

Abstract
The multifaceted adaptor protein β-arr1 (β-arrestin1) promotes activation of focal adhesion kinase (FAK) by the chemokine receptor CXCR4, facilitating chemotaxis. This function of β-arr1 requires the assistance of the adaptor protein STAM1 (signal-transducing adaptor molecule 1) because disruption of the interaction between STAM1 and β-arr1 reduces CXCR4-mediated activation of FAK and chemotaxis. To begin to understand the mechanism by which β-arr1 together with STAM1 activates FAK, we used site-directed spin-labeling EPR spectroscopy-based studies coupled with bioluminescence resonance energy transfer–based cellular studies to show that STAM1 is recruited to activated β-arr1 by binding to a novel surface on β-arr1 at the base of the finger loop, at a site that is distinct from the receptor-binding site. Expression of a STAM1-deficient binding β-arr1 mutant that is still able to bind to CXCR4 significantly reduced CXCL12-induced activation of FAK but had no impact on ERK-1/2 activation. We provide evidence of a novel surface at the base of the finger loop that dictates non-GPCR interactions specifying β-arrestin–dependent signaling by a GPCR. This surface might represent a previously unidentified switch region that engages with effector molecules to drive β-arrestin signaling.
Keywords: arrestin, arrestin signaling, CXC-chemokine receptor type 4 (CXCR4), electron paramagnetic resonance (EPR), G protein–coupled receptor (GPCR), PTK2 protein-tyrosine kinase 2 (PTK2), focal adhesion kinase (FAK), bioluminescence resonance energy transfer (BRET)
β-Arrestins (β-arrestin1 and β-arrestin2, also called arrestin-2 and arrestin-3, respectively) are multifaceted adaptor proteins that bind to ligand-activated and G protein–coupled receptor kinase (GRK) phosphorylated G protein–coupled receptors (GPCRs), thereby limiting the magnitude and duration of G protein–mediated signaling, leading to homologous receptor desensitization (1, 2). Once bound to a GPCR, β-arrestins can also promote distinct branches of signaling by interacting with various signaling molecules (3, 4). β-Arrestins are members of a family of four proteins that also includes the two visual arrestins (arrestin-1 and arrestin-4). Arrestins share a conserved fold characterized by two elongated domains, termed the N and C domains, that are connected by a hinge region (5–9). Αrrestins are mainly cytosolic proteins where they are maintained in an inactive state by several intramolecular interaction networks (5, 10). Agonist activation and phosphorylation of GPCRs promotes high-affinity arrestin binding characterized by a global conformational change leading to a fully activated arrestin (11–16). β-Arrestins might also assume receptor-specific conformations when bound to GPCRs, possibly endowing them with the ability to engage with discrete effector molecules (7, 16–18). However, this remains poorly understood.
The GPCR CXC motif chemokine receptor 4 (CXCR4) and its cognate chemokine CXCL12 (also called stromal cell-derived factor 1α) mediate chemotaxis during organ development, immune responses, stem cell mobilization, and metastatic cancer (19–23). CXCR4 is widely expressed on multiple cell types including lymphocytes, hematopoietic stem cells, epithelial cells, and cancer cells (24). CXCR4 is overexpressed in more than 23 human cancers, and its expression is associated with overall poor survival (22, 25–29). CXCR4 signaling is involved in several aspects of tumor progression and metastases (30, 31), yet the mechanisms governing CXCR4 signaling remain poorly understood.
Although GRKs and β-arrestins negatively regulate CXCR4 by canonical homologous desensitization (32), β-arrestins can also promote CXCR4 signaling. In particular, β-arr1 (β-arrestin 1) is necessary for the activation of focal adhesion kinase (FAK), but not Akt or ERK-1/2, by CXCR4 (33). A unique feature of FAK activation by β-arr1 is that it requires the assistance of endocytic adaptor protein STAM1 (signal-transducing adaptor molecule 1) (33). β-arr1, and to a lesser extent β-arr2, interacts directly with STAM1, and disruption of the interaction between them reduces FAK activation and chemotaxis by CXCR4 (33). Because binding to β-arr1 is enhanced by CXCR4 activation (34), it is likely that STAM1 prefers to bind to the receptor-bound, fully activated conformation of β-arr1. This is likely required to interact with and activate FAK, because a physical complex of β-arr1, STAM1, and FAK is formed following receptor activation, and they also co-localize at or near the cell periphery (33). Why β-arr1 requires STAM1 to activate FAK remains to be determined.
Once bound to GPCRs, β-arrestins undergo a global conformational change that enhances their ability to bind to effector molecules (15, 35–37). Because β-arr1 binding to activated and phosphorylated CXCR4 changes its ability to bind to STAM1, the interaction with STAM1 is likely driven by conformational changes in β-arrestin. STAM1 mainly binds to the N domain of β-arr1 between residues 25 and 161 (33), which also contains key contact sites for ligand-activated and phosphorylated GPCRs (10, 38). However, the precise structural or conformational determinants that dictate STAM1 binding remain unknown. Arrestins have several regions that become available for binding to effector molecules once activated by receptor binding. Importantly, the region between amino acid residues 89 and 97 of the N domain in β-arr2 referred to as arrestin switch region I (39) is an element that undergoes considerable movement from the basal state to the active state and might mediate agonist-dependent interactions with effector molecules (40). This region in β-arr2 contains two PXXP motifs, which are recognition sites for SH3 domains and likely mediate agonist-dependent binding to c-Src by GPCRs (39). The analogous region in β-arr1 contains only one PXXP motif, but it is unclear whether it can interact with SH3 domains. Notably, although STAM1 encodes an SH3 domain, it mainly interacts with β-arr1 via its coiled-coil region (34). Therefore, whether arrestin switch region I or a previously unidentified switch region binds to STAM1 remains to be determined.
Here, we investigated the structural and biophysical properties of the interaction between β-arr1 and STAM1 by EPR spectroscopy. Using continuous wave (CW) EPR, we mapped the STAM1-binding site to a discrete region at the base of the finger loop. To determine whether binding to this region causes a global or localized conformational change in β-arr1, we used another EPR approach known as double electron–electron resonance (DEER) spectroscopy that measures distances between two intramolecular spin labels attached at specific sites. The DEER data show that STAM1 binding induces movement of the finger loop, similar to GPCRs, but does not induce movement of two other regions known to undergo large movements upon GPCR binding (15). The finger loop is located between β-strands 5 and 6 and is considered to represent a major binding region for activated GPCRs (39, 40). Mutation of residues within the STAM1-binding site reduced agonist-dependent STAM1 binding but not CXCR4 binding, indicating that the STAM1-binding site does not overlap with the GPCR-binding site. Expression of a STAM1-deficient β-arr1–binding mutant that could still bind to CXCR4 reduced FAK activation, but not ERK-1/2, by CXCR4. We provide evidence of a novel region of β-arr1 specifying β-arrestin–dependent signaling.
Results
STAM1 induces local conformational changes in β-arrestin1
We used DEER spectroscopy to determine whether STAM1 binding induces conformational changes in β-arr1 by introducing spin labels at two reporter sites within β-arr1. The position of the spin labels was selected based on previous studies that revealed large conformational changes upon GPCR binding, including C-tail displacement, finger-loop extension, and middle-loop movement (15) (Fig. 1a). To report on C-tail displacement, the spin labels were placed on the C-tail (A392C) and the N domain (A12C). DEER-derived distance distributions are shown as overlays for the free (black) and STAM1-bound (blue) states (Fig. 1b). In the basal state, the C-tail is folded back onto the N domain, which helps to stabilize the inactive conformation of β-arr1 and is consistent with a small interspin distance between positions 12 and 392 (Fig. 1b). Upon GPCR binding, there is a large increase in the interspin distance, consistent with C-tail release from the N domain, as previously reported (15). However, STAM1 did not induce distance changes between spin labels at 12 and 392, indicating that the C-tail position is not affected by STAM1 binding (Fig. 1b). Middle-loop movement was assessed by placing both spin labels on the N domain at positions 136 and 167 (T136C and V167C) (Fig. 1a). STAM1 did not induce conformational changes in the middle loop, because no distance changes were observed between spin labels at positions 136 and 167 (Fig. 1c). This is in contrast to GPCR binding, which induced distance changes between spin labels in these positions, indicating the N domain moving away from the C domain (15, 16). By placing one spin label at site 167 (V167C) and the other at 68 (L68C), we were able to assess finger-loop movement (Fig. 1a). In contrast to C-tail displacement or middle-loop movement, STAM1 binding induced finger-loop extension (Fig. 1d). STAM1 induced a large distance change between these spin labels from 21 to 25 Å (Fig. 1d). This is similar to GPCR-binding, which also induces a distance change between positions 68 and 167, as previously shown (15).
Figure 1.

DEER measurements reveal conformational changes in β-arr1 induced by STAM1. a, structure of inactive β-arr1 (PDB code 1G4M (6)) highlighting the finger loop (orange), C-tail (cyan), and middle loop (blue). Residue pairs selected for DEER are connected by dotted lines. Each spin-labeled β-arr1 double cysteine mutant was analyzed by DEER with and without STAM1. Sample concentrations were as follows: 50 μm spin-labeled β-arr1 and 150 μm STAM1. b–d, DEER-derived distance probability distributions are shown as overlays for the free (gray area with black outline) and STAM1-bound (blue line) states. e–g, background-corrected dipolar evolution curves are plotted as overlays for the free (black line) and STAM1 bound states (blue line). Gray dots represent the raw data.
Shown in Fig. 1 (e–g) are the fits to the free (black) and the STAM1-bound (blue) background-corrected dipolar evolution data (gray dots) to indicate data quality to support the distance distribution data (41, 42). The attached spin labels on the β-arr1 double-cysteine mutants did not impact STAM1 binding, as assessed by pulldown assay (Fig. 2a), similar to what was previously observed with binding to GPCR (15). These data provide evidence that unlike GPCRs, STAM1 does not promote global conformational change in β-arr1, suggesting that STAM1 and GPCRs interact with the N domain differently.
Figure 2.

β-arr1 spin-labeled cysteine mutants maintained their ability to bind to STAM1, as assessed by pulldown experiments. Equimolar amounts of purified β-arr1-WT or indicated β-arr1 cysteine mutants (0.4 μm) were incubated with or without purified STAM1– (3 μm) for 20 min at 37˚C. Complexes were immobilized by incubation with Talon cobalt resin, and after washing, proteins were eluted in buffer with 200 mM imidazole and analyzed by immunoblotting. Representative blots are shown for β-arr1 double cysteine mutants (a) and single cysteine mutants (b and c).
STAM1-binding site maps to the base of the finger loop on β-arr1
To precisely map the STAM1-binding site on β-arr1, we employed site-directed spin-labeling CW EPR spectroscopy. EPR spectroscopy takes advantage of the fact that the spin labels have a paramagnetic center and when the label is attached to a unique cysteine on the surface of a protein, the spectrum reflects its mobility. Spin labels that are mobile display spectra that are narrow with sharp peaks; however, when a protein partner binds at or near the spin label or a conformational change constrains its mobility, the spectral lines broaden and decrease in amplitude (43, 44). This method has been previously used to successfully map the rhodopsin footprint on β-arr1 (38, 45). Because the N domain has previously been shown to bind STAM1 (34), we selected several reporter sites on the N domain to map the STAM1 footprint (Figs. 3a and 4a). We selected two reporter sites (Leu68 and Val167) on the front surface (GPCR-binding surface) of the N domain (Fig. 3a) that have been previously shown to be part of the GPCR-binding site (38, 45). On the opposite surface of the N domain or the back (non–GPCR-binding) surface, we selected eight reporter sites (Leu33, Tyr47, Leu71, Phe75, Arg76, Leu79, Val84, and Phe87) in three solvent-exposed regions (Fig. 4a). We first confirmed that each spin-labeled mutant was able to bind STAM1, similar to WT β-arr1, by a pulldown assay (Fig. 2, a–c). We then recorded the CW EPR spectra in the absence or presence of purified STAM1 or model GPCR. Rhodopsin, a prototypical GPCR, was used in these experiments because it is available in native membranes and is easily activated and phosphorylated (10, 46). Importantly, both β-arrestins bind to activated and phosphorylated rhodopsin (38, 47). The respective EPR spectra were normalized and overlaid to reveal spectral line-shape changes in the presence of GPCR or STAM1.
Figure 3.

Front surface N domain spin label mobility analysis of β-arr1 binding to STAM1 or GPCR. a, structure of inactive β-arr1 (PDB code 1G4M (6)) with spin label reporter sites 68 and 167 shown as red spheres. b, spectra for each spin-labeled β-arr1 in the absence (black) or presence of STAM1 (blue) are shown. Spectra obtained using 20 μm spin-labeled β-arr1 in the absence or presence of 400 μm STAM1 are overlaid. c, spectra in the absence or presence of 60 μm activated and phosphorylated rhodopsin (PRh*; red) in native disk membranes are overlaid. Spectra are normalized to the same center line height.
Figure 4.
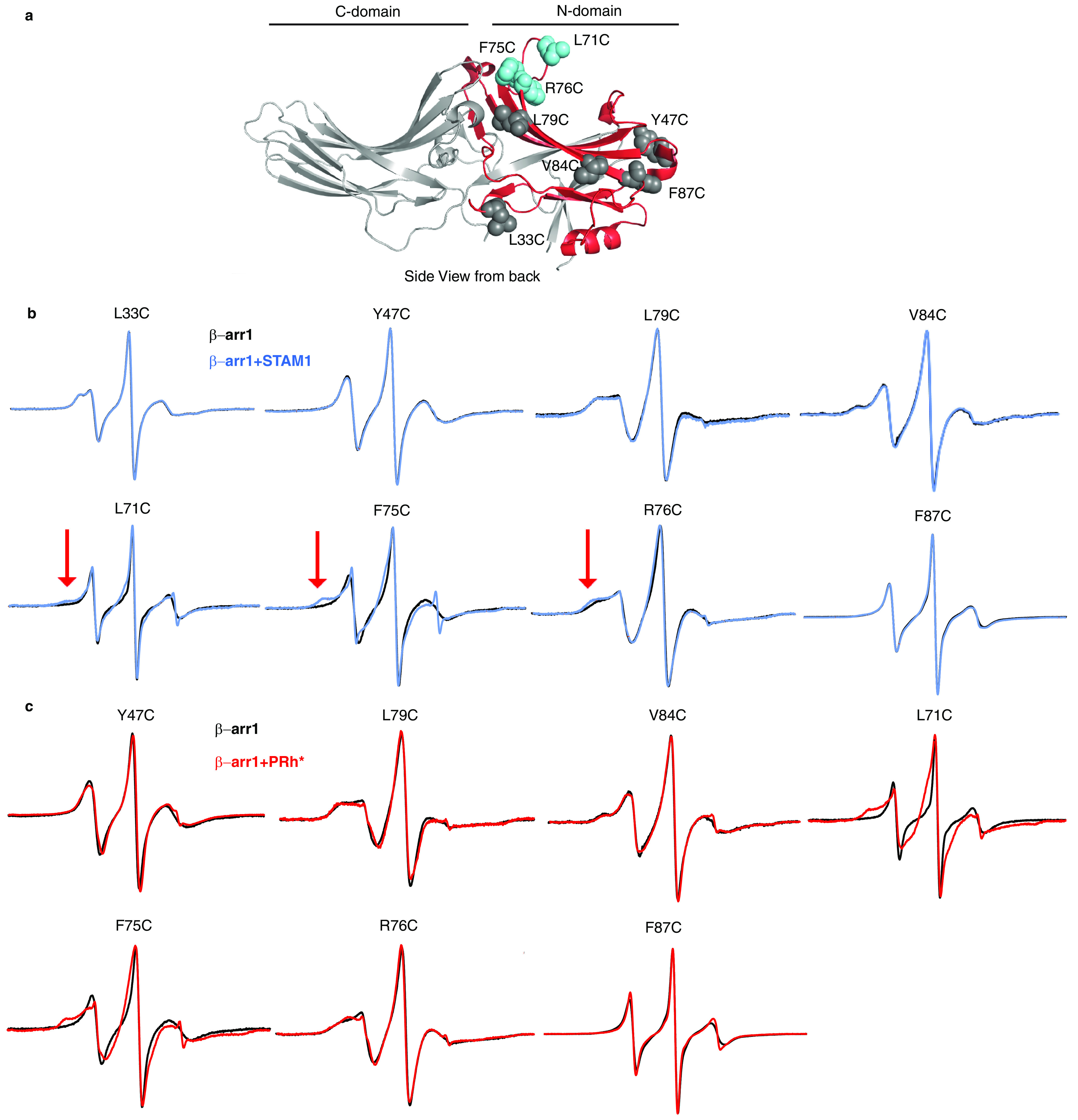
Back surface N domain spin label mobility analysis of β-arr1 binding to STAM1 or GPCR. a, Selected spin-labeled reporter sites are indicated in the β-arr1 crystal structure (PDB code 1G4M (6)). L33C, Y47C, L79C, V84C, and F87C are shown as gray spheres, and L71C, F75C, and R76C are shown as blue spheres. The previously determined primary STAM1-binding region on the N domain is shown in red. b, spectra for each spin-labeled β-arr1 in the absence (black) or presence of STAM1 (blue) are shown. Spectra obtained using 20 μm spin-labeled β-arr1 in the absence or presence of 400 μm STAM1 are overlaid. The red arrows indicate spectral line shape changes at the low field region caused by STAM1 binding. c, spectra in the absence or presence of 60 μm PRh* (red) in native disk membranes are overlaid. Spectra are normalized to the same center line height.
The overlaid EPR spectra of spin labels at position 68 or 167 from β-arr1 alone or associated with STAM1 were identical, indicating that the mobility of these sites was not impacted by association with STAM1 (Fig. 3b). In contrast, GPCR binding decreased the mobility of the spin labels at positions 68 and 167, as reflected in the overlaid spectra (Fig. 3c). These data suggest that these sites are part of or close to the GPCR-binding site (38, 48) but not the STAM1-binding site.
On the back (non–GPCR-binding) surface, the spectra from the majority of the sites did not show any changes in spin mobility upon association with STAM1, except sites 71, 75, and 76 (Fig. 4b). STAM1 association with β-arr1 notably decreases the spin label mobility at site 75 and to a lesser extent at sites 71 and 76, suggesting that these sites are part of or near to the STAM1-binding site. When GPCR associated with β-arr1, the largest decrease in spin label mobility was observed at sites 71 and 75 and to a lesser extent at site 76 (Fig. 4c). This is in agreement with the finger loop interacting with GPCRs, as showed by previous EPR (38, 45, 48) and structural (49–53) studies. A small decrease in spin label mobility was observed at sites 47 and 79, but not at sites 84 and 87. Sites 71, 75, and 76 are in the finger-loop region, which was hypothesized to serve as the activation sensor for GPCRs (39, 40). Our data suggest that STAM1 likely binds to the finger loop of β-arr1.
Because the spectral line shape changes at spin-labeled sites 71 and 75 were sufficient to quantitate changes caused by STAM1 binding, we took advantage of this to determine the affinity of the interaction between β-arr1 and STAM1 by performing a titration of spin-labeled L71C or F75C with increasing concentrations of STAM1 (Fig. 5). The concentration of labeled β-arr1 used in these experiments was 10 μm, well below the 100 μm concentration at which β-arrestins oligomerizes (54). The resultant data were fit to a one-site binding model, and the derived Kd values using spectral changes at L71C and F75C were 79.3 ± 29.2 and 52.4 ± 17.4 μm, respectively. The Kd values are similar, although the affinity for STAM1 is very low. This low Kd could be due to the use of inactive free β-arr1 in our studies, whereas STAM1 likely binds with higher affinity to an active conformation, i.e. the conformation of β-arr1 bound to ligand-activated and phosphorylated receptor (51–53).
Figure 5.

The affinity of STAM1 for β-arr1 determined by CW EPR spectroscopy. a and b, spectra obtained using 10 μm spin-labeled β-arr1 L71C (a) or F75C (b) in the presence of increasing concentrations of STAM1 are shown. c, graphs of STAM1 binding affinity curves for L71C and F75C. To quantify the unbound and bound population of spin-labeled β-arr1, EPR spectra were deconvoluted, and the resultant data points were fit to a one-site binding model.
Identification of STAM1-binding site on β-arr1 by mutagenesis
We next examined the role of the finger loop in STAM1 binding in cells. Although the finger loop undergoes an extension when STAM1 binds, as revealed by distance measurements between positions 68 and 167, CW EPR showed that these residues are not directly engaged by STAM1. Instead CW EPR revealed changes in spin label mobility at positions 71, 75, and 76, suggesting that these sites are either located at the STAM1-binding interface or undergo conformational changes induced by STAM1 binding. To address this, we substituted four consecutive amino acids to alanine residues (L73A/T74A/F75A/R76A) along the back surface of the finger loop (Fig. 6a) and examined the ability of this β-arr1 variant (β-arr1–4A) to bind to STAM1 by co-immunoprecipitation of epitope-tagged T7–STAM1 and FLAG-tagged WT or mutant β-arr1 co-expressed in HEK293 cells. T7–STAM1 was immunoprecipitated with an antibody against T7, followed by immunoblotting to detect the presence of β-arr1 in the immunoprecipitates. The presence of β-arr1–4A in the immunoprecipitates was significantly reduced compared with WT β-arr1 (Fig. 6b). To further validate these data, we performed an independent pulldown experiment with His-tagged STAM1 immobilized on cobalt resin and purified WT or mutant β-arr1. These experiments also showed that STAM1 binding to the 4A mutant was significantly reduced, as compared with WT β-arr1 (Fig. 6c). Collectively, these data provide evidence that the base of the finger loop (residues Leu73 to Arg76) is likely the major STAM1-binding site on β-arr1.
Figure 6.

Biochemical analysis of β-arr1 WT and finger-loop mutant (4A) binding to STAM1. a, sequence alignment of β-strand 5, finger loop, and β-strand 6 of bovine β-arr1 (residues 52–89), β-arr2 (residues 53–90), and visual arrestin (Arr1, residues 56–93). Boxed residues denote the four residues that were substituted to alanine residues in β-arr1. Letters highlighted in red denote residues that are identified to form two ionic locks to maintain the finger loop in an inactive bent conformation. b, cleared lysates from HEK293 cells co-transfected with T7–STAM1 and β-arr1–WT–FLAG, β-arr1–4A-FLAG, or empty vector (pCMV) were incubated with an anti-T7 antibody. Immunoprecipitates (IP) were analyzed by immunoblotting to detect bound β-arr1. Shown are representative immunoblots. Bound STAM1 was quantified by densitometric analysis. The graph represents the fraction of STAM1 bound to β-arr1–WT–FLAG from three independent experiments. The error bars represent the S.D. The data were analyzed by an unpaired t test. The p values are indicated. c, equimolar amounts of purified β-arr1–WT or β-arr1–4A (0.4 μm) were incubated with or without purified STAM1–HIS (3 μm) for 20 min at 37 °C. Complexes were captured by incubation with Talon cobalt resin, and after washing, the proteins were eluted in binding buffer supplemented with 200 mM imidazole and analyzed by immunoblotting. Representative blots are shown. Bound STAM1 was quantified by densitometric analysis. The graph represents the fraction of STAM1 bound to β-arr1 from three independent experiments. The error bars represent the S.D. The data were analyzed by an unpaired t test. The p values are indicated.
Because the STAM1-binding site is part of the finger loop, which interacts with GPCRs by inserting into the 7TM core (51–53), the STAM1-binding site might overlap with the GPCR-binding site, as the EPR data suggest (Fig. 4c). To explore this, we used BRET, which is commonly used to monitor protein–protein interactions in live cells (55–57). BRET measures the energy transfer in the presence of a substrate from a bioluminescent donor such as Renilla luciferase (Rluc), to a fluorescent acceptor, such as GFP. To examine the interaction between STAM1 and β-arr1, HEK293 cells were co-transfected with T7–STAM1 C-terminally tagged with Rluc (STAM1–Rluc) and WT or 4A-mutant β-arr1 tagged with GFP (β-arr1–WT–GFP or β-arr1–4A–GFP, respectively). The cells were stimulated with increasing concentrations of CXCL12, and the BRET ratio was calculated at each dose. As shown in Fig. 7a, there was a dose-dependent increase in BRET in cells expressing STAM1-Rluc and β-arr1–WT–GFP, in agreement with previous reports that the interaction between β-arr1 and STAM1 is CXCR4-dependent (34). In contrast, there was a significant rightward shift (increased EC50) in BRET in cells expressing β-arr1–4A–GFP, consistent with much weaker interaction between β-arr1–4A and STAM1 (Fig. 7a). Importantly, the dose-response curves of BRET between β-arr1–WT–GFP or β-arr1–4A–GFP with CXCR4–Rluc were similar (Fig. 7b). Therefore, mutations at the base of the finger loop disrupt β-arr1 binding to STAM1, but not the CXCR4 interaction.
Figure 7.

BRET analysis of β-arr1 binding to STAM1 or CXCR4 in cells. a and b, cells co-transfected with β-arr1–WT–GFP10 or β-arr1–4A–GFP10 with HA–CXCR4 and STAM1–Rluc (a) or CXCR4–Rluc (b) were stimulated with increasing concentrations of CXCL12 for 2 min before BRET measurements. DeepBlueC was added in the continuous presence of CXCL12, and BRET measurements were taken 30 min after the addition of the luciferase substrate. The data shown are from a representative experiment performed in triplicate ± S.D. The curves were fitted by nonlinear regression, assuming a single binding site (GraphPad Prism). The bar graphs represent the average EC50 values from four independent experiments. The error bars represent the S.D. The data were analyzed by unpaired t test. The p values are indicated.
We next examined whether expression of mutant β-arr1 affects CXCR4-dependent activation of FAK in cells. FAK activation was assessed by immunoblotting for the phosphorylation status of Tyr397, an autophosphorylation site that is phosphorylated upon CXCL12 activation (33). Expression of the STAM1 binding–deficient mutant significantly reduced FAK activation by CXCL12, similar to β-arr1-(25–161), which we have previously shown to inhibit activation of FAK by CXCL12 (33) (Fig. 8, a and b). Expression of WT β-arr1 did not affect CXCL12-induced phosphorylation of FAK, compared with empty vector control (Fig. 8, a and b). Activation of ERK-1/2 was not affected by expression of the STAM1 binding–deficient mutant, as we have previously shown for β-arr1-(25–161) (33) (Fig. 8, a and c). Thus, the interaction of β-arr1 with STAM1 specifically affects the activation of FAK by CXCR4.
Figure 8.

The β-arr1–4A mutant attenuates CXCR4-promoted autophosphorylation of FAK in cells. a, HeLa cells transfected with empty vector (pCMV), FLAG-tagged β-arr1 WT, mutant (4A), or N domain fragment (residues 25–161) were stimulated with 30 nm CXCL12 for 15 min. Whole cell lysates were analyzed by immunoblotting for the indicated proteins. Representative immunoblots are shown. b and c, bars represent the average levels of pTyr397–FAK (pFAK) (b) or pERK-1/2 (c) relative to pCMV-transfected cells stimulated with CXCL12 from four independent experiments. The error bars represent the S.D. The data were analyzed by two-way analysis of variance followed by Bonferroni's multiple comparison test. The p values between indicated groups are shown (b), and the asterisks indicate p < 0.05 in each transfection condition.
Discussion
Despite the fact that β-arrestin–dependent signaling by GPCRs is important biologically and therapeutically, the mechanistic basis for this signaling remains unknown. Our results explain how β-arr1 engages downstream effector molecules to direct GPCR signaling. Here, we identify a novel surface that controls β-arr1 signaling by a GPCR. Using biophysical and biochemical approaches, we provide evidence that STAM1 binds to a localized region of β-arr1 on the N domain at the base of the finger loop and β-strand 6, near but not overlapping with the part of the finger loop that binds to GPCRs (38, 51–53). This surface is selective for FAK signaling and requires binding to the adaptor protein STAM1 in an agonist-dependent manner. The binding of STAM1 may stabilize a unique receptor-evoked conformation that is selective for activating FAK in time and space. This paradigm of β-arrestin–dependent signaling is unique because it requires the assistance of another adaptor protein, which is not the case for many other β-arrestin–dependent signaling pathways (18, 35, 58, 59). Our data suggest that we can ascribe a distinctive conformational change that specifies a β-arrestin–dependent signaling outcome.
We provide evidence that STAM1 binds to a novel surface on β-arr1. The STAM1-binding site had been previously localized to the N domain (34), but its precise location remained unknown. Here we narrowed it to a small region at the base of the finger loop and β-strand 6 (Fig. 6a). CW EPR revealed large changes in spin label mobility at positions within this region (positions Leu71, Phe75 and Arg76) in the presence of STAM1, but not at other positions on the N domain, including position 68 at the tip of the finger loop (Figs. 3 and 4), which interacts with the core of GPCRs (51–53). The fact that most of the sites throughout the N domain did not show any changes suggests that the N domain does not undergo a significant conformational change. This supports the idea that the mobility changes at the base of the finger loop are due to proximity of STAM1 in the complex rather than structural changes in β-arr1. This is also supported by DEER, which revealed that STAM1 induces finger-loop extension, but does not promote C-tail displacement or middle loop rearrangement (Fig. 1, b–d), providing further evidence that STAM1 does not induce a global conformational change in β-arr1.
In orthogonal biochemical experiments with substitution of four consecutive residues with alanines (β-arr1–4A) significantly reduced STAM1 binding (Fig. 6, b and c), demonstrating that this region is the main binding site for STAM1. This region is solvent-exposed and likely mediates direct contact with the coiled-coil region of STAM1, the main binding site for β-arr1 (34). Although CW with rhodopsin also reduced mobility of spin labels at the same positions (positions 71, 75, and 76) as STAM1 (Fig. 4c), this could be because the finger loop inserts deep into the 7TM core, making multiple contacts, as is observed in a high-resolution complex of rhodopsin and visual arrestin (50). However, this might be unique to rhodopsin or certain GPCRs (15, 49), because our BRET data suggest that the STAM1-binding site does not overlap with the CXCR4-binding site (Fig. 7b). β-arr1–4A–GFP binding was reduced only to STAM-Rluc, not to CXCR4–Rluc (Fig. 7, a and b). It is possible that the finger loop has a shallow insertion point into the 7TM core of CXCR4, which might explain why β-arr1–4A binding to CXCR4 was not impacted (Fig. 7b). A high-resolution complex of the neurotensin receptor 1 and β-arr1 shows that the finger loop has a shallow insertion into the 7TM core (52), compared with other GPCRs (49–51, 53). Structural flexibility in the finger loop might account for differences in binding mode between certain GPCRs with the 7TM core (60). Therefore, the STAM1-binding site on the finger loop represents a unique highly localized site that does not overlap with the receptor-binding site.
The finger loop undergoes significant conformational rearrangement following receptor binding (49–53) and likely serves as the activation sensor for ligand-activated GPCRs (39, 40). We showed that the finger loop also engages at least one effector molecule. STAM1 preferentially binds to activated β-arr1 (34), which is consistent with a dose-dependent increase in BRET in cells transfected with WT β-arr1–GFP and STAM–Rluc (EC50 = 0.1 nm) (Fig. 7a). The fact that β-arr1–4A binding to CXCR4 was not impacted suggests that upon binding to CXCR4 this mutant β-arr1 likely undergoes the classical hallmark conformational changes that occur when β-arrestins interact with activated and phosphorylated GPCRs, indicating that loss of STAM1 binding is not due to structural changes in β-arr1–4A. The finger loop in inactive β-arr1 exhibits a bent conformation, which is held in place by ionic locks between charged residues on each arm of the finger loop with β-strands 5 and 6 (6, 61) (Fig. 6a). These ionic locks break when β-arrestins are activated, ultimately leading to finger-loop extension and binding to the receptor core, thereby exposing the STAM1-binding site. Because we used inactive β-arr1 in the EPR studies, this could explain the low affinity we observed (Fig. 5), because STAM1 likely binds with higher affinity to the active β-arr1 conformation. How STAM1 binds to the base of the finger loop while β-arr1 is engaged with the 7TM core remains to be determined. Conceivably, STAM1 may bind to β-arr1, exhibiting a tail conformation in which β-arr1 binds to the phosphorylated C-tail of CXCR4 without engaging with the 7TM core (62, 63). This could facilitate STAM1 binding without interference from additional contacts β-arr1 might make when in a 7TM core position. Further work will be required to address each of these two possibilities.
Expression of the β-arr1–4A mutant reduces FAK activation by CXCR4 (Fig. 8). This provides further evidence that the interaction between STAM1 and β-arr1 mediates FAK signaling downstream of CXCR4. Although β-arr1–4A is effectively recruited to CXCR4 upon CXCL12 stimulation (Fig. 7b), it is likely unable to recruit STAM1 and activate FAK. This is in agreement with previous results in which expressing minigenes from either β-arr1 or STAM1 that disrupt the interaction between β-arr1 and STAM1 reduce CXCR4 promoted activation of FAK (33). β-arr1 has been reported to interact directly with FAK (33), and it has been previously shown that STAM1, β-arr1, and FAK exist in an agonist-dependent complex and co-localize in cells (33). STAM1 may stabilize a β-arr1 conformation that binds to FAK in a way that would release its autoinhibition, leading to cis-mediated autophosphorylation at Tyr397 (64,65). The molecular determinants in β-arr1 or STAM1 that promote FAK activation remain to be identified. FAK likely interacts with a different surface of β-arr1 that is not engaged by STAM1. There are several switch regions or elements in β-arrestins that change conformation upon activation and might explain agonist-dependent interactions with effectors molecules (39), but it is possible that STAM1 stabilizes a unique β-arr1 conformation that facilitates interaction with and activation of FAK (64, 65). Because STAM1 binding does not affect signaling to other effectors, such as ERK-1/2, it may stabilize a unique β-arr1 conformation specific to FAK activation (Fig. 8). In addition to conformational bias, STAM1 may also specify location bias for FAK activation. Because STAM1 is likely recruited to the CXCR4–β-arr1 complex at or near the plasma membrane, FAK activation and signaling at this location is consistent with its role in CXCR4-mediated cell migration (33).
Although we narrowed the STAM1-binding site to the C-terminal base of the finger loop (Leu73, Thr74, Phe75, and Arg76) (Fig. 6), we cannot rule out the possibility of contributions from the C domain. Previously, we showed that separated C domain β-arr1-(261–418) also binds STAM1, albeit with relatively weak affinity as compared with the separated N domain (34). Therefore, it is possible that the C domain might also contribute to STAM1 binding (34). Consistent with this possibility, the BRET signal between β-arr1–4A-GFP and STAM1–Rluc was not completely abolished, suggesting some binding between β-arr1–4A and STAM1, although it was observed only at the two highest doses of CXCL12 (Fig. 7a). Sites on the C domain might become exposed or better positioned to bind to STAM1 following the interdomain twist that is expected to occur following β-arr1 binding to CXCR4. Therefore, multiple contacts might be required to stabilize β-arr1 in a conformation that favors FAK signaling.
Despite the debate concerning arrestin-mediated signaling (66, 67), our results support a role for β-arrestin1–mediated signaling by GPCRs. In summary, we demonstrate here that a non–GPCR-binding partner engages a specific surface on β-arr1 that facilitates distinct β-arrestin–dependent signaling. The binding site for STAM1 is localized to the base of the finger loop at a site that does not overlap with the GPCR-binding site. This interaction specifies CXCR4-mediated FAK activation, but not ERK-1/2, suggesting that STAM1 stabilizes a unique β-arrestin1 conformation that can discriminate between engagement with diverse effector molecules. FAK is typically linked to integrin receptor signaling and plays a key role in the regulation of cell adhesion (68–71). We have previously shown that FAK is necessary for CXCR4-mediated chemotaxis (33). Because CXCR4-mediated chemotaxis is essential for the metastatic potential of tumor cells (24, 28, 29, 72), targeting this novel surface on β-arr1 with small molecules to inhibit STAM1 binding might be effective at selectively reducing chemotactic signaling to treat cancers associated with CXCR4 (22, 23, 72).
Materials and methods
Cell culture, antibodies, and reagents
HEK293 cells were from Microbix (Toronto, Canada), and HeLa cells were from American Type Culture Collection (Manassas, VA). The cells were maintained in DMEM (catalog no. D5796) (Sigma) supplemented with 10% fetal bovine serum (Omega). The rabbit monoclonal anti-STAM1 (catalog no. 12434-1-AP) and anti-FAK (catalog no. 12636-1-AP) antibodies were from Protein Tech (Rosemont, IL). The rabbit polyclonal anti-pTyr397-FAK antibody (catalog no. 44-624G) was from Life Technologies Inc. The rabbit monoclonal anti-βarrestin1 (catalog no. 12697), anti–ERK-1/2 (catalog no. 4695), and anti-pERK-1/2 (catalog no. 4370) antibodies were from Cell Signaling Technologies (Danvers, MA). The goat polyclonal anti-T7 tag antibody (catalog no. ab9138) and the mouse monoclonal anti–glyceraldehyde-3-phosphate dehydrogenase antibodies (catalog no. ab9482) were from Abcam. The mouse monoclonal anti-FLAG antibody (catalog no. F4049) was from Sigma–Aldrich. The mouse monoclonal anti-HA antibody (catalog no. 901514) was from BioLegend (San Diego, CA). CXCL12 was from Protein Foundry (Milwaukee, WI). Cycloheximide (catalog no. C7698) was from Sigma–Aldrich. Coelenterazine 400a (DeepBlue C) (catalog no. 10125) was from Biotium (Fremont, CA).
DNA constructs
The CXCR4–Rluc plasmid was a kind gift from Nikolaus Heveker (Hôpital Sainte-Justine, Montréal, Canada). The β-arr1–GFP10 plasmid was a kind gift from Michel Bouvier (Département de Biochimie, Université de Montréal, Montréal, Canada). β-arr1 single cysteine mutants used in EPR experiments were generated by QuikChange PCR using a cysteine-less β-arr1 mutant (45), in which all seven native cysteines were substituted with other residues to maintain existing intramolecular interactions (C59V, C125S, C140L, C150V, C242V, C251V, and C269S). The cysteine-less mutant is fully functional for receptor binding (48). The double cysteine mutant plasmids were described previously (15). The FLAG–β-arr1-(25–161) plasmid was described previously (34). The β-arr1–4A mutants were generated by QuikChange PCR using the β-arr1-FLAG or β-arr1–GFP10 as templates. The sequences were as follows for the forward and reverse primers, respectively: 5′-GGATGTCCTGGGTGCGGCTGCTGCCAAGGACCTGTTTG-3′ and 5′-CAAACAGGTCCTTGGCAGCAGCCGCACCCAGGACATCC-3′. The T7–STAM1–Rluc plasmid was generated by PCR amplification of T7–STAM1, which was subcloned into HindIII and ApaI restriction sites of pRluc-N1(h) (BioSignal, Packard). STAM1–His was generated by PCR amplification of STAM1 from T7–STAM1, which was subcloned into pQE30 (Qiagen). HA–CXCR4 in pcDNA3 was described previously (59, 60). All plasmids were confirmed by dideoxy sequencing.
Expression and purification of recombinant proteins
β-arrestin1 was expressed in Escherichia coli and purified as previously described (15, 73, 74). Briefly, BL-21(DE3) cells from frozen cell stocks were grown in LB broth supplemented with ampicillin (100 mg/liter) at 30 °C with vigorous shaking. The cultures were induced with 0.1 m isopropyl 1-thio-β-d-galactopyranoside for 4 h at 30 °C. After lysis and ammonium sulfate precipitation, the protein pellet was dissolved in 1× column buffer (10 mm Tris, 2 mm EDTA, 2 mm EGTA, pH 7.5) and proceeded to sequential chromatography on heparin–Sepharose and Q-Sepharose (GE Life Sciences) columns. The β-arrestin1 purity was then verified by 10% SDS-PAGE gel. Protein concentration was then determined by a Pierce protein assay with BSA as a standard. Proteins were concentrated using Amicon Ultra-0.5 centrifugal concentrators.
STAM1–HIS in pQE30 vector was expressed in E. coli M15 pREP4 and purified as described (75, 76). Briefly, M15 pREP4 cells from STAM1–HIS frozen cells stock were grown in LB broth containing 100 mg/liter ampicillin and kanamycin at 37 °C with vigorous shaking. The culture was then induced by 1 mm isopropyl 1-thio-β-d-galactopyranoside to an A600 0.6. After lysis and sonication, the soluble proteins in the supernatant were incubated with Cobalt Talon resins (GoldBio) at 4 °C in the presence of 10 mm imidazole. STAM1–HIS protein was eluted by imidazole gradient (from 30 to 300 mm) in the column buffer (20 mm Tris, 150 mm NaCl, 0.1% Triton, pH 7), followed by separation on a FastFlow Q-Sepharose column (GE Healthcare). The purity of STAM1–His was verified by 10% SDS-PAGE and gel code blue staining. Protein concentration was determined by a Pierce protein assay with BSA as a standard. Proteins were concentrated with Amicon Ultra-0.5 centrifugal concentrators.
Rhodopsin was isolated from bovine rod outer segments, as previously described (77). Rhodopsin was phosphorylated by endogenous GRK1 in purified rod outer segments, yielding 2.6 phosphates/rhodopsin.
Spin labeling
Purified recombinant β-arrestin1 single cysteine mutants were incubated with a 10-fold molar excess of the sulfhydryl-specific 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (Toronto Research Chemicals) overnight at 4 °C under gentle agitation. Recombinant β-arrestin1 double cysteine mutants were incubated with a 40-fold molar excess of 4-maleimido-TEMPO (MAL-6, Sigma–Aldrich) overnight at 4 °C under gentle agitation. Excess spin labels were removed by extensive dialysis into buffer containing 50 mm MOPS, 100 mm NaCl, pH 7.0. The protein was concentrated to the final desired concentration with Amicon Ultra-0.5 centrifugal concentrators (molecular mass cutoff, 10,000 Da).
EPR spectroscopy
CW EPR spectroscopy was carried out at X-band on a Bruker ELEXSYS E500 fitted with a super high Q cavity. The samples were contained in a glass capillary, and spectra were recorded at room temperature over 100 G at a microwave power of 10 milliwatt with a scan time of 42 s, and typically the signal was averaged 20–25 times. For the titration experiments, the signal was averaged 50–100 times.
To quantify the unbound and bound population of spin-labeled β-arr1 to STAM1, the multicomponent EPR spectra were deconvoluted using spectral subtraction methods. The apo spectrum was manually subtracted from each composite spectrum to obtain each STAM1-bound spectrum. Double integration of each resulting spectrum is proportional to the number of spins affected by STAM1 binding and thus when compared with the double integration value of the composite spectrum, which is proportional to the total number of spins in the sample, yields the percentage of proteins in each sample affected by STAM binding. These percentage values were plotted and fit to a one-site binding model in SigmaPlot to obtain Kd values.
DEER spectroscopy data were collected on a Q-band Bruker ELEXSYS 580 equipped with an EN5107D2 resonator with overcoupling at 80K. The samples contained 20% deuterated glycerol as a cryoprotectant and were flash-frozen in a dry ice and acetone mixture. Acquired raw data after phase correction were background-corrected, plotted, and analyzed using LongDistances software program (78) provided by C. Altenbach (University of California, Los Angeles, CA). Distance distributions were determined by fitting the corrected dipolar evolution data using the algorithms included in the LongDistances program.
Co-immunoprecipitation
HEK293 cells grown in 10-cm plates were co-transfected with T7–STAM1 and β-arr1-WT, β-arr1–4A, or empty vector (pCMV10) using polyethylenimine (PEI). Twenty-four hours post-transfection, the cells were lysed in immunoprecipitation buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% Nonidet P-40, and protease inhibitors) by incubation at 4 °C for 30 min. The Cell lysates were then sonicated and centrifuged by Eppendorf microcentrifuge at 13,000 rpm for 30 min. Cleared cell lysates were incubated with anti-T7 tag antibody overnight at 4 °C. The next morning, equal amounts of cleared cell lysates were incubated with 20 μl of a 50% slurry of protein G–agarose resin for 1 h at 4 °C. The samples were washed three times with 750 μl of ice-cold immunoprecipitation buffer before elution with 20 μl of SDS sample buffer at room temperature. The samples were analyzed by 10% SDS-PAGE and Western blotting using primary and secondary antibodies. The bands were quantified by densitometry using ImageJ (National Institutes of Health, Bethesda, MD). The results from three independent experiments were statistically analyzed by unpaired t test using GraphPad Prism software, p < 0.05 was considered significant.
His-tag pulldown
Equimolar amounts of WT or mutant purified β-arr1 were preincubated with or without purified STAM1–His proteins in 50 μl of binding buffer (20 mm Tris-HCl, 150 mm NaCl, pH 7.5) for 20 min at 37 °C. Then the protein mixture was applied to 20 μl of cobalt resin beads equilibrated with binding buffer in the presence of 50 μm imidazole. After 1 h of gentle rotation, each sample was washed three times with ice-cold binding buffer and eluted by binding buffer supplemented with 200 mm imidazole. Pulled down proteins were analyzed by 10% SDS-PAGE and immunoblotting.
BRET assay
For CXCR4 and β-arr1 BRET, HEK293 cells grown in 10-cm dishes were co-transfected with CXCR4–Rluc and β-arr1–GFP10 or β-arr1–4A-GFP10 using PEI. For STAM1 and β-arr1 BRET, HEK293 cells were co-transfected with HA–CXCR4, STAM1–Rluc, and β-arr1–GFP10. Twenty-four hours post-transfection, the cells were lifted from the plate with trypsin, counted, and diluted to 5 × 105 cells/ml in DMEM containing 5% fetal bovine serum. 100 μl of cell suspension was seeded into each well of the clear-bottomed 96-well plate. After incubating for 24 h, the cells were serum-starved in DMEM (phenol red–free) supplemented with 20 mm HEPES, pH 7.4, for 3 h. The medium was aspirated and replaced with 80 μl of Dulbecco's PBS supplemented with 20 mm HEPES, pH 7.4. The cells were stimulated with increasing concentrations of CXCL12 (10−7, 10−8, 10−9, 10−10, 10−11, 10−12, and 10−13 m) in triplicate. Immediately after the addition of CXCL12, 50 μm luciferase substrate coelenterazine 400A (DeepBlue C) was added and incubated for 15 min at 37 °C. BRET2 measurements were performed with BioTek Cytation 5 cell imaging multimode reader, which measures Rluc donor emission at 410 nm and acceptor GFP10 emission at 515 nm. BRET ratio was calculated by dividing the emission at 515 nm by the emission at 410 nm.
Signaling assay
HeLa cells grown on 6-cm dishes were transiently transfected with empty vector (pCMV-10), β-arrestin1–WT–FLAG, β-arrestin1–4A–FLAG, or FLAG–β-arrestin1-(25–161) using PEI. Twenty-four hours post-transfection, the cells were dislodged from the surface of the plate by trypsin and counted, and equal numbers of cells were seeded onto 6-well plates. The next day, the cells were serum-starved in DMEM supplemented with 20 mm HEPES for 3 h. The medium was aspirated and replaced with the same medium containing vehicle or 30 nm CXCL12 for 15 min at 37 °C. The cells were washed once with ice-cold PBS and lysed in 300 μl of 2× sample buffer (8% SDS, 10% glycerol, 5% β-mercaptoethanol, 37.5 mm Tris, pH 6.5, 0.003% bromphenol blue). The samples were sonicated, the debris were pelleted by centrifugation at 13,000 rpm, and equal amounts of supernatants were analyzed by 10% SDS-PAGE and immunoblotting. The blots were quantified using the ImageJ (National Institutes of Health). The average level of pTyr397–FAK or pERK-1/2 was normalized to total FAK or ERK-1/2, respectively. The results from four independent experiments were analyzed by two-way analysis of variance with Bonferrini's multiple comparison test using GraphPad Prism software (p < 0.05 was considered significant).
Data availability
All data associated with this work are contained within the article.
Acknowledgments
We are grateful to James Buhrmaster and Mudassir Ali for generating the T7–STAM–Rluc and STAM1–HIS plasmids, respectively. We are grateful to Dr. Michel Bouvier for expert advice on receptor-arrestin BRET and for providing the GFP-tagged β-arr1 plasmid and Dr. Nikolaus Heveker for providing the CXCR4–Rluc plasmid. We thank Dr. John McCorvy for help with setting up BRET experiments and Dr. Brian Smith for use of the Biotek Cytation 5 cell imaging multimode reader.
Author contributions—Y. Z., V. V. G., and A. M. conceptualization; Y. Z., V. V. G., C. S. K., and A. M. data curation; Y. Z., V. V. G., C. S. K., and A. M. formal analysis; Y. Z. and A. M. supervision; Y. Z. and A. M. funding acquisition; Y. Z. and V. V. G. investigation; Y. Z. and V. V. G. methodology; Y. Z., V. V. G., and A. M. writing-original draft; Y. Z. and A. M. project administration; Y. Z., V. V. G., C. S. K., and A. M. writing-review and editing; V. V. G., S. A. V., C. S. K., and A. M. resources.
Funding and additional information—This work was supported by National Institutes of Health Grant GM106727 (to A. M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- BRET
- bioluminescence resonance energy transfer
- CXCR4
- CXC motif chemokine receptor 4
- FAK
- focal adhesion kinase
- GPCR
- G protein–coupled receptor
- GRK
- GPCR kinase
- CW
- continuous wave
- DEER
- double electron–electron resonance
- ERK
- extracellular signal-regulated kinase
- Rluc
- Renilla luciferase
- HA
- hemagglutinin
- DMEM
- Dulbecco's modified Eagle's medium
- PEI
- polyethylenimine.
References
- 1. Carman C. V., and Benovic J. L. (1998) G-protein–coupled receptors: turn-ons and turn-offs. Curr. Opin. Neurobiol. 8, 335–344 10.1016/S0959-4388(98)80058-5 [DOI] [PubMed] [Google Scholar]
- 2. Gurevich V. V., and Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 10.1016/j.tips.2003.12.008 [DOI] [PubMed] [Google Scholar]
- 3. Shenoy S. K., and Lefkowitz R. J. (2003) Multifaceted roles of β-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 375, 503–515 10.1042/BJ20031076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- 5. Hirsch J. A., Schubert C., Gurevich V. V., and Sigler P. B. (1999) A Model for arrestin's regulation: the 2.8 Å crystal structure of visual arrestin. Cell 97, 257–269 10.1016/S0092-8674(00)80735-7 [DOI] [PubMed] [Google Scholar]
- 6. Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., and Schubert C. (2001) Crystal structure of β-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure 9, 869–880 10.1016/S0969-2126(01)00644-X [DOI] [PubMed] [Google Scholar]
- 7. Milano S. K., Pace H. C., Kim Y.-M., Brenner C., and Benovic J. L. (2002) Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 41, 3321–3328 10.1021/bi015905j [DOI] [PubMed] [Google Scholar]
- 8. Zhan X., Gimenez L. E., Gurevich V. V., and Spiller B. W. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 10.1016/j.jmb.2010.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., and Gurevich V. V. (2005) Crystal structure of cone arrestin at 2.3Å: evolution of receptor specificity. J. Mol. Biol. 354, 1069–1080 10.1016/j.jmb.2005.10.023 [DOI] [PubMed] [Google Scholar]
- 10. Gurevich V. V., and Benovic J. L. (1993) Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 268, 11628–11638 [PubMed] [Google Scholar]
- 11. Charest P. G., Terrillon S., and Bouvier M. (2005) Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep. 6, 334–340 10.1038/sj.embor.7400373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nuber S., Zabel U., Lorenz K., Nuber A., Milligan G., Tobin A. B., Lohse M. J., and Hoffmann C. (2016) β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664 10.1038/nature17198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim M., Vishnivetskiy S. A., Van Eps N., Alexander N. S., Cleghorn W. M., Zhan X., Hanson S. M., Morizumi T., Ernst O. P., Meiler J., Gurevich V. V., and Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412 10.1073/pnas.1216304109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim Y. J., Hofmann K. P., Ernst O. P., Scheerer P., Choe H. W., and Sommer M. E. (2013) Crystal structure of pre-activated arrestin p44. Nature 497, 142–146 10.1038/nature12133 [DOI] [PubMed] [Google Scholar]
- 15. Zhuo Y., Vishnivetskiy S. A., Zhan X., Gurevich V. V., and Klug C. S. (2014) Identification of receptor binding-induced conformational changes in non-visual arrestins. J. Biol. Chem. 289, 20991–21002 10.1074/jbc.M114.560680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shukla A. K., Manglik A., Kruse A. C., Xiao K., Reis R. I., Tseng W. C., Staus D. P., Hilger D., Uysal S., Huang L. Y., Paduch M., Tripathi-Shukla P., Koide A., Koide S., Weis W. I., et al. (2013) Structure of active β-arrestin-1 bound to a G-protein–coupled receptor phosphopeptide. Nature 497, 137–141 10.1038/nature12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gurevich V. V., and Gurevich E. V. (2012) Synthetic biology with surgical precision: targeted reengineering of signaling proteins. Cell Signal. 24, 1899–1908 10.1016/j.cellsig.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhan X., Perez A., Gimenez L. E., Vishnivetskiy S. A., and Gurevich V. V. (2014) Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell Signal. 26, 766–776 10.1016/j.cellsig.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peled A., Petit I., Kollet O., Magid M., Ponomaryov T., Byk T., Nagler A., Ben-Hur H., Many A., Shultz L., Lider O., Alon R., Zipori D., and Lapidot T. (1999) Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 283, 845–848 10.1126/science.283.5403.845 [DOI] [PubMed] [Google Scholar]
- 20. Tachibana K., Hirota S., Iizasa H., Yoshida H., Kawabata K., Kataoka Y., Kitamura Y., Matsushima K., Yoshida N., Nishikawa S., Kishimoto T., and Nagasawa T. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594 10.1038/31261 [DOI] [PubMed] [Google Scholar]
- 21. Nagasawa T., Hirota S., Tachibana K., Takakura N., Nishikawa S., Kitamura Y., Yoshida N., Kikutani H., and Kishimoto T. (1996) Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638 10.1038/382635a0 [DOI] [PubMed] [Google Scholar]
- 22. Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verástegui E., and Zlotnik A. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 10.1038/35065016 [DOI] [PubMed] [Google Scholar]
- 23. Hurley J. H., and Stenmark H. (2011) Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu. Rev. Biophys. 40, 119–142 10.1146/annurev-biophys-042910-155404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Teicher B. A., and Fricker S. P. (2010) CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 16, 2927–2931 10.1158/1078-0432.CCR-09-2329 [DOI] [PubMed] [Google Scholar]
- 25. Li Y. M., Pan Y., Wei Y., Cheng X., Zhou B. P., Tan M., Zhou X., Xia W., Hortobagyi G. N., Yu D., and Hung M. C. (2004) Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6, 459–469 10.1016/j.ccr.2004.09.027 [DOI] [PubMed] [Google Scholar]
- 26. Orimo A., Gupta P. B., Sgroi D. C., Arenzana-Seisdedos F., Delaunay T., Naeem R., Carey V. J., Richardson A. L., and Weinberg R. A. (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 10.1016/j.cell.2005.02.034 [DOI] [PubMed] [Google Scholar]
- 27. Zhang X. H., Wang Q., Gerald W., Hudis C. A., Norton L., Smid M., Foekens J. A., and Massagué J. (2009) Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 10.1016/j.ccr.2009.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balkwill F. (2004) The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 14, 171–179 10.1016/j.semcancer.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 29. Hung C. S., Su H. Y., Liang H. H., Lai C. W., Chang Y. C., Ho Y. S., Wu C. H., Ho J. D., Wei P. L., and Chang Y. J. (2014) High-level expression of CXCR4 in breast cancer is associated with early distant and bone metastases. Tumour Biol. 35, 1581–1588 10.1007/s13277-013-1218-9 [DOI] [PubMed] [Google Scholar]
- 30. Shi J., Wei Y., Xia J., Wang S., Wu J., Chen F., Huang G., and Chen J. (2014) CXCL12-CXCR4 contributes to the implication of bone marrow in cancer metastasis. Future Oncol. 10, 749–759 10.2217/fon.13.193 [DOI] [PubMed] [Google Scholar]
- 31. Balkwill F. (2004) Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 10.1038/nrc1388 [DOI] [PubMed] [Google Scholar]
- 32. Busillo J. M., and Benovic J. L. (2007) Regulation of CXCR4 signaling. Biochim. Biophys. Acta 1768, 952–963 10.1016/j.bbamem.2006.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alekhina O., and Marchese A. (2016) β-Arrestin1 and signal-transducing adaptor Molecule 1 (STAM1) cooperate to promote focal adhesion kinase autophosphorylation and chemotaxis via the chemokine receptor CXCR4. J. Biol. Chem. 291, 26083–26097 10.1074/jbc.M116.757138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malik R., and Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 10.1091/mbc.e10-02-0169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Coffa S., Breitman M., Hanson S. M., Callaway K., Kook S., Dalby K. N., and Gurevich V. V. (2011) The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One 6, e28723 10.1371/journal.pone.0028723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumari P., Srivastava A., Banerjee R., Ghosh E., Gupta P., Ranjan R., Chen X., Gupta B., Gupta C., Jaiman D., and Shukla A. K. (2016) Functional competence of a partially engaged GPCR–β-arrestin complex. Nat. Commun. 7, 13416 10.1038/ncomms13416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee M. H., Appleton K. M., Strungs E. G., Kwon J. Y., Morinelli T. A., Peterson Y. K., Laporte S. A., and Luttrell L. M. (2016) The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668 10.1038/nature17154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vishnivetskiy S. A., Gimenez L. E., Francis D. J., Hanson S. M., Hubbell W. L., Klug C. S., and Gurevich V. V. (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J. Biol. Chem. 286, 24288–24299 10.1074/jbc.M110.213835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Q., Perry N. A., Vishnivetskiy S. A., Berndt S., Gilbert N. C., Zhuo Y., Singh P. K., Tholen J., Ohi M. D., Gurevich E. V., Brautigam C. A., Klug K. S., Gurevich V. V., and Iverson T. M. (2017) Structural basis of arrestin-3 activation and signaling. Nat. Commun. 8, 1427 10.1038/s41467-017-01218-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Q., Iverson T. M., and Gurevich V. V. (2018) Structural basis of arrestin-dependent signal transduction. Trends Biochem. Sci. 43, 412–423 10.1016/j.tibs.2018.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klug C. S., and Feix J. B. (2008) Methods and applications of site-directed spin labeling EPR spectroscopy. Methods Cell Biol. 84, 617–658 10.1016/S0091-679X(07)84020-9 [DOI] [PubMed] [Google Scholar]
- 42. Jeschke G. (2012) DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 10.1146/annurev-physchem-032511-143716 [DOI] [PubMed] [Google Scholar]
- 43. Hubbell W. L., Gross A., Langen R., and Lietzow M. A. (1998) Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8, 649–656 10.1016/S0959-440X(98)80158-9 [DOI] [PubMed] [Google Scholar]
- 44. Hubbell W. L., Cafiso D. S., and Altenbach C. (2000) Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7, 735–739 10.1038/78956 [DOI] [PubMed] [Google Scholar]
- 45. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., and Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 10.1073/pnas.0600733103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gray-Keller M. P., Detwiler P. B., Benovic J. L., and Gurevich V. V. (1997) Arrestin with a single amino acid substitution quenches light-activated rhodopsin in a phosphorylation-independent fashion. Biochemistry 36, 7058–7063 10.1021/bi963110k [DOI] [PubMed] [Google Scholar]
- 47. Celver J., Vishnivetskiy S. A., Chavkin C., and Gurevich V. V. (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043–9048 10.1074/jbc.M107400200 [DOI] [PubMed] [Google Scholar]
- 48. Hanson S. M., Cleghorn W. M., Francis D. J., Vishnivetskiy S. A., Raman D., Song X., Nair K. S., Slepak V. Z., Klug C. S., and Gurevich V. V. (2007) Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J. Mol. Biol. 368, 375–387 10.1016/j.jmb.2007.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H. E., Zhang C., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou X. E., He Y., de Waal P. W., Gao X., Kang Y., Van Eps N., Yin Y., Pal K., Goswami D., White T. A., Barty A., Latorraca N. R., Chapman H. N., Hubbell W. L., Dror R. O., et al. (2017) Identification of phosphorylation codes for arrestin recruitment by G protein–coupled receptors. Cell 170, 457–469.e13 10.1016/j.cell.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Staus D. P., Hu H., Robertson M. J., Kleinhenz A. L. W., Wingler L. M., Capel W. D., Latorraca N. R., Lefkowitz R. J., and Skiniotis G. (2020) Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 579, 297–302 10.1038/s41586-020-1954-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang W., Masureel M., Qu Q., Janetzko J., Inoue A., Kato H. E., Robertson M. J., Nguyen K. C., Glenn J. S., Skiniotis G., and Kobilka B. K. (2020) Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308 10.1038/s41586-020-1953-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yin W., Li Z., Jin M., Yin Y. L., de Waal P. W., Pal K., Yin Y., Gao X., He Y., Gao J., Wang X., Zhang Y., Zhou H., Melcher K., Jiang Y., et al. (2019) A complex structure of arrestin-2 bound to a G protein–coupled receptor. Cell Res. 29, 971–983 10.1038/s41422-019-0256-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Q., Zhuo Y., Kim M., Hanson S. M., Francis D. J., Vishnivetskiy S. A., Altenbach C., Klug C. S., Hubbell W. L., and Gurevich V. V. (2014) Self-association of arrestin family members. Handb. Exp. Pharmacol. 219, 205–223 10.1007/978-3-642-41199-1_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hamdan F. F., Percherancier Y., Breton B., and Bouvier M. (2006) Monitoring protein–protein interactions in living cells by bioluminescence resonance energy transfer (BRET). Curr. Protoc. Neurosci., chapter 5, unit 5.23 10.1002/0471142301.ns0523s34 [DOI] [PubMed] [Google Scholar]
- 56. Bonneterre J., Montpas N., Boularan C., Gales C., and Heveker N. (2016) Analysis of arrestin recruitment to chemokine receptors by bioluminescence resonance energy transfer. Methods Enzymol. 570, 131–153 10.1016/bs.mie.2015.09.003 [DOI] [PubMed] [Google Scholar]
- 57. Marullo S., and Bouvier M. (2007) Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol. Sci. 28, 362–365 10.1016/j.tips.2007.06.007 [DOI] [PubMed] [Google Scholar]
- 58. Zhan X., Kook S., Gurevich E. V., and Gurevich V. V. (2014) Arrestin-dependent activation of JNK family kinases. Handb. Exp. Pharmacol. 219, 259–280 10.1007/978-3-642-41199-1_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Song X., Coffa S., Fu H., and Gurevich V. V. (2009) How does arrestin assemble MAPKs into a signaling complex?. J. Biol. Chem. 284, 685–695 10.1074/jbc.M806124200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Scheerer P., and Sommer M. E. (2017) Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 45, 160–169 10.1016/j.sbi.2017.05.001 [DOI] [PubMed] [Google Scholar]
- 61. Sente A., Peer R., Srivastava A., Baidya M., Lesk A. M., Balaji S., Shukla A. K., Babu M. M., and Flock T. (2018) Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 25, 538–545 10.1038/s41594-018-0071-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thomsen A. R. B., Plouffe B., Cahill T. J. III, Shukla A. K., Tarrasch J. T., Dosey A. M., Kahsai A. W., Strachan R. T., Pani B., Mahoney J. P., Huang L., Breton B., Heydenreich F. M., Sunahara R. K., Skiniotis G., et al. (2016) GPCR–G protein–β-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 10.1016/j.cell.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nguyen A. H., Thomsen A. R. B., Cahill T. J. 3rd, Huang R., Huang L. Y., Marcink T., Clarke O. B., Heissel S., Masoudi A., Ben-Hail D., Samaan F., Dandey V. P., Tan Y. Z., Hong C., Mahoney J. P., et al. (2019) Structure of an endosomal signaling GPCR–G protein–β-arrestin megacomplex. Nat. Struct. Mol. Biol. 26, 1123–1131 10.1038/s41594-019-0330-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ceccarelli D. F., Song H. K., Poy F., Schaller M. D., and Eck M. J. (2006) Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 281, 252–259 10.1074/jbc.M509188200 [DOI] [PubMed] [Google Scholar]
- 65. Lietha D., Cai X., Ceccarelli D. F., Li Y., Schaller M. D., and Eck M. J. (2007) Structural basis for the autoinhibition of focal adhesion kinase. Cell 129, 1177–1187 10.1016/j.cell.2007.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Luttrell L. M., Wang J., Plouffe B., Smith J. S., Yamani L., Kaur S., Jean-Charles P. Y., Gauthier C., Lee M. H., Pani B., Kim J., Ahn S., Rajagopal S., Reiter E., Bouvier M., et al. (2018) Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 11, eaat7650 10.1126/scisignal.aat7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grundmann M., Merten N., Malfacini D., Inoue A., Preis P., Simon K., Rüttiger N., Ziegler N., Benkel T., Schmitt N. K., Ishida S., Müller I., Reher R., Kawakami K., Inoue A., et al. (2018) Lack of β-arrestin signaling in the absence of active G proteins. Nat. Commun. 9, 341 10.1038/s41467-017-02661-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brunton V. G., MacPherson I. R., and Frame M. C. (2004) Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitry. Biochim. Biophys. Acta 1692, 121–144 10.1016/j.bbamcr.2004.04.010 [DOI] [PubMed] [Google Scholar]
- 69. Schlaepfer D. D., Hauck C. R., and Sieg D. J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71, 435–478 10.1016/S0079-6107(98)00052-2 [DOI] [PubMed] [Google Scholar]
- 70. Schaller M. D. (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta 1540, 1–21 10.1016/S0167-4889(01)00123-9 [DOI] [PubMed] [Google Scholar]
- 71. Parsons J. T. (2003) Focal adhesion kinase: the first ten years. J. Cell Sci. 116, 1409–1416 10.1242/jcs.00373 [DOI] [PubMed] [Google Scholar]
- 72. Burger J. A., and Kipps T. J. (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107, 1761–1767 10.1182/blood-2005-08-3182 [DOI] [PubMed] [Google Scholar]
- 73. Hanson S. M., and Gurevich V. V. (2006) The differential engagement of arrestin surface charges by the various functional forms of the receptor. J. Biol. Chem. 281, 3458–3462 10.1074/jbc.M512148200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gurevich V. V., and Benovic J. L. (2000) Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 315, 422–437 10.1016/s0076-6879(00)15859-8 [DOI] [PubMed] [Google Scholar]
- 75. Malik R., Soh U. J., Trejo J., and Marchese A. (2012) Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein–coupled receptor signaling. J. Biol. Chem. 287, 9013–9027 10.1074/jbc.M111.336792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bornhorst J. A., and Falke J. J. (2000) Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 326, 245–254 10.1016/s0076-6879(00)26058-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., and Gurevich V. V. (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J. Biol. Chem. 282, 32075–32083 10.1074/jbc.M706057200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Toledo Warshaviak D., Khramtsov V. V., Cascio D., Altenbach C., and Hubbell W. L. (2013) Structure and dynamics of an imidazoline nitroxide side chain with strongly hindered internal motion in proteins. J. Magn. Reson. 232, 53–61 10.1016/j.jmr.2013.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data associated with this work are contained within the article.