wavelengths.
Statistical analysis
Results are expressed as mean ± SEM. t test was used to analyze the differences between two groups, and one-way ANOVA with Newman Keuls post hoc analysis was used to analyze the differences between multiple groups. p < 0.05 was considered significant. Data were analyzed using Prism 5.0 (GraphPad Software).
Results
Characterization of ethanol effects on hypothalamic and microglial exosomes during the developmental period
We investigated whether exosomes can initiate or promote the apoptotic death of hypothalamic β-endorphin neurons following ethanol treatment. Exosomes were collected from (1) MBH tissues of PND6 rats, which were fed daily with an ethanol-containing diet or control diet, or (2) primary microglia cells, which were challenged with ethanol or vehicle control for 24 h. Nanoparticle tracking analysis determined that in vivo MBH exosomes from AD rats had a mean particle diameter of 138.7 nm, a mode of 82 nm, and a concentration of 2.78 × 1011 particles/ml (Fig. 1A). For cultured microglia, the mean particle diameters were 114 nm for the control, 114 nm and 167.7 nm for ethanol, the modes were for 84.8 nm for the control and 105.3 nm for ethanol, and concentrations were 6.94 × 1011 particles/ml for the control and 1.26 × 1012 particles/ml for ethanol (Fig. 1B,C). It should be noted that some exosomes appeared to be clumped together, forming distortions in the video and may be a reason why larger particle sizes were observed during nanoparticle tracking analysis. TEM analysis of exosomes showed that the particle size ranges from 30 to 150 nm for MBH exosomes and 65 to 113 nm for primary microglia, and had the characteristic cup shape typically observed in exosomes under TEM (Fig. 1D–F). These vesicles were similar in shape and dimension to the previously described exosomes (Wu et al., 2015). Primary microglia-derived exosomes were visible under fluorescent microscopy when stained with BODIPY TR ceramide (data not shown). Exosomes from MBH were assayed by Western blot to confirm their purity against rat liver lysate, which served as a positive control. These exosomes were positive for exosome marker protein CD-63 and were negative for possible vesicular contaminates cytochrome c (mitochondrial-specific protein), GM130 (Golgi-specific protein), and calnexin (endoplasmic reticulum-specific protein) (Fig. 1G). Exosomes from primary microglia show no contamination of BSA (Fig. 1H). Exosomes from ethanol- and control-treated animals showed detectable levels of CD9, HSP70, and CD63 in MBH (Fig. 1I) and in primary microglial cell cultures (Fig. 1J). Interestingly, 50 mm ethanol-treated microglial cultures (EE1 and EE2) showed elevated levels of CD63 compared with the control (EC1 and EC2) (Fig. 1J). These data demonstrate that the extracellular vesicles collected from MBH tissue and microglial cells in cultures are exosomes.
Figure 1.
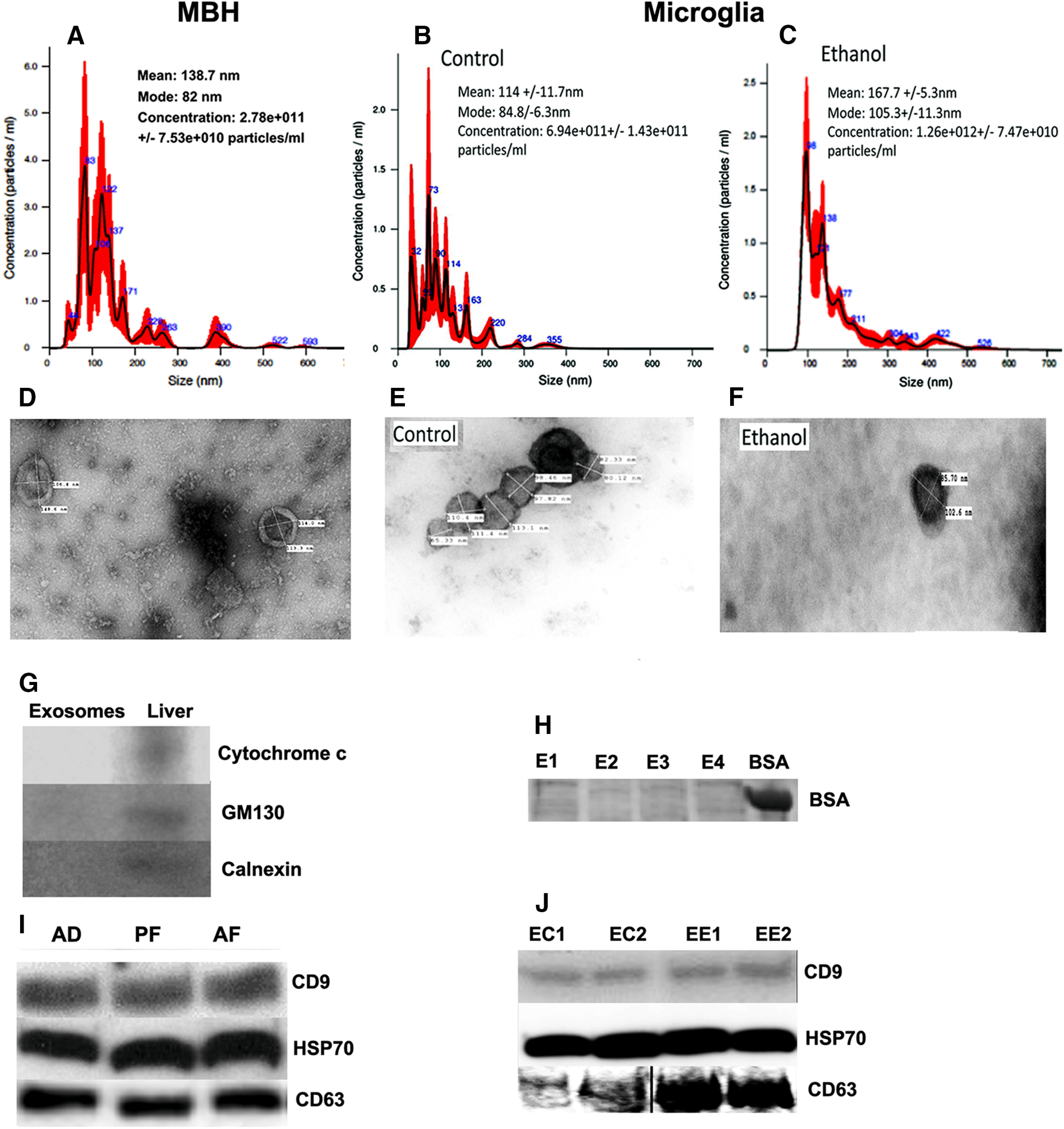
Characterization of hypothalamic and microglial exosomes; effects of alcohol. Nanoparticle tracking analysis, TEM, and Western blot verification of exosomes prepared from the tissue of MBH obtained from postnatal day 6 rats, which were fed daily with ethanol-containing milk formula (AF), pair-fed isocaloric milk formula (PF), or left undisturbed in the litter with mother (AD) (A, D, G, I); or from primary cultures of microglial cells treated with 50 mm ethanol for 24 h (B, C, E, F, H, J). For nanoparticle tracking analysis, the calculated size distribution is depicted as a mean (black line) with SE (red shaded area). Mean particle size, mode particle size, and concentration of particles in exosomes collected from MBH from AD rats (A) or microglia treated with vehicle control (B) or 50 mm ethanol (C). Representative images of exosomes from MBH of AD rats (D) and from control (E) or ethanol-treated microglia (F) under TEM. The width and length of each exosome are represented on the individual exosome. Representative bands for the protein levels of cellular proteins, mitochondria marker cytochrome c, Golgi apparatus marker GM130, and endoplasmic reticulum marker calnexin in MBH exosomes or in liver lysates (G) as determined by Western blot analysis. No BSA band was found in microglial-derived exosomes (H). Exosome marker proteins CD9, HSP70, and CD63 were detectable in Western blot analysis of exosomes prepared from MBH tissues of AD, PF, and AD rats (I) or from control (EC1, EC2) or ethanol (EE1, EE2)-treated microglial cultures (J). An equal amount of protein lysate was used from each group. Data represent three or more independent experiments.
Exosomes are produced from multiple cell types within the brain and transport cargo from one cell to another for communication. Each exosome's cargo contains a cell-specific protein, which identifies its biogenesis (Schey et al., 2015). We determined whether ethanol exposure altered the cargo containing microglial-specific protein CD13 by Western blot measurements. MBH exosomes from AF-treated rats exhibited augmented CD13 levels relative to AD controls (Fig. 2A; F(2,15) = 4.302, p = 0.0334). Similarly, exosomes from ethanol-treated microglial cultures (EE1 and EE2) showed elevated levels of CD13 compared with control (EC1 and EC2) (Fig. 2D; t = 5.572, df = 6, p = 0.0014). CD13 is also a marker for brain pericytes (Smyth et al., 2018) and has aminopeptidase enzymatic activity (Potolicchio et al., 2005). Therefore, we quantified exosomal aminopeptidase activity. In concert with the Western blot data (Fig. 2A), AF-treated rats exhibited significantly greater aminopeptidase activity than AD and PF controls (Fig. 2B; time × treatment interaction, F(6,48) = 2.841, p = 0.0190; time, F(3,48) = 81.21, p < 0.001; treatment, F(2,16) = 7.095, p = 0.0062). Further, administration of minocycline, an antibiotic with significant inhibitory effects on microglial activity, completely attenuated CD13 activity in AF-treated rats (Fig. 2C; time × treatment interaction, F(6,30) = 5.75, p = 0.0004; time F(3,30) = 11.59, p < 0.001; treatment, F(2,10) = 0.70, p = 0.5192). Furthermore, in primary cultures of microglia, ethanol dose-dependently increased the aminopeptidase activity of exosomes (Fig. 2E; time × treatment interaction, F(15,120) = 43.94, p ≤ 0.0001; time, F(3,120) = 458.51, p < 0.0001; treatment, F(5,120) = 2452.87, p < 0.0001). We also measured concentrations of MMP2, another marker to assess the activity of exosomes, in exosomes released from primary microglia treated with different doses of ethanol. Ethanol dose-dependently increased the levels of MMP2 in exosomes released from microglial cultures (Fig. 2F; F(3,16) = 102.7, p ≤ 0.0001). These data suggest that PAE increases exosome production and activity at least from microglial cells in the hypothalamus.
Figure 2.

Alcohol effects in the hypothalamic and microglial exosomes. In in vivo studies, exosomal activity was determined by measuring CD13 protein levels in the MBH of AD, PF, and AF offspring (A), and CD13 activity levels (at 60, 120, 240, and 360 min) in the MBH for AD, PF, and AF without (B) and with minocycline (C). Data are mean ± SEM (n = 4-7/group). *p <0.05, AF versus AD and PF. **p < 0.01, AF versus AD. #p < 0.01, AF versus PF. In in vitro studies, the activity of exosomes was determined by measuring the level of CD13 proteins 24 h after 0 or 50 mm ethanol treatment (D), CD13 activity levels at various time points (0-240 min) with various doses of ethanol (0-100 mm; E), and changes of MMP2 protein levels 24 h after treatment with various doses of ethanol (F) in microglial cells in primary cultures. Data are mean ± SEM (n = 4-6/group). **p < 0.01, ethanol versus control. *p < 0.05, 25 mm ethanol versus control. ***p < 0.001, 50 mm and 100 mm ethanol versus control.
Roles of exosomes in ethanol-induced β-endorphin neuronal apoptosis
We next determined whether hypothalamic exosomes contribute to ethanol-induced apoptosis of β-endorphin neurons. First, we measured the effect of a spingomyelinase inhibitor GW4869, known to reduce exosome formation and release (Essandoh et al., 2015; Menck et al., 2017), on ethanol-activated microglial cells in primary cultures. Nanoparticle tracking analysis data demonstrated that most exosomes were <200 nm, the average particle concentration was higher in exosomes released from ethanol-treated microglia than in the control, and GW4869 significantly reduced ethanol-increased exosome release (Figs. 3C–E, 3F; F(2,6) = 18.14, p = 0.0029). Using the PAE animal model, we have previously shown that ethanol increases apoptosis of β-endorphin neurons in the hypothalamus (Sarkar et al., 2007; Kuhn and Sarkar, 2008). In this study, we determined the role of exosomes by treating postnatally ethanol-treated (AF) rats with GW4869. As previously shown, AF-treated rats had significantly less β-endorphin neurons in the arcuate nucleus compared with AD and PF controls. Pretreatment with GW4869 did not affect the number of β-endorphin neurons in AD and PF control rats but reduced the ethanol-induced loss of β-endorphin neurons in AF rats (Fig. 3A,B; F(5,31) = 5.420, p = 0.0011). These data suggest the possibility that exosomes of undefined cellular origin might be involved in ethanol apoptotic action on β-endorphin neurons. Further studies were conducted to determine the role of microglial exosomes in ethanol apoptotic action using primary cultures of microglia and β-endorphin neurons We first traced the uptake of labeled exosomes in β-endorphin neuronal cells. When fluorescently labeled exosomes from microglia were added to neuronal cultures, a significant number of labeled exosomes (green) were found to be deposited on β-endorphin neurons (red) (Fig. 3G), suggesting uptake of exosomes in these neurons. We also determined the ability of ethanol-activated exosomes from microglia to enhance β-endorphin neuronal death. The result indicated a significant increase in apoptosis of β-endorphin neurons on incubation with exosomes, released by the ethanol-treated microglia compared with those released from vehicle-treated microglia (Fig. 3H; F(3,20) = 52.92, p ≤ 0.0001).
Figure 3.
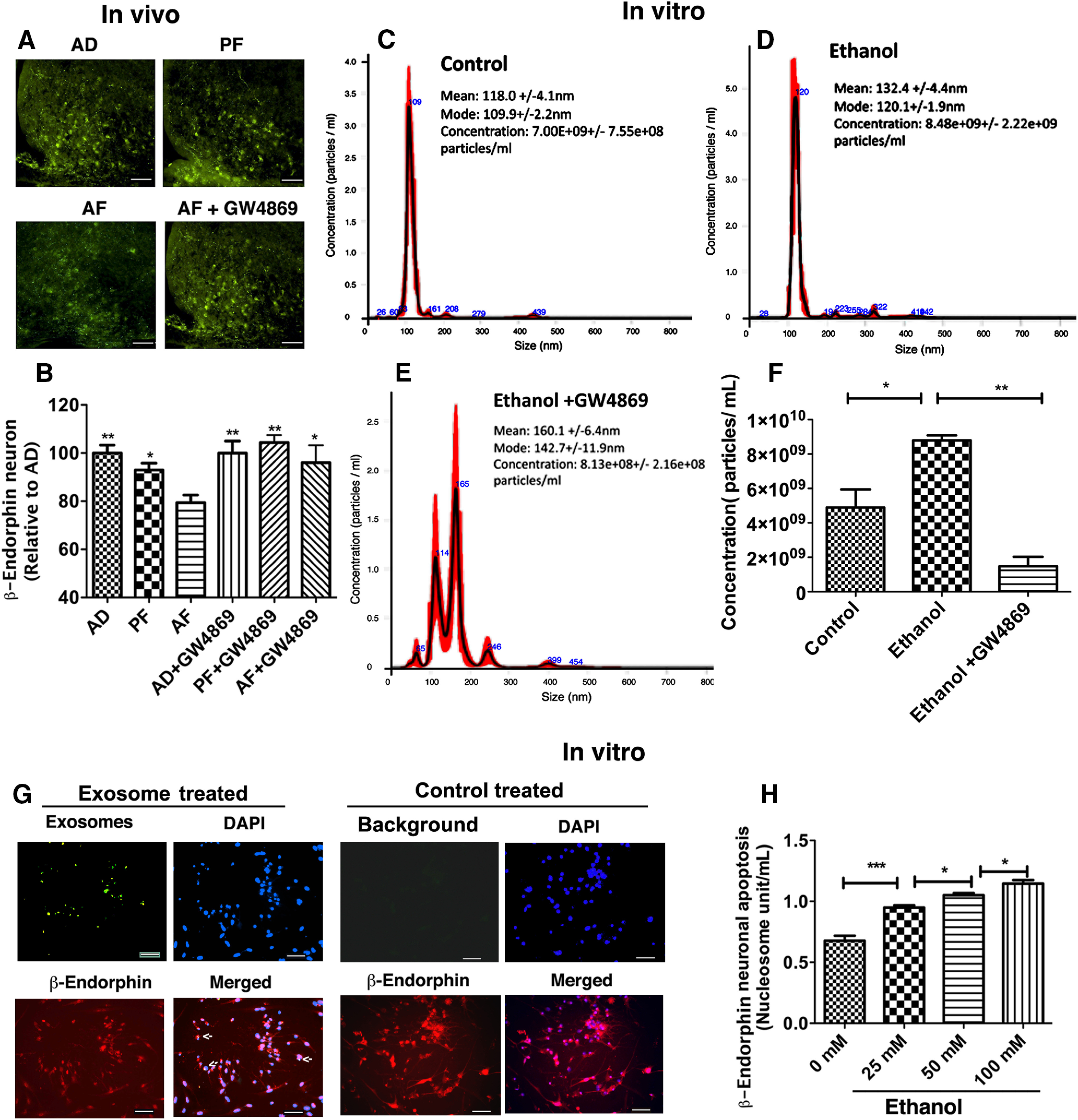
Involvement of exosomes in ethanol-induced β-endorphin neuronal apoptosis. Effect of PAE with or without GW4869 on β-endorphin neuronal number in the arcuate nucleus (A, B), exosome release from microglia in primary cultures (C–F), exosome deposition on β-endorphin neurons in primary cultures (G), and β-endorphin neuronal apoptosis following addition of ethanol-activated exosomes in primary cultures (H). Representative images of (A) β-endorphin-positive staining (green) along with a histogram (B) representing the mean ± SEM values in AD, PF, AF, and AF+GW4869 treated rat pups (n = 5 or 6). Scale bars, 200 µm. *p< 0.05; **p< 0.01; compared with AF. Showing data of nanoparticle tracking analysis, the calculated size distribution is depicted as a mean (black line) with SE (red shaded area). Mean particle size, mode particle size, and concentration of particles collected from microglial exosomes treated with vehicle control (C), ethanol (D), or ethanol and GW4869 (E). F, Histogram represents the mean ± SEM values (n = 3) of exosome release as determined by nanoparticle tracking analysis in microglial cultures following treatment with ethanol with or without GW4869. Shown is a significant number of fluorescently labeled exosomes (green) on β-endorphin neurons (red) in primary cultures (G). Nucleus was stained with DAPI (blue). Arrows indicate uptake of exosomes in these neurons. Scale bars, 50 µm. β-endorphin neuronal apoptosis as determined by nucleosome unit following incubation with microglial exosomes treated with various doses of ethanol for a period of 24 h in primary cultures (H). Data are mean ± SEM (n = 4-7/group). *p< 0.05, ***p< 0.001.
Effects of ethanol on exosomal cargo content as determined by proteomic analysis
Since our study found that ethanol-activated microglial exosomes increased the apoptotic death of β-endorphin neurons, we characterized by proteomics analysis those elements that make up exosomal cargo and might have played a role in the neuronal death. The proteomic dataset included NCBI GeneInfo identifiers, log2 ratios, and p values for identified proteins, and those parameters were added to IPA using the core analysis platform. The mass-spectrometry analysis identified 460 and 280 proteins that were activated (red) and deactivated (green), respectively (Fig. 4A). Among those proteins, many of them are chemokines, apoptosis regulatory proteins, MAPK, phagosomes, epigenetic regulatory proteins, and RNA polymerases, which are known to regulate the expression of noncoding RNA and tRNA and complements (Fig. 4B). Therefore, our proteomic analysis suggested an interesting list of proteins that might have played a role during ethanol-induced neuronal death. There are a few reports that identified a role of the complement system in the CNS that dysfunctions during the development (Lee et al., 2019). Proteomic analysis of our data identified that one of the hub proteins regulating the complement system is C1q (Fig. 4C).
Figure 4.
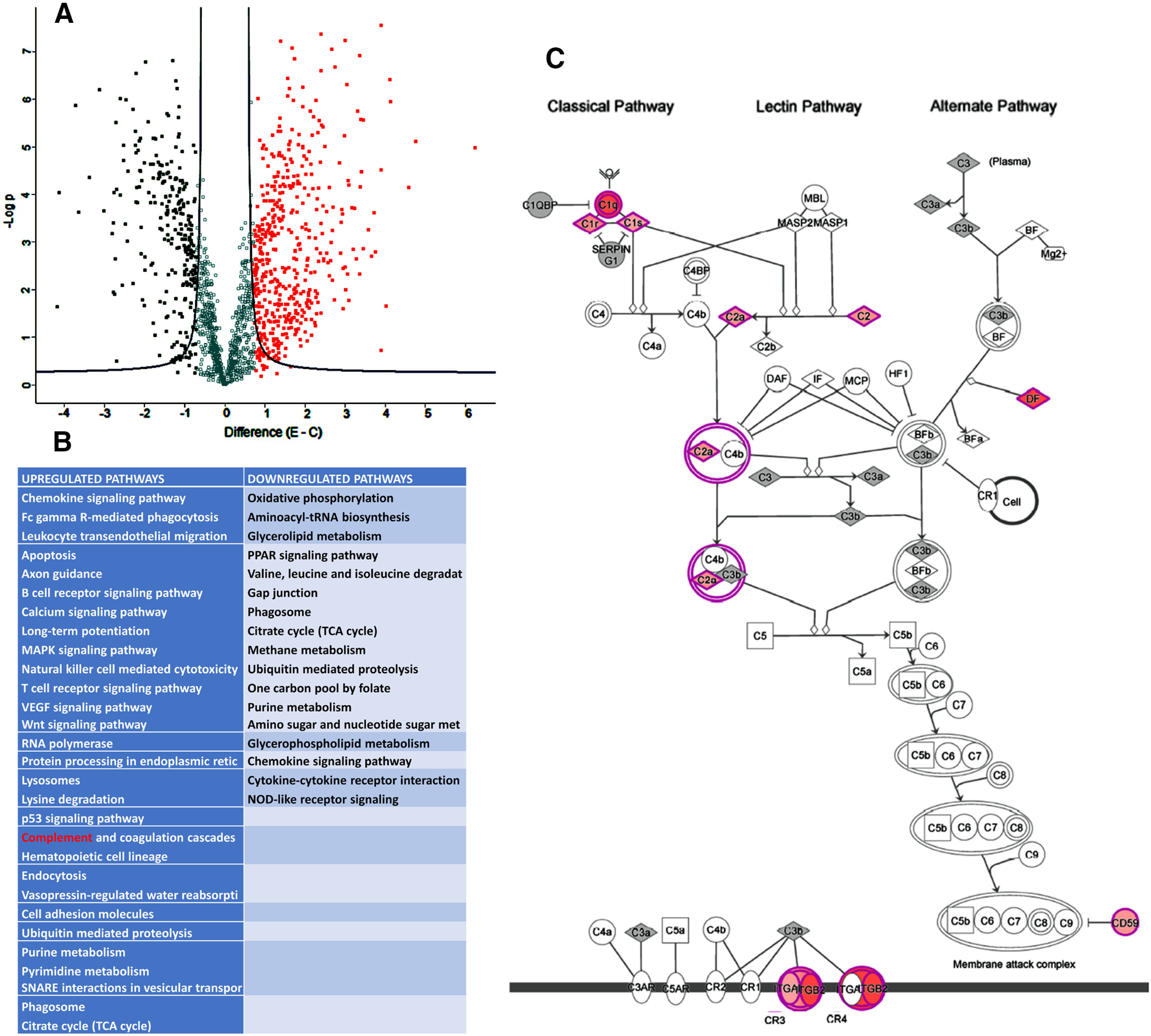
Proteomics characterization of exosome cargo from microglial cells treated with and without ethanol. Exosomes were prepared from primary microglia cells treated with 50 mm ethanol (E) or vehicle control (C) for 24 h and used for LC-MS/MS analysis. Comparative proteomic data were presented in a volcano plot, which was constructed by plotting the negative log of the p value on the y axis. Each dot represents the proteomes from three biological replicates per group. A, Volcano plot showed significant downregulated and upregulated proteins as determined by proteomic analysis in both ethanol and vehicle control. B, IPA identified the significantly affected canonical pathways, including complement pathways (highlighted in red), in ethanol-treated exosomes. C, The complement proteins, which are affected by ethanol (increase highlighted in pink; hub gene highlighted in red) in the complement pathway.
Regulation of ethanol-induced changes in complement system in the hypothalamus
In the CNS, microglia are the dominant source of C1q and chronic neuroinflammation, or ethanol exposure augments C1q release (Sebastian et al., 2011; Silverman et al., 2016; Fonseca et al., 2017). We determined whether PAE affected C1q expression and deposition on β-endorphin neurons in the arcuate nucleus. First, we used Western blot measurements of C1q protein levels in MBH tissue in alcohol-fed (AF) and control-fed (AD and PF) offspring with or without inhibiting microglial activation with minocycline (Mino). Data show increased levels of C1q in the MBH tissue of AF rats compared with those in PF and AD rats (Fig. 5A; F(2,15) = 9.517, p < 0.0021). Minocycline treatment prevented ethanol-induced increases in C1q levels in AF rats since the levels of C1q were similar in AD, PF, and AF groups treated with the microglial activation blocker (Fig. 5B; F(2,15) = 0.3708, p = 0.6963). Immunofluorescent staining revealed that AF-treated rats exhibited significantly greater C1q expression than AD and PF controls, represented by greater fluorescence intensity (Fig. 5C,D; F(5,31) = 4.860, p = 0.0021). Furthermore, Pearson's correlation coefficient showed that C1q and β-endorphin colocalization was greater in AF-treated rats (Fig. 5F; F(5,31) = 4.761, p = 0.0024). Since microglial activation can augment C1q expression, we determined whether ethanol-induced microglial activation was involved in increased C1q staining and colocalization in AF-treated rats by inhibiting microglial activation with minocycline. Inhibition of microglial activation prevented ethanol-induced increases in C1q expression and colocalization on β-endorphin neurons (Fig. 5D–F). These data suggest that PAE activates microglia to produce C1q that then deposits on β-endorphin neurons.
Figure 5.
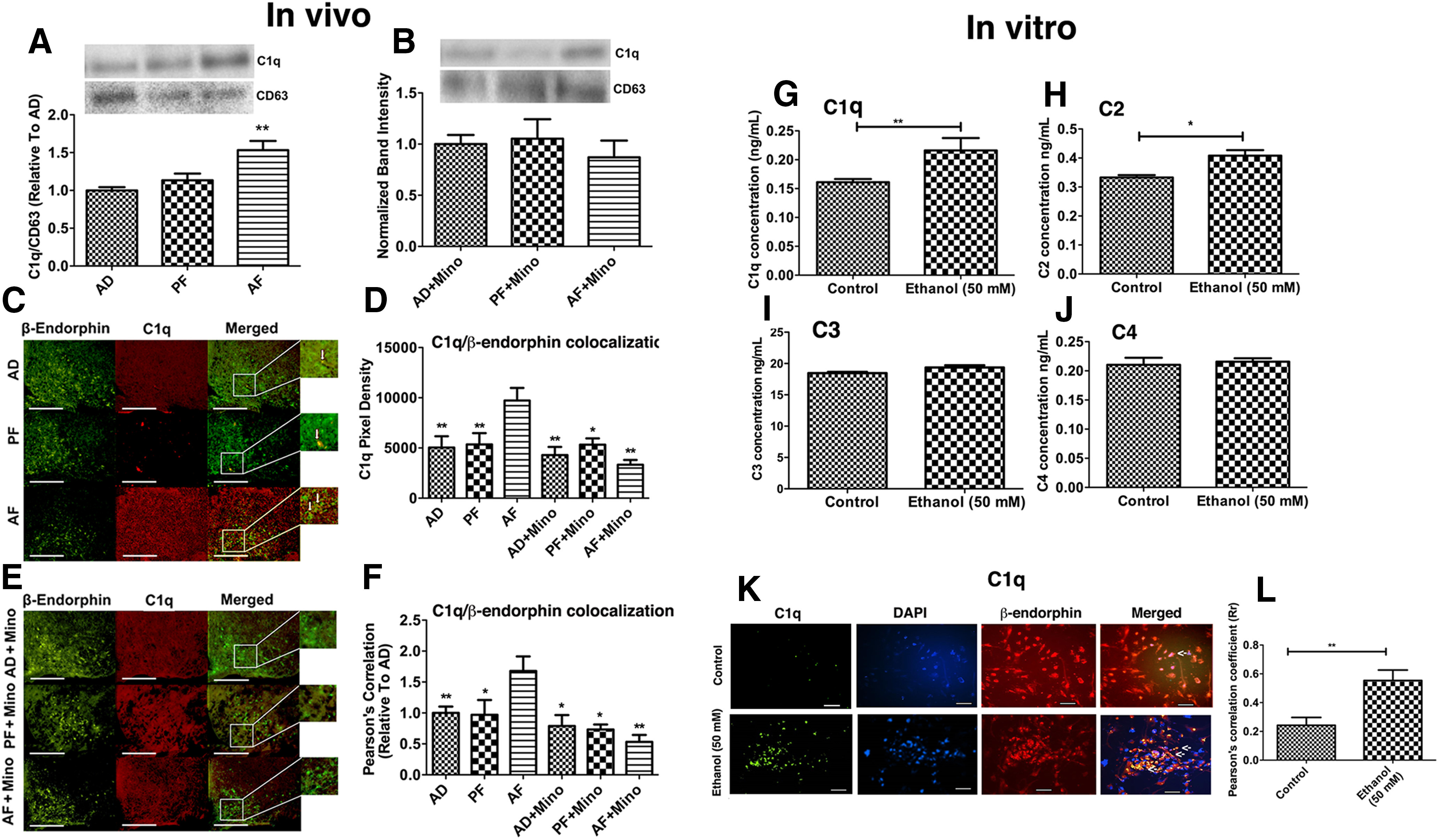
Regulation of ethanol-induced changes in complement system in the hypothalamus. The effects of PAE with or without minocycline administration on C1q expression (A, B)and colocalization on β-endorphin neurons in the MBH (C–F), ethanol-induced changes in the levels of complements in exosomes derived from microglial cells in cultures (G–J), and ethanol-induced changes in deposition of microglia derived exosomal C1q on β-endorphin neurons in cultures (K, L). A, B, Bar graphs represent C1q protein levels along with representative bands for C1q and CD63 (used for normalizing C1q protein levels) (Kavanagh et al., 2017) measured by Western blot analysis. Histograms represent the mean ± SEM in AD, PF, AF, AD+Mino, PF+Mino, and AF+Mino rat pups on PND 6 (n = 5-8). **p< 0.01, AF versus AD, PF. C–F, Representative images of C1q-positive staining (red) and β-endorphin-positive staining (green), along with merged images and a zoomed-in picture to demonstrate colocalization. Scale bars, 200 µm. Histograms represent the mean ± SEM values of pixel density (D) or Pearson's correlation of C1q colocalization with β-endorphin (F) in AD, PF, AF, and AD+Mino, PF+Mino, and AF+Mino rat pups on PND 6 (n = 5-7). **p < 0.01 versus AF. *p < 0.01 versus AF. G–J, Histograms represent complement protein levels measured by ELISA in exosomes derived from microglial cells treated with vehicle (control) or 50 mm ethanol for 24 h. N = 4-6. *p < 0.05. **p < 0.01. Representative images of C1q-positive staining C1q-positive staining (Green) and β-endorphin-positive staining (Red) in β-endorphin neuronal culture after 24 h of incubation with exosomes harvested from control or ethanol-treated (50 mm) microglia cultures. Nucleus was stained with DAPI (blue). Arrows indicate C1q proteins inside the cells. Scale bars, 50 µm. Histograms represent the mean ± SEM values of Pearson's correlation of C1q colocalization with β-endorphin in cells of control and alcohol-treated groups. N = 6. **p < 0.01.
In in vitro studies, we analyzed C1q and other complement protein C2, C3, and C4 levels in exosomes from microglia culture treated with vehicle or 50 mm ethanol. The results show a significant increase in C1q and C2 protein levels in exosomes, released by ethanol-activated microglia (Fig. 5G, t = 2.722, df = 9, p = 0.0235; Fig. 5H, t = 3.669, df = 8, p = 0.0063). Other complement proteins (C3 and C4) were not responsive to the ethanol challenge (Fig. 5I, t = 1.647 df = 14, p = 0.1218; Fig. 5J, t = 0.3948 df = 10, p = 0.7013). We also analyzed C1q deposition in exosome-treated cultured β-endorphin neurons by immunohistochemical methods. The deposition of C1q was increased in the β-endorphin neuronal culture after 24 h of incubation with exosomes, harvested from ethanol-treated microglia cultures (Fig. 5K,L; t = 3.433, df = 12, p = 0.0050). These data support the finding of in vivo studies and suggest that ethanol increases deposition of microglial derived C1q in β-endorphin neurons.
Role of the C1q-activated complement pathway in ethanol-induced cell death of β-endorphin neurons
Since an increased amount of C1q protein is deposited in β-endorphin neurons following ethanol treatment, the question arose whether the C1q protein is responsible for the stimulation of downstream complement pathways involving C3, C4, C5b9/membrane attack complex (MAC), ROS, and apoptosis in β-endorphin neurons. To address this question, we first used the in vivo model of PAE where we block C1q activation by C1NH and studied ethanol-activated apoptosis in β-endorphins. The immunofluorescence data shown in Figure 6A, B, (F(3,29) = 27.57, p < 0.0001) indicate that the colocalization of terminal complement complex C5b9 on β-endorphin neurons was elevated in the AF group compared with AD and PF groups. C1NH treatment prevented alcohol-induced increases in C5b9 levels in β-endorphin neurons of AF rats. In addition, C1NH treatment prevented an ethanol-activated increase in the colocalization of apoptotic marker caspase 3 in β-endorphin neurons in AF rats (Fig. 6C,D; F(3,24) = 8.463, p = 0.0005). It is interesting to note that very few colocalizations of C5b9 or Caspase3 were found on β-endorphin neurons in AD and PF animals. Hence, these data support the possibility that alcohol treatment activates a complement pathway to induce apoptotic cell death in β-endorphin neuron.
Figure 6.
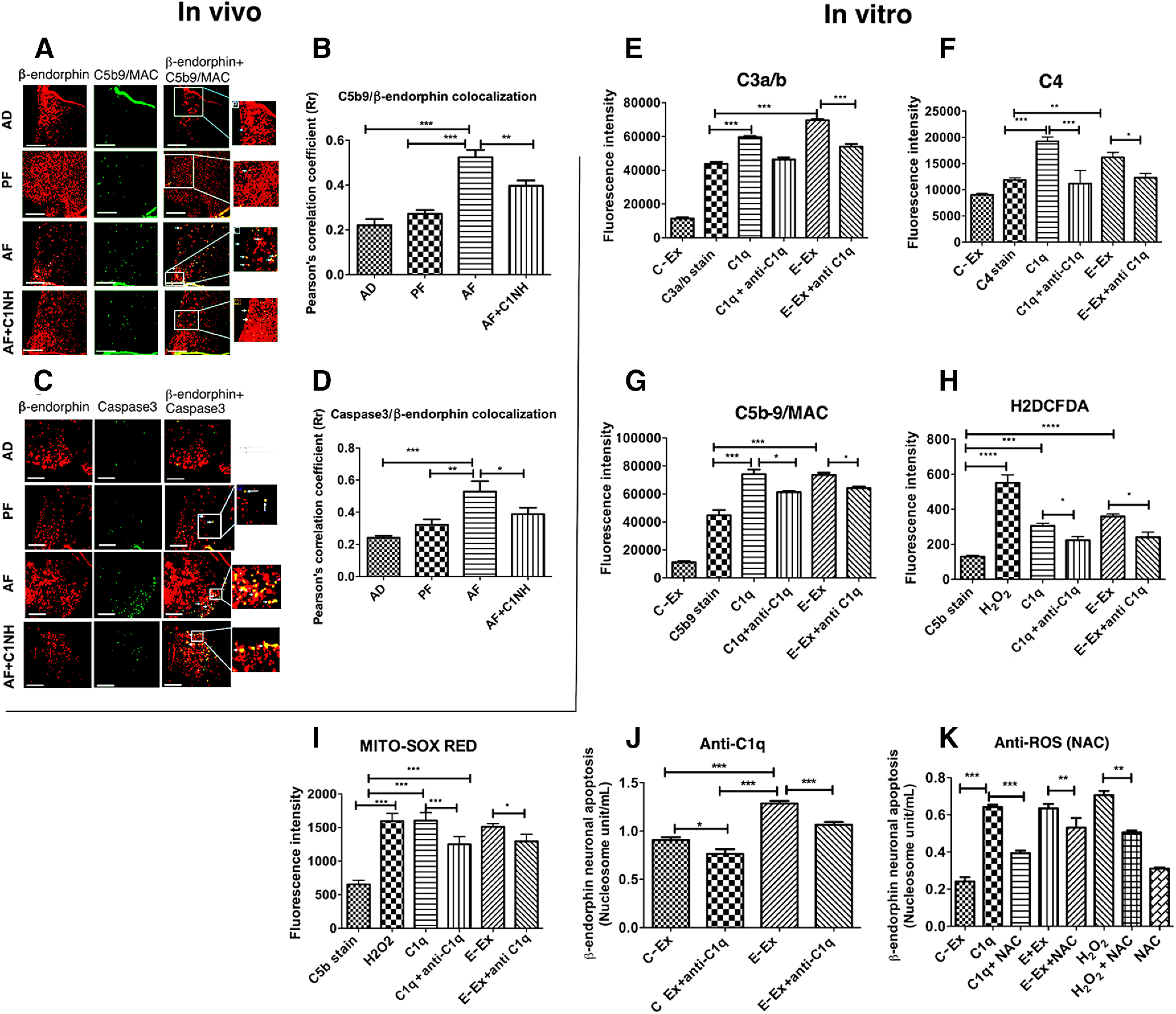
Role of the C1q-activated complement pathway in ethanol-induced cell death of β-endorphin neurons in the hypothalamus. A, B, Representative images of β-endorphin-positive staining (red) and C5b9-positive staining (green), along with merged images and zoomed pictures to demonstrate colocalization (A). Scale bars, 50 μm. Histograms represent the mean ± SEM values of Pearson's correlation of C5b9 colocalization with β-endorphin (B) in AD-, PF-, AF-, and AD+C1NH (100 U/kg)-treated rat pups on PND 6 (n = 7-9). C, D, Representative images of β-endorphin-positive staining (red) and caspase 3-positive staining (green), along with merged images and zoomed pictures to demonstrate colocalization (C). Scale bars, 50 μm. Bar graphs representing mean ± SEM values of Pearson's correlation of caspase 3 colocalization with β-endorphin (D) in AD-, PF-, AF-, and AD+C1NH-treated rat pups on PND6 (n = 7). E–G, Flowcytometric analysis of C3a/b (E), C4 (F), and C5b-9 expression (G) on β-endorphin neurons in primary cultures treated with ethanol-activated (50 mm) microglial exosomes or recombinant C1q protein (500 ng) with or without anti-C1q (C1q neutralizing antibody; 1:25 dilution) for 2 h. H, I, Flowcytometric analysis of cellular ROS by H2DCFDA (H) or mitochondrial ROS by MITO-SOX RED (I) on β-endorphin neurons treated with H2O2 (40 μm) for 30 min, ethanol-activated (50 mm) microglial exosomes, or recombinant C1q protein (500 ng) with or without anti-C1q (C1q neutralizing antibody; 1:25 dilution) for 2 h. J, K, Effects of anti-C1q (C1q neutralizing antibody; 1;25 dilution; J) or anti-ROS NAC (10 μm; K) in control-treated microglial exosomes (C-Ex) or ethanol-treated (50 mm) microglial exosomes (E-Ex)-induced changes in β-endorphin neuronal apoptosis as determined by nucleosome assay in primary cultures of β-endorphin neuronal cells. Data are mean ± SEM (N = 4-6). *p< 0.05, **p< 0.01, ***p< 0.001.
The in vivo model is a multicellular model system, and it is difficult to identify the downstream signaling process of a unique cell population following a challenge. Hence, we used primary microglial cells and primary β-endorphin cells to investigate whether the C1q protein carried by exosomal cargo from ethanol-activated microglia is responsible for the stimulation of downstream complement pathways involving C3, C4, and C5b9/MAC, leading to apoptotic death of β-endorphin neurons. Flow cytometric analysis of β-endorphin neurons treated with ethanol-activated microglial exosomes or recombinant C1q showed an increase in the levels of C3a/b (Fig. 6E; F(5,30) = 1007, p < 0.0001), C4 (Fig. 6F; F(5,17) = 35.84, p < 0.0001), and MAC/C5b9 (Fig. 6G; F(5,28) = 242.7, p < 0.0001), which was suppressed when these neurons were coadministered with a neutralizing antibody of C1q (a blocker of C1q). These data suggest that C1q is important in the activation of complement pathways, including the MAC/C5b9 in β-endorphin neurons.
We also analyzed the release of reactive super oxygen species in β-endorphin neurons treated with H2O2, C1q, and exosomes, and we found that all of them increased the ROS production in these cells (Fig. 6H; F(5,22) = 33.66, p < 0.0001). Conversely, neutralizing anti-C1q antibody treatment reduced the ROS generation by H2O2-, C1q-, and ethanol-treated microglial exosomes (Fig. 6I). We also analyzed mitochondrial reactive super oxygen species in β-endorphin neurons treated with H2O2-, C1q-, and ethanol-treated microglial exosomes. Like the cellular ROS, the level of mitochondrial ROS in β-endorphin neurons was elevated by H2O2-, C1q-, and ethanol-activated microglial exosomes, while anti-C1q antibody prevented the mitochondrial ROS production by H2O2-, C1q-, and ethanol-activated exosomes (Fig. 6I; F(5,26) = 73.89, p < 0.0001). These data identify a role of ROS in C1q-altered effects on β-endorphin neurons.
Finally, the involvement of C1q and ROS in β-endorphin neuronal apoptosis induced by ethanol-activated microglial exosomes was investigated. Monoclonal anti-C1q antibody was used to neutralize the effect of this complement in exosomes. We found that exosomes, from both control-treated or ethanol-treated microglial cultures, induced β-endorphin neuronal apoptosis, which was effectively reduced on incubation with neutralizing anti-C1q antibody (Fig. 6J; F(3,12) = 58.76, p < 0.0001). We also tested the role of ROS in C1q-mediated apoptosis of β-endorphin neurons by determining the effects of NAC, a ROS inhibitor, on the apoptotic death of β-endorphin neurons by inhibiting ROS generation. We found that NAC treatment prevented the apoptotic death of β-endorphin neurons induced by H2O2 treatment, suggesting that the inhibitor is effective in preventing the neurotoxic effect of ROS on β-endorphin neurons (Fig. 6K; F(7,25) = 34.73, p < 0.0001). NAC also prevented C1q-induced apoptotic death of β-endorphin neurons, supporting the view that C1q activates ROS to induce β-endorphin neuronal death. NAC treatment also prevented the apoptotic death of β-endorphin neurons induced by exosomes from ethanol-treated microglial culture. Together, these data support the view that ROS production in β-endorphin neurons, at least partly, mediates the C1q-increased neuronal apoptosis (Fig. 7).
Figure 7.

A schematic diagram showing a proposed mechanism by which ethanol-activated microglial exosomes uses complement C1q system to induce β-endorphin-producing POMC neuronal death. C1q deposition on POMC neurons may increase expression of C3a/b and C4 and MAC/C5b9 proteins in POMC neurons and may induce cell death in these cells. C1q deposition may also drive ROS production and induce oxidative stress to cause apoptotic-type cell death of POMC neurons. A complement blocker (anti-C1q) or an antioxidant NAC protects POMC neurons from ethanol-induced cellular death.
Discussion
Exosomes are small vesicles (30-150 nm in diameter) that have been shown to play a role in communication between microglia and neurons (Bahrini et al., 2015; Drago et al., 2017). Furthermore, neuroinflammation or neurodegenerative diseases can alter this communication by changing the cargo of microglial exosomes to promote apoptosis in recipient neurons (Sharples et al., 2008; Kumar et al., 2017). What was not determined previously is whether microglial exosomes contribute to ethanol-induced apoptosis of β-endorphin neurons. In this study, we first confirmed that the vesicles we collected from the MBH and microglial cell cultures were exosomes by nanoparticle tracking analysis and TEM (Wu et al., 2015). Additionally, these vesicles were not contaminated by vesicle-producing organelles, as they were negative for Golgi apparatus protein GM130 and endoplasmic reticulum protein calnexin, nor did they contain mitochondrial cytochrome c or normal cell culture contaminant BSA. These exosomes were positive for exosome marker protein CD-63, CD9, and HSP70 (Perez-Gonzalez et al., 2012; Kavanagh et al., 2017). Therefore, these vesicles collected from the MBH of AD-, PF-, and AF-treated rats or ethanol- or vehicle-treated microglial cultures were primarily exosomes. Since PAE is known to activate microglia, we probed for a microglial-specific marker, CD13 (Potolicchio et al., 2005), and found that PAE increased CD13 protein expression in exosomes. CD13 is also a marker for brain pericytes (Smyth et al., 2018). However, we found that ethanol treatment increased CD13 levels in primary cultures of microglia. One potential concern with the MBH exosome data is that they contained both astrocytes and neuronal markers (data not shown), suggesting the possibility that MBH exosomes derived from multiple cell types. These suggest that microglia are one of the cell types in CNS that contributes to the increased exosomal cargo following PAE. Changes in the CD13 protein were also confirmed by augmented CD13 activity in AF-treated MBH exosomes. Next, if the augmented exosome release from MBH following PAE was because of microglial activation, then these effects should be nullified following minocycline treatment. Indeed, minocycline administration normalized the CD13 protein and activity from AF-treated rat MBH exosomes. Additionally, ethanol dose-dependently increased the CD13 protein and activity from microglial cultures. Ethanol also increased the level of MMP2, another marker for exosome activity, from primary microglial cultures. These data suggest that PAE increased the amounts and activity of microglial exosomes.
Postnatal ethanol exposure also activates microglia to induce β-endorphin neuronal death. To evaluate whether MBH exosomes affected ethanol-induced β-endorphin neuronal loss, we inhibited exosome production with GW4869. A previous study reported that exosome secretion is modulated by the sphingolipid-metabolizing enzymes SMPD2, SMPD3, and SMS2. GW4869 has been shown to decrease the secretion of exosomes by inhibiting SMPD2/3 (Menck et al., 2017). GW4869 has been shown to significantly reduce exosome production as well as prevent exosome-induced apoptosis for neurons in an Alzheimer's disease model (Wang et al., 2012; Dinkins et al., 2014). However, a recent study showed a compensatory increase in the secretion of micro-vesicles (Menck et al., 2017). Moreover, inhibition of neutral sphingomyelinase with this agent could have effects on cell viability that may be independent of exosomes (Candalija et al., 2014), In our study, GW4869 administration prevented the loss of β-endorphin neurons in the arcuate nucleus of AF-treated rats without producing any effects in PF and AD rats. We argued that, if the GW4869 effect was primarily because of cell viability independent of exosomes, GW4869 should have altered β-endorphin neurons in AD and PF rats. We also showed an increased number of apoptotic β-endorphin neurons on incubation with exosomes, released by the ethanol-treated microglia. Hence, it is plausible that ethanol-induced apoptosis of β-endorphins is due, at least in part, to exosomes from microglia.
We also characterized the elements that made up exosomal cargo and might have played a role in β-endorphin neuronal death by proteomics. This analysis identified an interesting group of proteins that belong to the complement system. It has been reported that dysregulation of the complement system can be found in the early stages of neurologic diseases, but the effect of ethanol exposure on the complement system was not determined. Here we have presented data suggesting that complement proteins are involved. AF treatment significantly increased the expression of C1q in the arcuate nucleus relative to AD and PF controls. However, C1q must be deposited on neurons in order for the classical complement pathway to begin and produce the MAC to lyse cells. Therefore, we determined whether C1q colocalized with β-endorphin neurons in the arcuate nucleus using Pearson's correlation coefficient, which is independent of any form of preprocessing and relatively safe from user bias (Dunn et al., 2011). We found that C1q and β-endorphin signals were colocalized significantly more in AF-treated rats than in AD and PF controls. Since microglia have been shown to be the dominant producer of C1q (Fonseca et al., 2017), we determined whether the increase in C1q expression was because of ethanol-induced microglial activation. Therefore, inhibition of microglial activation with minocycline normalized both C1q fluorescence intensity and colocalization in AF-treated rats. These data show that PAE activates microglia to produce C1q, which gets deposited on β-endorphin neurons. We also determined the changes in complement protein levels in exosomes following ethanol treatment in the primary microglial culture and found ethanol-elevated C1q and related C2 complement proteins. The deposition of C1q was also increased in primary β-endorphin neuronal cells following ethanol treatment. These data support the finding of in vivo studies and suggest that ethanol increases deposition of microglial derived C1q in β-endorphin neurons.
Deposition of C1q is critical to complement mediated apoptosis, as activation of the C1 complex is required to generate the MAC. First, the C1 complex cleaves complement components C2 and C4. Their cleavage products, C2a and C4b, bind together to form the C3 convertase and adhere to the cell. C3 convertase can then cleave C3 into C3a and C3b, C3b binds to the C3 convertase to form the C5 convertase, and the C5 convertase cleaves C5, which leads to the formation of the MAC and cell lysis (Rus et al., 2005). We tested involvement of some of these downstream C1q signaling pathways. Our flow cytometric analysis of ethanol-activated microglial exosomes or recombinant C1q-treated β-endorphin neuronal cells showed an increase of C3a/b and C4 deposition and MAC/C5b9 formation in β-endorphin neurons.
Another reason why C1q deposition is important is because C3a/b on microglia induces phagocytosis of neurons (Arcuri et al., 2017). Indeed, blockade of C1q deposition by a blocker C1NH or anti-C1q-neutralizing antibody prevented MAC formation and β-endorphin neuronal death in vivo and in vitro, respectively. MAC is a transmembrane protein channel created during the activated stage of the terminal complement pathway and induces a necrotic-type cell death by triggering Ca2+ influx, mitochondrial damage, and an ATP depletion that leads to a rapid necrotic-type cell lysis (Kim et al., 1987; Papadimitriou et al., 1991). Death receptors are well known for induction of apoptosis and cellular death. Liu et al. (2020) showed that rats that binge drink in adolescence have high expression of TNF receptor superfamily death receptor 3 and Fas receptors, which can trigger caspase-mediated cell death cascades in the hippocampus by forming death-inducing signaling complexes. Moreover, it has been shown that exogenous treatment of SKOV3 cells with C1q and individual globular head modules can induce upregulation of TNF-α and Fas and ultimately trigger death cascade. These data support the view that C1q can act as an ROS initiator (Kaur et al., 2016). Other studies claimed that C1q can bind to mitochondria through the globular C1q receptor to drive ROS production and neuronal apoptosis following hypoxia-mediated damage or cortical ischemia (Dedio et al., 1998; Ten et al., 2010). Hence, we also determined whether this complement is involved in the cellular and mitochondrial ROS production and that it can ultimately cause neuronal death. Our data presented in this study show that C1q activates both cellular and mitochondrial ROS, and a C1q blocker prevents the effects of ethanol-activated microglial exosomes on ROS production and β-endorphin neuronal apoptosis. A ROS blocker also prevents C1q and ethanol-activated microglial exosome-induced β-endorphin neuronal apoptosis. Therefore, C1q deposition is required for complement-induced cell lysis of β-endorphin neurons.
In conclusion, PAE activates microglia and its exosomal cargo containing high level of C1q. The increased production of C1q also corresponds with increased deposition on β-endorphin neurons and may play a role in ethanol-induced death of these neurons. C1q deposition on β-endorphin neurons may increase C3a/b and C4 deposition and MAC/C5b9 formation in β-endorphin neurons and may induce a necrotic-type cell death in these cells. C1q deposition may also drive ROS production and induce oxidative stress to cause apoptotic death of β-endorphin neurons. Therefore, the complement system on exosomal cargo of microglia contributes to the PAE-induced death of β-endorphin neurons in the hypothalamus. These data suggest that the complement system may have potential to prevent some of the deleterious effects of fetal alcohol exposure; however, more research is needed to confirm this possibility.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institutes of Health Grant R37AA08757 to D.K.S. and Grant 5R37 AA08757-S1 to M.A.C. We thank Haiyan Zhang, Peter Lobel, and Nadia Rachdaoui for advice and help in conducting proteomics assays and data analyses; Oleta Sandiford for Nanosight analysis; and Stacey Pontoriero for editorial assistance.
References
- Arcuri C, Fioretti B, Bianchi R, Mecca C, Tubaro C, Beccari T, Franciolini F, Giambanco I, Donato R (2017) Microglia-glioma cross-talk: a two way approach to new strategies against glioma. Front Biosci (Landmark Ed) 22:268–309. 10.2741/4486 [DOI] [PubMed] [Google Scholar]
- Bahrini I, Song JH, Diez D, Hanayama R (2015) Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci Rep 5:7989. 10.1038/srep07989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco F, Fumagalli M, Pravettoni E, D'Ambrosi N, Volonte C, Matteoli M, Abbracchio MP, Verderio C (2005) Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res Brain Res Rev 48:144–156. 10.1016/j.brainresrev.2004.12.004 [DOI] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK (2010) Role of microglia in ethanol's apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin Exp Res 34:1835–1842. 10.1111/j.1530-0277.2010.01271.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK (2013) Cyclic adenosine monophosphate and brain-derived neurotrophic factor decreased oxidative stress and apoptosis in developing hypothalamic neuronal cells: role of microglia. Alcohol Clin Exp Res 37:1370–1379. 10.1111/acer.12104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candalija A, Cubí R, Ortega A, Aguilera J, Gil C (2014) Trk receptors need neutral sphingomyelinase activity to promote cell viability. FEBS Lett 588:167–174. 10.1016/j.febslet.2013.11.032 [DOI] [PubMed] [Google Scholar]
- Chastain LG, Franklin T, Gangisetty O, Cabrera MA, Mukherjee S, Shrivastava P, Jabbar S, Sarkar DK (2019) Early life alcohol exposure primes hypothalamic microglia to later-life hypersensitivity to immune stress: possible epigenetic mechanism. Neuropsychopharmacology 44:1579–1588. 10.1038/s41386-019-0326-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Kuhn P, Chaturvedi K, Boyadjieva NI, Sarkar DK (2006) Ethanol induces apoptotic death of developing β-endorphin neurons via suppression of cyclic adenosine monophosphate production and activation of transforming growth factor-β1-linked apoptotic signaling. Mol Pharmacol 69:706–717. 10.1124/mol.105.017004 [DOI] [PubMed] [Google Scholar]
- Chettimada S, Lorenz DR, Misra V, Dillon ST, Reeves RK, Manickam C, Morgello S, Kirk GD, Mehta SH, Gabuzda D (2018) Exosome markers associated with immune activation and oxidative stress in HIV patients on antiretroviral therapy. Sci Rep 8:7227. 10.1038/s41598-018-25515-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Cortens JP, Beavis RC (2004) Open source system for analyzing, validating, and storing protein identification data. J Proteome Res 3:1234–1242. 10.1021/pr049882h [DOI] [PubMed] [Google Scholar]
- Crenshaw BJ, Kumar S, Bell CR, Jones LB, Williams SD, Saldanha SN, Joshi S, Sahu R, Sims B, Matthews QL (2019) Alcohol modulates the biogenesis and composition of microglia-derived exosomes. Biology 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K (2009) Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol 44:115–127. 10.3390/biology8020025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A, Boyadjieva NI, Pastorcic M, Reddy BV, Sarkar DK (1994) cAMP and ethanol interact to control apoptosis and differentiation in hypothalamic β-endorphin neurons. J Biol Chem 269:26697–26705. [PubMed] [Google Scholar]
- Dedio J, Jahnen-Dechent W, Bachmann M, Müller-Esterl W (1998) The multiligand-binding protein gC1qR, putative C1q receptor, is a mitochondrial protein. J Immunol 160:3534–3542. [PubMed] [Google Scholar]
- Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E (2014) Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging 35:1792–1800. 10.1016/j.neurobiolaging.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago F, Lombardi M, Prada I, Gabrielli M, Joshi P, Cojoc D, Franck J, Fournier I, Vizioli J, Verderio C (2017) ATP modifies the proteome of extracellular vesicles released by microglia and influences their action on astrocytes. Front Pharmacol 8:910. 10.3389/fphar.2017.00910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH (2011) A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300:C723–C742. 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Andaloussi S, Mäger I, Breakefield XO, Wood MJ (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12:347–357. 10.1038/nrd3978 [DOI] [PubMed] [Google Scholar]
- Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, Wang Y, Zingarelli B, Peng T, Fan GC (2015) Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta 1852:2362–2371. 10.1016/j.bbadis.2015.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C (2009) Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol 183:4733–4744. 10.4049/jimmunol.0803590 [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14:48. 10.1186/s12974-017-0814-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KG, Verma P, Yoon E, Yu W, Weinberg J (2008) Prenatal alcohol exposure increases vulnerability to stress and anxiety-like disorders in adulthood. Ann NY Acad Sci 1144:154–175. 10.1196/annals.1418.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraci N, Leonardi T, Gessler F, Vega B, Pluchino S (2016) Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci 17:171. 10.3390/ijms17020171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JS, Miller MW (2001) Proliferation and death of cultured fetal neocortical neurons: effects of ethanol on the dynamics of cell growth. J Neurocytol 30:391–401. 10.1023/a:1015013609424 [DOI] [PubMed] [Google Scholar]
- Janas AM, Sapoń K, Janas T, Stowell MH, Janas T (2016) Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim Biophys Acta 1858:1139–1151. 10.1016/j.bbamem.2016.02.011 [DOI] [PubMed] [Google Scholar]
- Kaur A, Sultan SH, Murugaiah V, Pathan AA, Alhamlan FS, Karteris E, Kishore U (2016) Human C1q induces apoptosis in an ovarian cancer cell line via tumor necrosis factor pathway. Front Immunol 7:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh EL, Lindsay S, Halasz M, Gubbins LC, Weiner-Gorzel K, Guang MH, McGoldrick A, Collins E, Henry M, Blanco-Fernández A, Gorman PO, Fitzpatrick P, Higgins MJ, Dowling P, McCann A (2017) Protein and chemotherapy profiling of extracellular vesicles harvested from therapeutic induced senescent triple negative breast cancer cells. Oncogenesis 6:e388. 10.1038/oncsis.2017.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Camey DF, Shin ML (1987) Nucleated cell killing by complement: effects of C5b-9 channel size and extracellular Ca2+ on the lytic process. J Immunol 138:1530–1536. [PubMed] [Google Scholar]
- Kuhn P, Sarkar DK (2008) Ethanol induces apoptotic death of β-endorphin neurons in the rat hypothalamus by a TGF-β1-dependent mechanism. Alcohol Clin Exp Res 32:706–714. 10.1111/j.1530-0277.2008.00627.x [DOI] [PubMed] [Google Scholar]
- Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, Kumar A, Thom SR, Faden AI (2017) Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation 14:47. 10.1186/s12974-017-0819-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JD, Coulthard LG, Woodruff TM (2019) Complement dysregulation in the central nervous system during development and disease. Semin Immunol 45:101340. 10.1016/j.smim.2019.101340 [DOI] [PubMed] [Google Scholar]
- Liu W, Vetreno RP, Crews FT (2020) Hippocampal TNF-death receptors, caspase cell death cascades, and IL-8 in alcohol use disorder. Mol Psychiatry. Advance online publication. Retrieved Mar 5, 2020. doi: 10.1038/s41380-020-0698-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SE, Cramer JA, West JR, Sohrabji F (1999a) Alcohol exposure during the first two trimesters equivalent alters granule cell number and neurotrophin expression in the developing rat olfactory bulb. J Neurobiol 41:414–423. [DOI] [PubMed] [Google Scholar]
- Maier SE, Miller JA, West JR (1999b) Prenatal binge-like alcohol exposure in the rat results in region-specific deficits in brain growth. Neurotoxicol Teratol 21:285–291. 10.1016/S0892-0362(98)00056-7 [DOI] [PubMed] [Google Scholar]
- May PA, Chambers CD, Kalberg WO, Zellner J, Feldman H, Buckley D, Kopald D, Hasken JM, Xu R, Honerkamp-Smith G, Taras H, Manning MA, Robinson LK, Adam MP, Abdul-Rahman O, Vaux K, Jewett T, Elliott AJ, Kable JA, Akshoomoff N, et al. (2018) Prevalence of fetal alcohol spectrum disorders in 4 US communities. JAMA 319:474–482. 10.1001/jama.2017.21896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menck K, Sönmezer C, Worst TS, Schulz M, Dihazi GH, Streit F, Erdmann G, Kling S, Boutros M, Binder C, Gross JC (2017) Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J Extracell Vesicles 6:1378056. 10.1080/20013078.2017.1378056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW. (1995) Generation of neurons in the rat dentate gyrus and hippocampus: effects of prenatal and postnatal treatment with ethanol. Alcohol Clin Exp Res 19:1500–1509. 10.1111/j.1530-0277.1995.tb01014.x [DOI] [PubMed] [Google Scholar]
- Morton MC, Neckles VN, Seluzicki CM, Holmberg JC, Feliciano DM (2018) Neonatal subventricular zone neural stem cells release extracellular vesicles that act as a microglial morphogen. Cell Rep 23:78–89. 10.1016/j.celrep.2018.03.037 [DOI] [PubMed] [Google Scholar]
- Mulcahy LA, Pink RC, Carter DR (2014) Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Miyoshi K, Narita M, Suzuki T (2007) Involvement of microglia in the ethanol-induced neuropathic pain-like state in the rat. Neurosci Lett 414:21–25. 10.1016/j.neulet.2006.10.062 [DOI] [PubMed] [Google Scholar]
- Papadimitriou JC, Ramm LE, Drachenberg CB, Trump BF, Shin ML (1991) Quantitative analysis of adenine nucleotides during prelytic phase of cell death mediated by C5b-9. J Immunol 147:212–217. [PubMed] [Google Scholar]
- Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E (2012) The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem 287:43108–43115. 10.1074/jbc.M112.404467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potolicchio I, Carven GJ, Xu X, Stipp C, Riese RJ, Stern LJ, Santambrogio L (2005) Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J Immunol 175:2237–2243. 10.4049/jimmunol.175.4.2237 [DOI] [PubMed] [Google Scholar]
- Prada I, Furlan R, Matteoli M, Verderio C (2013) Classical and unconventional pathways of vesicular release in microglia. Glia 61:1003–1017. 10.1002/glia.22497 [DOI] [PubMed] [Google Scholar]
- Rus H, Cudrici C, Niculescu F (2005) The role of the complement system in innate immunity. Immunol Res 33:103–112. 10.1385/IR:33:2:103 [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Kuhn P, Marano J, Chen C, Boyadjieva N (2007) Alcohol exposure during the developmental period induces β-endorphin neuronal death and causes alteration in the opioid control of stress axis function. Endocrinology 148:2828–2834. 10.1210/en.2006-1606 [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Boyadjieva NI, Chen CP, Ortigüela M, Reuhl K, Clement EM, Kuhn P, Marano J (2008) Cyclic adenosine monophosphate differentiated beta-endorphin neurons promote immune function and prevent cancer growth. Proc Natl Acad Sci USA 105:9105–9110. 10.1073/pnas.0800289105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar DK, Gangisetty O, Wozniak JR, Eckerle JK, Georgieff MK, Foroud TM, Wetherill L, Wertelecki W, Chambers CD, Riley E, Zymak-Zakutnya N, Yevtushok L (2019) Persistent changes in stress-regulatory genes in pregnant women or children exposed prenatally to alcohol. Alcohol Clin Exp Res 43:1887–1897. 10.1111/acer.14148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schey KL, Luther JM, Rose KL (2015) Proteomics characterization of exosome cargo. Methods 87:75–82. 10.1016/j.ymeth.2015.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feildstein AF, Stahl GL, Takahashi K, Nagy LE (2011) Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem 286:35989–35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples RA, Vella LJ, Nisbet RM, Naylor R, Perez K, Barnham KJ, Masters CL, Hill AF (2008) Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J 22:1469–1478. 10.1096/fj.07-9357com [DOI] [PubMed] [Google Scholar]
- Shrivastava P, Cabrera MA, Chastain LG, Boyadjieva NI, Jabbar S, Franklin T, Sarkar DK (2017) Mu-opioid receptor and delta-opioid receptor differentially regulate microglial inflammatory response to control proopiomelanocortin neuronal apoptosis in the hypothalamus: effects of neonatal alcohol. J Neuroinflammation 14:83. 10.1186/s12974-017-0844-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman SM, Kim BJ, Howell GR, Miller J, John SWM, Wordinger RJ, Clark AF (2016) C1q propagates microglial activation and neurodegeneration in the visual axis following retinal ischemia/reperfusion injury. Mol Neurodegener 11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LC, Rustenhoven J, Scotter EL, Schweder P, Faull RL, Park TI, Dragunow M (2018) Markers for human brain pericytes and smooth muscle cells. J Chem Neuroanat 92:48–60. 10.1016/j.jchemneu.2018.06.001 [DOI] [PubMed] [Google Scholar]
- Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, Sivasankar B, Morgan BP, Silverstein S, Stark R, Polin R, Vannucci SJ, Pinsky D, Starkov AA (2010) Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci 30:2077–2087. 10.1523/JNEUROSCI.5249-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama H, Hatano K, Suzuki H, Ichise M, Momosaki S, Kudo G, Ito F, Kato T, Yamaguchi H, Katada K, Sawada M, Ito K (2008) In vivo imaging of microglial activation using a peripheral benzodiazepine receptor ligand: [11C]PK-11195 and animal PET following ethanol injury in rat striatum. Ann Nucl Med 22:417–424. 10.1007/s12149-008-0136-1 [DOI] [PubMed] [Google Scholar]
- Tseng AM, Chung DD, Pinson MR, Salem NA, Eaves SE, Miranda RC (2019) Ethanol exposure increases miR-140 in extracellular vesicles: implications for fetal neural stem cell proliferation and maturation. Alcohol Clin Exp Res 43:1414–1426. 10.1111/acer.14066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turola E, Furlan R, Bianco F, Matteoli M, Verderio C (2012) Microglial microvesicle secretion and intercellular signaling. Front Physiol 3:149. 10.3389/fphys.2012.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, Porto-Carreiro I, Simoes S, Raposo G (2006) Exosomes: a common pathway for a specialized function. J Biochem 140:13–21. 10.1093/jb/mvj128 [DOI] [PubMed] [Google Scholar]
- Wang G, Dinkins M, He Q, Zhu G, Poirier C, Campbell A, Mayer-Proschel M, Bieberich E (2012) Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): potential mechanism of apoptosis induction in Alzheimer disease (AD). J Biol Chem 287:21384–21395. 10.1074/jbc.M112.340513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Riley EP, Charness ME (2019) Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorder. Lancet Neurol 18:760–770. 10.1016/S1474-4422(19)30150-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Deng W, Klinke DJ 2nd (2015) Exosomes: improved methods to characterize their morphology, RNA content, and surface protein biomarkers. Analyst 140:6631–6642. 10.1039/c5an00688k [DOI] [PMC free article] [PubMed] [Google Scholar]