

Abstract
Autophagy has been shown to maintain neural system homeostasis during stroke. However, the molecular mechanisms underlying neuronal autophagy in ischemic stroke remain poorly understood. This study aims to investigate the regulatory mechanisms of the pathway consisting of MEG3 (maternally expressed gene 3), microRNA-378 (miR-378), and GRB2 (growth factor receptor-bound protein 2) in neuronal autophagy and neurological functional impairment in ischemic stroke. A mouse model of the middle cerebral artery occluded–induced ischemic stroke and an in vitro model of oxygen-glucose deprivation–induced neuronal injury were developed. To understand the role of the MEG3/miR-378/GRB2 axis in the neuronal regulation, the expression of proteins associated with autophagy in neurons was measured by Western blotting analysis, and neuron death was evaluated using a lactate dehydrogenase leakage rate test. First, it was found that the GRB2 gene, up-regulated in middle cerebral artery occluded–operated mice and oxygen-glucose deprivation–exposed neurons, was a target gene of miR-378. Next, miR-378 inhibited neuronal loss and neurological functional impairment in mice, as well as neuronal autophagy and neuronal death by silencing of GRB2. Confirmatory in vitro experiments showed that MEG3 could specifically bind to miR-378 and subsequently up-regulate the expression of GRB2, which in turn suppressed the activation of Akt/mTOR pathway. Taken together, these findings suggested that miR-378 might protect against neuronal autophagy and neurological functional impairment and proposed that a MEG3/miR-378/GRB2 regulatory axis contributed to better understanding of the pathophysiology of ischemic stroke.
Keywords: maternally expressed gene 3, microRNA-378, growth factor receptor–bound protein 2 (GRB2), Akt/mTOR signaling pathway, neuronal autophagy, neurological functional impairment, ischemic stroke, stroke, ischemia, neurological disease, autophagy
Stroke, a principal cause of disability and the second leading cause of death in the world, affects an estimated 17 million people each year, of which most are ischemic stroke (1, 2). Cerebrovascular disease is considered the first leading cause of death in China and the main cause of disability in adolescents in line with the recent report of the global relative burden of stroke (3, 4). Data from China and other countries show an increase in the incidence of ischemic stroke in men compared with women less than 80 years old; however, most stroke patients over 80 years old are women (5, 6). After suffering an ischemic stroke, patients often have a poor prognosis, failing to regain independence and requiring long-term care (7). This is attributed to irreversible tissue damage caused by the stroke incident, which often triggers complex cellular biochemical events, ultimately causing necrosis, apoptosis, and autophagy of neurons in the ischemic brain (8). Understanding the mechanisms underlying stroke-induced neuronal autophagy and neurological functional impairment might be beneficial to the development of new therapeutic targets against ischemic stroke effects.
An increasing amount of evidence indicates that microRNAs (miRNAs) play key roles in neural pathological and physiological changes (9, 10). Many reports of stroke-associated miRNA expression profiles conducted both in vivo and in vitro have surfaced, because dysregulation of miRNAs has been associated with neurological diseases, making this class of RNAs ideal drug candidates for stroke therapies because of their simultaneous regulation of multiple target genes (11, 12). Interestingly, Zhang et al. (13) have reported that overexpression of miR-378 could protect neurons against oxygen-glucose deprivation (OGD)– or middle cerebral artery occluded (MCAO)–induced ischemic injury by targeting caspase-3. GRB2 (growth factor receptor-bound protein 2) is reported to be a target of miR-378, involving the cardiac fibrosis after myocardial infarction while long noncoding RNA (lncRNA) and pro-cardiac fibrotic lncRNA can bind to miR-378 to impair miR-378–dependent inhibition of GRB2 (14). GRB2 is found to be enriched in the mitogen-activated protein kinase, phosphatidylinositol 3-kinase–Akt, mTOR pathways on the basis of Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. Aggravation or alleviation of OGD-induced neuronal ischemic injury may be associated with the Akt/mTOR pathway-mediated autophagy (15). Another lncRNA, maternally expressed gene 3 (MEG3), is down-regulated in both MCAO mice and OGD-cultured HT22 cells and has been found to attenuate hypoxia-induced apoptosis of HT22 cells (16). In this study, we found that miR-378 could target GRB2, which was up-regulated in ischemic stroke samples shown by microarray GSE9391, whereas MEG3 was predicted to bind or occur in a complex with miR-378. Therefore, we investigated the interplay between MEG3, miR-378, and GRB2 in conjunction with the Akt/mTOR pathway in neuronal autophagy and neurological functional impairment caused by ischemic stroke.
Results
miR-378 decreased but GRB2 increased in brain tissues of MCAO-operated mice and OGD-exposed neurons
By analyzing the microarray profile GSE9391, the top 50 differentially expressed genes with the largest fold change are shown in a heat map (Fig. 1A). Among those genes, we found that GRB2 was up-regulated in patients with stroke (Fig. 1A). GRB2 has been reported to have important functions in neurogenesis (17, 18). KEGG analysis suggested that GRB2 was involved in mitogen-activated protein kinase, phosphatidylinositol 3-kinase–AKT, and mTOR pathways, and the relationships between those pathways and neurogenesis have been reported in many studies (19, 20). However, the literature regarding the role of GRB2 in ischemic stroke is sparse. Thus, we selected GRB2 as the focus of subsequent experiments.
Figure 1.

High GRB2 expression and low miR-378 expression are identified in brain tissues of MCAO-operated mice and OGD-exposed neurons. A, GRB2 expression in stroke-related microarray profile GSE9391 (brain tissues from stroke cases and normal control tissues from contralateral hemisphere). B, immunofluorescence images of co-localization of GRB2, MAP-2, and GFAP in neurons and dual positive rate of GFAP+/GRB2+ and MAP2+/GRB2+ (GFAP+/GRB2+ refers to the ratio of GFAP+/GRB2+ cells to GFAP+ cells, and MAP2+/GRB2+ refers to the ratio of MAP2+/GRB2+ cells to MAP2+ cells). C, relative mRNA level of GRB2 in sham-operated and MCAO-operated mice (3 days post-MCAO), as well as control and OGD-exposed neurons determined by RT-qPCR. D, relative protein level of GRB2 in sham-operated and MCAO-operated mice (3 days post-MCAO), as well as control and OGD-exposed neurons measured by Western blotting analysis. E, the predicted miR-378 binding sites in the 3´-UTR of GRB2. F, relative luciferase activity of GRB2-WT and GRB2-MUT in the HEK-293T cells transfected with miR-378 mimic determined by Dual-Luciferase reporter assay. G, miR-378 expression in sham-operated and MCAO-operated mice (3 days post-MCAO), as well as control and OGD-exposed neurons determined by RT-qPCR. H, miR-378 expression in sham-operated and MCAO-operated mice (3 days post-MCAO) detected by ISH; measurement data are expressed as means ± standard deviation, and comparison between two groups was conducted by unpaired t test, whereas that among multiple groups was done by one-way analysis of variance with Tukey's post hoc test. *, p < 0.05 versus the normal group (sham-operated mice); #, p < 0.05 versus the control group (untreated neurons). DAPI, 4[prime],6[prime]-diamino-2-phenylindole. MCAO, middle cerebral artery occluded; OGD, oxygen-glucose deprivation.
The neurons were isolated from the cerebral cortex of neonatal suckling mice. The neurons adhered to the well after 1 day of culture. At days 4 and 5, the neurites were gradually elongated and further extended outwards. At the same time, the neurites gradually intertwined and formed a neural network. Neurons were considered mature after 6 days of culture. Immunofluorescence was subsequently conducted to identify mouse neurons with the neuron marker MAP-2 and astrocyte marker glial fibrillary acidic protein (GFAP). The percentage of MAP-2–positive cells was above 95%, which can be used for subsequent cell experiments. GRB2 was expressed in MAP-2–positive neurons but hardly expressed in GFAP-positive neurons (Fig. 1B). Reverse transcription–quantitative PCR (RT-qPCR) and Western blotting assays were conducted to determine the expression pattern of GRB2 in the brain tissues and neurons in the settings of in vivo and in vitro models. In our experiment, mRNA and protein levels of GRB2 were increased significantly in brain tissues of MCAO-operated mice 3 days postoperation (p < 0.05) when compared with those in the normal mice. Compared with control neurons, mRNA and protein levels of GRB2 were consistently increased in OGD-exposed neurons (p < 0.05) (Fig. 1, C and D).
A TargetScan.org (http://www.targetscan.org/vert_72/) target prediction site shows a potential binding site for miR-378 within the 3´-UTR of the GRB2 gene (Fig. 1E). Hence, we selected miR-378 for further study. The results of Dual-Luciferase reporter assay were conducted to verify this binding relationship in HEK-293T cells. The results showed no significant differences in the luciferase activity of GRB2 MUT–3´-UTR between the NC and miR-378 mimic groups (p > 0.05). The luciferase activity of the GRB2 WT–3´-UTR in the HEK-293T cells transfected with miR-378 mimic was significantly lower than that in the HEK-293T cells transfected with NC (p < 0.05) (Fig. 1F). Therefore, miR-378 could specifically bind to GRB2. Next, RT-qPCR and Western blotting assays were conducted to determine the expression pattern of miR-378 in the brain tissues and neurons in the settings of in vivo and in vitro models. miR-378 was found to be expressed at lower levels in brain tissues of MCAO-operated mice 3 days postoperation, as well as in OGD-exposed neurons (p < 0.05) (Fig. 1G). Through ISH assay, miR-378 was found to be consistently down-regulated in the brain tissues of MCAO-operated mice (Fig. 1H). These data collectively suggested the down-regulation of miR-378 and up-regulation of GRB2 in the settings of both in vivo and in vitro models for ischemic stroke.
GRB2 is a target of miR-378
To verify the regulation of miR-378 and GRB2 in ischemic stroke, miR-378+/− and GRB2+/− mice were generated and were used for miR-378 and GRB2 knockdown experiments. Next, the expression of miR-378 in the transgenic mice was determined by RT-qPCR. Compared with WT mice, miR-378 expression in the brain tissues of miR-378+/− mice was down-regulated, and GRB2 mRNA levels in the brain tissues of GRB2+/− mice were also reduced (p < 0.05) (Fig. 2, A and B). The knockdown efficiency of miR-378 was 85.4%, and that of GRB2 was 91.2%. Hence miR-378+/− and GRB2+/− mice can be used in subsequent studies. Next, the GRB2 protein level in the brain tissues of GRB2+/− mice was determined by Western blotting assay and detected using immunohistochemical staining. Compared with WT mice, GRB2 protein level was reduced in the GRB2+/− mice (Fig. 2, C and D). Meanwhile, the miR-378 expression in brain tissues of miR-378+/− mice was detected by ISH assay, the results of which displayed that miR-378 expression was significantly reduced in miR-378+/− mice as compared with WT mice (Fig. 2E). The aforementioned results suggested the successful knockdown of miR-378 and GRB2 in the transgenic mice, which could be used for the following experiments.
Figure 2.
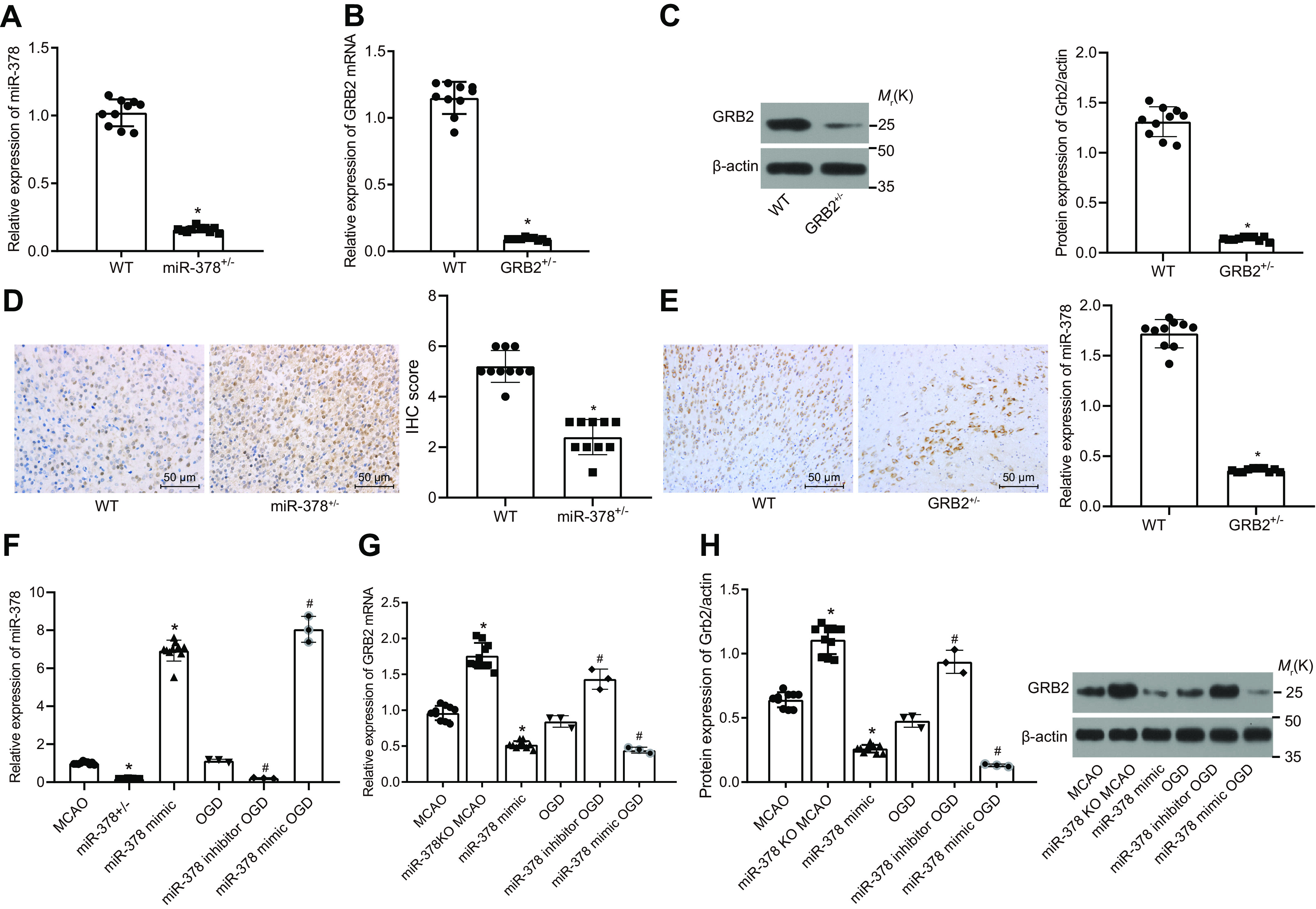
miR-378 negatively regulates GRB2 gene expression. A, efficiency of miR-378 knockdown in mice determined by RT-qPCR. B, efficiency of GRB2 knockdown in mice determined by RT-qPCR. C, relative protein level of GRB2 in the GRB2+/− mice measured by Western blotting analysis. D, protein level of GRB2 in the GRB2+/− mice detected using immunohistochemical staining. E, miR-378 expression in miR-378+/− mice detected using ISH. F, miR-378 expression in the brain tissues of MCAO-operated mice (3 days post-MCAO) and OGD-exposed neurons following miR-378 knockdown or overexpression determined by RT-qPCR. G and H, relative mRNA (G) and protein (H) levels of GRB2 in the brain tissues of MCAO-operated mice (3 days post-MCAO) and OGD-exposed neurons following miR-378 knockdown or overexpression determined by RT-qPCR and Western blotting analysis, respectively; In F–H, miR-378 knockdown was achieved using miR-378+/− mice and miR-378 inhibitor in neurons; miR-378 overexpression was achieved using infection with lentivirus expressing miR-378 mimic in mice and miR-378 mimic treatment in neurons. Measurement data are expressed as means ± standard deviation, and comparison among multiple groups was analyzed by one-way analysis of variance with Tukey's post hoc test. *, p < 0.05 versus the MCAO group (MCAO-operated mice), NC group (neurons transfected with scrambled sequence and exposed to OGD), or WT group (WT mice); #, p < 0.05 versus the OGD group (OGD-exposed neurons). IHC, immunohistochemical.
A mouse model was developed by conducting MCAO in the miR-378+/− and GRB2+/− mice, whereas an in vitro model was induced in the neurons by exposure to OGD. The expression of miR-378 as well as the mRNA and protein levels of GRB2 was determined in the brain tissues MCAO-operated mice and OGD-exposed neurons by RT-qPCR and Western blotting assay. The miR-378 expression was reduced, whereas mRNA and protein levels of GRB2 in brain tissues were up-regulated in the miR-378+/− MCAO group compared with the MCAO group (p < 0.05). The miR-378 expression was increased, but mRNA and protein levels of GRB2 were down-regulated in the miR-378 mimic MCAO group compared with those in the MCAO group (p < 0.05) (Fig. 2, F–H). When compared with the OGD model group, miR-378 expression was down-regulated, whereas mRNA and protein levels of GRB2 in neurons were up-regulated in the miR-378 inhibitor OGD group (p < 0.05), and miR-378 was increased, but mRNA and protein levels of GRB2 were down-regulated in the miR-378 mimic OGD group (p < 0.05). These results confirmed that miR-378 negatively regulated GRB2 expression.
miR-378/GRB2/Akt/MTOR pathway participates in neuronal autophagy
To confirm the effects of GRB2 and miR-378 on neurological impairment, cerebral infarct size, and neuronal autophagy in ischemic stroke mice, we used miR-378+/− and GRB2+/− mice and infected WT mice with lentiviruses harboring a miR-378 mimic and then established the 1-h MCAO model. Following MCAO induction, neurological deficit scores for MCAO-operated mice were evaluated using the Longa grade point standard at 6, 12, and 24 h after MCAO. Compared with the sham-operated mice, the neurological deficit scores of MCAO-operated mice were increased 24 h post-MCAO, indicating that the MCAO model was successfully established, and neurological function of mice was impaired. The neurological deficit score (24 h post-MCAO) of mice in the miR-378+/− MCAO group was increased significantly, and scores (24 h post-MCAO) of mice in the miR-378 mimic MCAO and GRB2+/− MCAO groups were reduced compared with that of mice in the MCAO group (p < 0.05) (Fig. 3A). The results of a pole test showed that Tturn and Ttotal of MCAO mice were increased on the 1st and 3rd days in contrast to those of sham mice. Tturn and Ttotal of the miR-378+/− MCAO group were elevated, whereas Tturn and Ttotal of the miR-378 mimic MCAO and GRB2+/− MCAO groups were decreased than those in the MCAO group (p < 0.05). The results of foot fault test indicated that compared with the sham group, the numbers of right foot faults in the MCAO group on the 1st and 3rd days were increased (p < 0.05). The number of right foot faults in the miR-378+/− MCAO group was increased, but those in the miR-378 mimic MCAO and GRB2+/− MCAO group were reduced when compared with the MCAO group (p < 0.05) (Fig. 3C). These data indicated that miR-378 knockdown aggravated neurological impairment in the ischemic stroke model induced by MCAO, whereas miR-378 overexpression and GRB2 knockdown retarded neurological impairment.
Figure 3.

GRB2 silencing or miR-378 overexpression represses neurological impairment, cerebral infarct size, and neuron loss in ischemic stroke mice. miR-378 knockdown was achieved using miR-378+/− mice, and miR-378 overexpression was achieved using infection with lentivirus expressing miR-378 mimic in mice. GRB2 knockdown was achieved using GRB2+/− mice. A, neurological deficit scores of MCAO-operated mice evaluated using the Longa grade point standard 6, 12, and 24 h after the surgery. B, LCBF change during MCAO process. C, the results of the pole test and foot fault test of MCAO-operated mice. D, the effects of GRB2 and miR-378 on cerebral infarct size in MCAO-operated mice (3 days post-MCAO). E, the effects of GRB2 and miR-378 on number of neurons in anterior cerebral cortex tissues in MCAO-operated mice (3 days post-MCAO); measurement data are expressed as means ± standard deviation, and comparison among multiple groups was analyzed by one-way analysis of variance with Tukey's post hoc test. *, p < 0.05 versus the sham group (sham-operated mice); #, p < 0.05 versus the MCAO group (MCAO-operated mice). CBF, cerebral blood flow.
We measured LCBF during MCAO process. LCBF was 100% under the normal condition and gradually reduced to 20% during MCAO process. LCBF was restored to 100% 1 h after the removal of the monofilament. Under the same treatment, LCBF in the miR-378+/− MCAO, GRB2+/−MCAO, and miR-378 mimic MCAO groups showed no differences compared with that in the MCAO group (p > 0.05) (Fig. 3B). After operation of MCAO for 1 h, cerebral infarct size was recorded, and mice in the MCAO group showed a larger LCBF size, but the miR-378 mimic MCAO and GRB2+/− MCAO groups revealed smaller size than the MCAO group (p < 0.05) (Fig. 3D). These results suggested that miR-378 knockdown increased the cerebral infarct size in ischemic stroke, whereas miR-378 overexpression and GRB2 knockdown had the opposite effect, reducing cerebral infarct size, which was not caused by LCBF.
Compared with the sham group, neuron loss in the brain tissues in the MCAO group was increased on the 3rd day (p < 0.05). miR-378+/− MCAO mice showed increased neuron loss in the brain tissues, whereas the miR-378 mimic MCAO and GRB2+/− MCAO mice showed reduced neuron loss compared with WT MCAO mice on the 3rd day postsurgery (p < 0.05) (Fig. 3E). Overall, these results showed that miR-378 knockdown increased neuron loss in an ischemic stroke scenario, whereas miR-378 overexpression and consequently GRB2 knockdown reduced neuron loss.
To evaluate the effects of GRB2 and miR-378 on neuron autophagy and apoptosis, miR-378 was overexpressed or inhibited, and/or GRB2 was silenced in the primary neurons, which were used for developing OGD models. MDC observation (Fig. 4A) under a fluorescence microscope revealed that the OGD group showed increased intensity of punctate green fluorescence compared with the control group. The miR-378 inhibitor OGD group revealed increased intensity of punctate green fluorescence, whereas the miR-378 mimic OGD, GRB2 siRNA OGD, and miR-378 inhibitor + GRB2 siRNA OGD groups indicated decreased intensity of punctate green fluorescence compared with the OGD model group. The green fluorescence intensity in the miR-378 inhibitor + GRB2 siRNA OGD group was lower than that in the miR-378 inhibitor OGD group and higher than that in the GRB2 siRNA OGD group. LDH leakage rate detection indicated that the OGD model group showed increased neuron death rate (p < 0.05) compared with the control group (Fig. 4, B and C). We found that the miR-378 inhibitor OGD group showed increased neuron death rate at the 24th h (p < 0.05), whereas the miR-378 mimic OGD, GRB2 siRNA OGD, and miR-378 inhibitor + GRB2 siRNA OGD groups indicated increased neuron survival rate and reduced death rate compared with the OGD model group (p < 0.05). The death rate of neurons in the miR-378 inhibitor + GRB2 siRNA OGD group were significantly different from those in the miR-378 inhibitor OGD and GRB2 siRNA OGD groups (p < 0. 05). These results demonstrated that miR-378 knockdown increased formation of autophagic vacuoles and neuron apoptosis in ischemic stroke, whereas miR-378 overexpression and GRB2 knockdown reduced formation of autophagic vacuoles and neuron apoptosis.
Figure 4.

miR-378 reduces neuron autophagy by down-regulating GRB2. A, neuron autophagy detected after MDC staining (400×). B, neuronal death rate measured by LDH leakage rate. C, the protein levels of LC3-II/(LC3-I + LC3-II) and Beclin I normalized to β-actin in MCAO-operated mice (3 days post-MCAO) by Western blotting analysis. D, the protein levels of LC3-II/(LC3-I + LC3-II) and Beclin I normalized to β-actin in OGD-exposed neurons by Western blotting analysis; measurement data are expressed as means ± standard deviation, and comparison among multiple groups was analyzed by one-way analysis of variance with Tukey's post hoc test. *, p < 0.05 versus the control group (untreated neurons); #, p < 0.05 versus the OGD group (OGD-exposed neurons); &, p < 0.05 versus the miR-378 inhibitor OGD group (neurons treated with miR-378 inhibitor and exposed to OGD); ∼, p < 0.05 versus the GRB2 siRNA OGD group (neurons treated with GRB2 siRNA and exposed to OGD).
The results of Western blotting analysis obtained from in vivo experiments indicated that compared with the sham group, the MCAO group showed an increase in the protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) (p < 0.05). In contrast to the MCAO model mice, miR-378+/− MCAO mice showed an increase in protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) in brain tissue on the 3rd day (p < 0.05), but the miR-378 mimic MCAO and GRB2+/− MCAO mice indicated a decrease in protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) in brain tissues (p < 0.05) (p < 0.05) (Fig. 4, B and C). Compared with the control group, the OGD model group showed an increase in protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) (p < 0.05). Based on the data of in vitro experiments, when compared with the OGD model group at the 24th h, the miR-378 inhibitor OGD group revealed increased protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) (p < 0.05), whereas the miR-378 mimic OGD, GRB2 siRNA OGD, and miR-378 inhibitor + GRB2 siRNA OGD groups showed reduced protein levels of Beclin I and LC3-II/(LC3-I + LC3-II) (p < 0.05). The levels of autophagy-associated protein in the miR-378 inhibitor + GRB2 siRNA OGD group was significantly different from that in the miR-378 inhibitor and GRB2 siRNA OGD groups (p < 0. 05) (Fig. 4D). These findings showed that miR-378 knockdown increased neuronal autophagy in ischemic stroke, whereas miR-378 overexpression and GRB2 knockdown reduced neuronal autophagy.
The protein levels of GRB2, Beclin, LC3-I, and LC3-II and the extents of Akt, mTOR, and S6 phosphorylation in the brain tissues from mice were determined by Western blotting analysis. As shown in Fig. 5A, the MCAO group exhibited increases in the protein level of Beclin and LC3-II/(LC3-I + LC3-II) ratio compared with the sham group (p < 0.05). Compared with the MCAO group, the protein level of Beclin and LC3-II/(LC3-I + LC3-II) ratio were much higher in the miR-378+/− MCAO group. However, the protein level of Beclin and LC3-II/(LC3-I + LC3-II) ratio were decreased in the miR-378 mimic MCAO and GRB2+/−MCAO groups relative to the MCAO group (p < 0.05), which was consistent with the results of obtained from experiments on primary neurons. The MCAO group showed an increase in the protein level of GRB2 and a decrease in the extents of Akt, mTOR and S6 phosphorylation (p < 0.05). Compared with the MCAO model mice, the protein level of GRB2 was elevated, but the extents of Akt, mTOR, and S6 phosphorylation were reduced in the miR-378+/− MCAO mice (p < 0.05). The miR-378 mimic MCAO and GRB2+/− MCAO groups showed a reduction in the protein level of GRB2 and increases in the extents of Akt, mTOR, and S6 phosphorylation in contrast to the miR-378+/− MCAO group (p < 0.05). In addition, the extents of Akt and S6 phosphorylation in the brain tissues from mice were detected using immunohistochemical staining. The results revealed changes in the extents of Akt and S6 phosphorylation consistent with those determined by Western blotting analysis (Fig. 5B).
Figure 5.
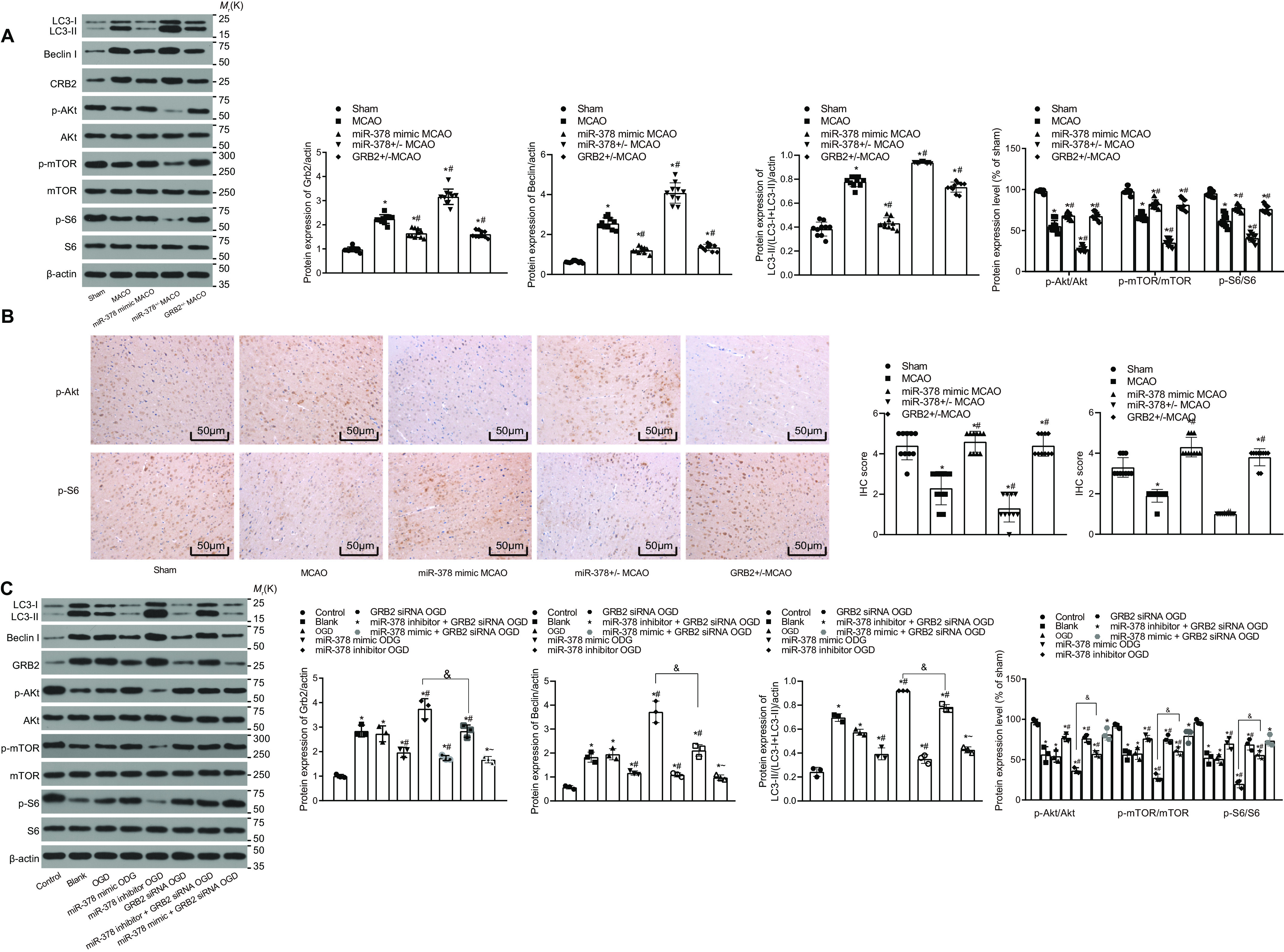
miR-378 activates the Akt/mTOR pathway by down-regulating GRB2. A, protein levels of GRB2, Akt, mTOR, and S6 normalized to β-actin and the extent of Akt, mTOR, and S6 phosphorylation in the brain tissues of MCAO-operated mice (3 days post-MCAO). B, the levels of phosphorylated Akt and S6 in the brain tissues of MCAO-operated mice detected using immunohistochemical staining (200×). C, protein levels of GRB2, Akt, mTOR, and S6 normalized to β-actin and the extent of Akt, mTOR, and S6 phosphorylation in OGD-exposed neurons; measurement data are expressed as means ± standard deviation, and comparison among multiple groups was analyzed by one-way analysis of variance with Tukey's post hoc test. *, p < 0.05 versus the sham group (sham-operated mice) or control group (untreated neurons); #, p < 0.05 versus the MCAO group (MCAO-operated mice) or OGD group (OGD-exposed neurons); &, p < 0.05 versus the miR-378 inhibitor OGD group (neurons treated with miR-378 inhibitor and exposed to OGD); ∼, p < 0.05 versus the GRB2 siRNA OGD group (neurons treated with GRB2 siRNA and exposed to OGD).
Next, the protein levels of GRB2, Beclin, LC3-I, and LC3-II, and the extent of Akt, mTOR, and S6 phosphorylation were determined in the in vitro setting. Compared with the control group, the protein level of Beclin and the LC3-II/(LC3-I + LC3-II) ratio were increased in the OGD group (p < 0.05). Compared with the OGD group, the protein levels of Beclin and LC3-II/(LC3-I + LC3-II) ratio were much higher in the miR-378 inhibitor OGD group, whereas those levels were decreased in the miR-378 mimic OGD, GRB2 siRNA OGD, and miR-378 inhibitor + GRB2 siRNA OGD groups (p < 0.05). In addition, the expression of those autophagy-related proteins demonstrated significant difference in the miR-378 inhibitor + GRB2 siRNA OGD group compared with the miR-378 inhibitor OGD and GRB2 siRNA OGD groups (p < 0.05). Moreover, the Beclin protein level and LC3-II/(LC3-I + LC3-II) ratio were lowered in the miR-378 mimic + GRB2 siRNA OGD group relative to the miR-378 inhibitor + GRB2 siRNA OGD group. Compared with the control group, the OGD model group showed elevated protein levels of GRB2 and reduced extents of Akt, mTOR, and S6 phosphorylation (p < 0.05). The miR-378 inhibitor OGD group revealed elevated protein levels of GRB2 and reduced the extent of Akt, mTOR, and S6 phosphorylation, whereas the miR-378 mimic OGD, GRB2 siRNA OGD, miR-378 inhibitor + GRB2 siRNA OGD, and miR-378 mimic + GRB2 siRNA OGD groups showed reduced protein levels of GRB2 and increased extent of Akt, mTOR, and S6 phosphorylation versus the OGD model group (p < 0.05). No significant difference was witnessed in those levels between the GRB2 siRNA OGD and miR-378 mimic + GRB2 siRNA OGD groups (Fig. 5C). Combining the results of the MCAO and OGD models, we found that silencing of miR-378 promoted the autophagy level of in brain tissues, whereas either miR-378 overexpression or GRB2 silencing slowed down the autophagy level. Furthermore, miR-378 knockdown decreased, whereas miR-378 overexpression and GRB2 knockdown enhanced Akt, mTOR, and S6 phosphorylation, suggesting that miR-378 knockdown might disrupt the activation of AKT/mTOR pathway by targeting GRB2.
MEG3 elevates GRB2 expression by binding to miR-378
It has been reported that MEG3 is expressed in both the cytoplasm and the nucleus (21, 22). As shown in Fig. 6A, MEG3 was up-regulated in the MCAO-operated mice compared with the sham-operated mice (p < 0.05). Consistently, MEG3 was up-regulated in OGD-exposed neurons compared with the control neurons (p < 0.05).
Figure 6.
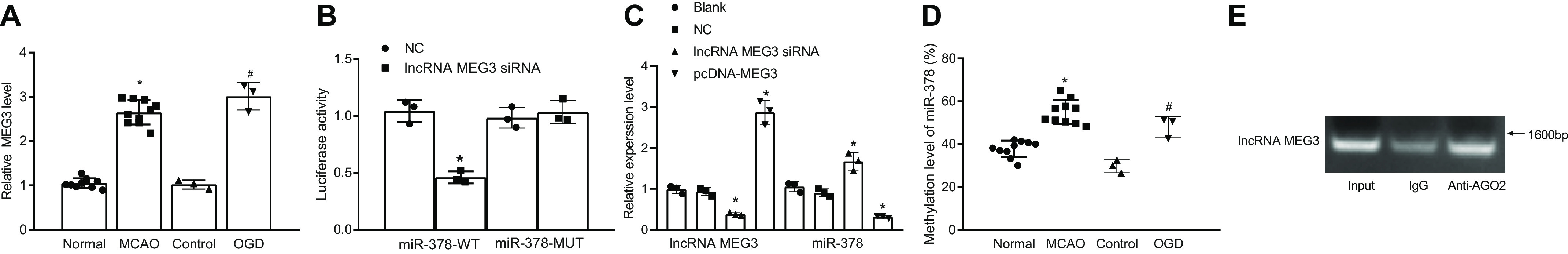
Down-regulation of miR-378 inhibits the activation of the Akt/mTOR pathway through up-regulation of GRB2. A, MEG3 expression in brain tissues of sham-operated and MCAO-operated mice, control neurons, and OGD-exposed neurons. B, the predicted sequence containing miR-378 binding site on MEG3 and the luciferase activity of miR-378–WT and miR-378–MUT in the HEK-293T cells in the presence of MEG3. C, miR-378 expression in OGD-exposed neurons following MEG3 silencing or overexpression. D, miR-378 methylation level in brain tissues of MCAO-exposed mice and OGD-exposed neurons detected by MS-PCR. E, the binding relation between MEG3 and miR-378 in OGD-exposed neurons analyzed by RIP assay. *, p < 0.05 versus the normal group (sham-operated mice) or NC group (neurons transfected with scramble sequence and exposed to OGD); #, p < 0.05 versus the control group (untreated neurons).
Based on the lncRNA–miRNA interaction prediction by the RNA22 database (RRID:SCR_016507), MEG3 had a binding interaction with miR-378. The predicted binding site of MEG3 and miR-378 is shown in Fig. 6B. The results of Dual-Luciferase reporter assay showed no significant differences in the luciferase activity of miR-378–Mut–plasmid between the NC and lncRNA MEG3-cDNA groups (p > 0.05). The luciferase activity of the miR-378–WT–plasmid was significantly lower in the lncRNA MEG3-cDNA group than in the NC group (p < 0.05). Therefore, MEG3 had a binding relation with miR-378.
RT-qPCR was conducted to determine the expression of miR-378 in the OGD model group after neurons were transfected with MEG3 siRNA and pcDNA-MEG3. Compared with the blank and NC groups, the lncRNA MEG3 siRNA group showed a decline in the MEG3 expression and an increase in the miR-378 expression, but pcDNA-MEG3 indicated an increase in the MEG3 expression and a decrease in the miR-378 expression (p < 0.05) (Fig. 6C).
The results of MS-PCR indicated that the MCAO-operated mice showed elevated levels of miR-378 methylation compared with the sham-operated mice, whereas the OGD model group revealed an elevated level of miR-378 methylation compared with the control group (p < 0.05) (Fig. 6D). These data suggested that the down-regulation of miR-378 was associated with the level of miR-378 methylation.
RNA-binding protein immunoprecipitation (RIP) was employed to detect whether AGO2 interacted with MEG3 in OGD models. The results of the RIP (Fig. 6E) showed that an antibody against AGO2 could precipitate MEG3, indicating that MEG3 could form complexes with AGO2. AGO proteins play many roles in the miRNA pathway, which participate in miRNA assembling via producing ac-pre-miRNA. At the same time, AGO proteins participate as effector proteins in the RNA-induced silencing complex, which coordinates mRNA degradation, destabilization, or transcriptional repression (23). MEG3 could form complexes with AGO2 to competitively binding to miR-378 and reduce the unbound miR-378, thus upregulating GRB2 expression and inhibiting the activation of the Akt/mTOR pathway, ultimately promoting autophagy in ischemic stroke mice. Taken together, down-regulation of miR-378 in the MCAO-operated mice might be attributed to miR-378 methylation and the ceRNA mechanism in a complex with MEG3.
Discussion
lncRNAs may form regulatory complex with proteins or modulate miRNA function through acting as endogenous sponges to mediate gene expression, and miRNAs have been demonstrated to bind and regulate lncRNA stability (24). This study evaluated the interacting epigenetic change of lncRNA MEG3 and miR-378 in ischemic stroke of mice subjected to MCAO and neuron model of OGD and the interaction between miR-378 and GRB2.
In vivo MCAO animal models as well as in vitro OGD neuronal and endothelial cell models have been widely used to mimic ischemic injury, and the autophagy pathway has been shown to be active in these models (25, 26). Our results reported expression change of miR-378 during MCAO-induced cerebral ischemia and in OGD-cultured neuronal cell line in periods of post hypoxia. miR-378 was down-regulated in both brain tissues of MCAO-exposed mice and OGD-exposed neurons. Multiple studies have shown that miR-378 plays multifunctional roles in brain diseases (27, 28). Thus, miR-378 overexpression could protect neurons against OGD- or MCAO-induced ischemic injury via attenuating apoptosis, cerebral infarction, and neuronal loss by negatively regulating the apoptosis executioner caspase-3 (13). A pathway analysis showed that miR-378 dysregulation triggering apoptosis was implicated in myocardial signaling networks (29).
As one of the two main degradation pathways in eukaryotes, autophagy is significant for cellular homeostasis and is involved in a broad range of human diseases (30). Interestingly, Akt/mTOR pathway-mediated autophagy has been shown to aggravate OGD-induced neuronal ischemic injury (15). The autophagy marker, microtubule-associated LC3-I, is expressed in the cytoplasm, whereas another protein, Beclin I, has a dominant function in the mediation of autophagy because of its interaction with several partners (31, 32). The ratio of LC3-II/LC3-I is associated with the level of autophagosome formation, and an absence of increased LC3-II level may lead to the activation of autophagy (33). Autophagy has been shown to be activated following cerebral ischemia/reperfusion and plays a double-edged sword for neuronal survival after cerebral ischemia (34). Elevated p-Akt (S473) levels appear to have a neuroprotective role in OGD-induced ischemic injury in hippocampal cells (35). Akt signaling negatively regulates macrophage proliferation in vivo through induction of miR-378 (36). The results of our Dual-Luciferase reporter assay showed that miR-378 could target GRB2 and negatively regulated its expression. GRB2 is a small ubiquitously expressed protein implicated in autophagy (37) and functions as an adaptor protein involved in signal transduction and communication (38). Our study demonstrated that miR-378 overexpression could reduce OGD- or MCAO-induced autophagy in neurons and brain tissues, hence contributing to neuroprotection. More data obtained from rescue experiments suggested that miR-378 overexpression reduced neuronal autophagy and death by down-regulating GRB2.
Intervention in the MEG3-miR-378 interaction presents a new target for the therapeutic treatment of ischemic insults. In addition, AGO2 protein, binding with a miRNA, is a key component of RNA-induced silencing complex, functioning in mRNA degradation, destabilization, or transcriptional repression (39). By detecting the miR-378 promoter methylation and AGO2 interactions with MEG3, we found that MEG3 could form a complex with AGO2 to completely bind to miRNA and reduce unbound miR-378, ultimately impairing miR-378–induced silencing of GRB2. miR-378a promoter methylation may be responsible for miR-378a down-regulation in hepatic stellate cells in liver fibrosis (40). Taken together, down-regulation of miR-378 in ischemic stroke might be attributed to miR-378 methylation and the ceRNA mechanism. However, the mechanism of methylation-induced silencing of miR-378 would be another interesting topic in future studies. Previously, MEG3 has been confirmed as a neuronal death promoter in ischemia (41). In addition, MEG3 silencing is demonstrated to ameliorate neurological impairment, reduce infarct area, and reduce neuronal apoptosis, suggesting protection against cerebral ischemia–reperfusion injury (42). Also, MEG3 knockdown attenuated OGD-induced apoptosis (16). Therefore, we speculate that MEG3 reduced miR-378 to up-regulate GRB2 expression and inactive the Akt/mTOR pathway, ultimately promoting autophagy in ischemic stroke mice.
In conclusion, miR-378 down-regulates GRB2 to activate the Akt/mTOR pathway, and consequently contributes to the attenuation of neuronal autophagy and neurological functional impairment. In addition, the autophagy should be moderate to meet neuroprotection, considering that either stimulating or inhibiting this process could lead to neuronal death. Further studies are necessary to optimize the model establishment, transfection efficiency, and cell growth conditions, which may in turn open a new window into clinical therapeutic strategies for ischemic stroke.
Experimental procedures
Gene Expression Omnibus microarray data analysis
The ischemic stroke–related gene expression profile (GSE9391) and a microarray probe set annotation were downloaded from the Gene Expression Omnibus database (RRID:SCR_005012) and detected by the Atlas Human 1.2 Array (catalog no. 7850-1). This microarray consists of three normal brain tissue samples and three brain tissue samples of ischemic stroke. The differentially expressed genes were screened using the limma package in the R language, and a heat map of those genes was constructed using the pheatmap package. The pathways related to GRB2 were analyzed by the KEGG database (RRID:SCR_012773).
Establishment of MCAO models
A total of 40 C57BL/6J mice, 10 miR-378+/− C57BL/6J mice, and 10 GRB2+/− C57BL/6J mice were provided by SPF Biotechnology Co., Ltd. (Beijing, China). The mice were housed in a specific pathogen-free environment with controlled temperature (21 ± 1 °C) and relative humidity (40–60%) under a 12-h light/dark cycle. Next, we prepared the MCAO model as previously described (41, 43). Healthy adult male C57BL/6J mice at the age of 6 months were injected with 1% pentobarbital sodium (0.06 g/kg) intraperitoneally. A 1-cm longitudinal median incision was made from the mandible to the sternal stalk. Then double ligation of the proximal end of the common carotid artery was performed. The internal carotid artery was blocked by a miniature artery clamp. The thread (tip diameter, 0.23 mm; trunk diameter, 0.18 mm) tip was inserted into the common carotid artery. The orifice of the external carotid artery was ligated by using 5-0 silk sutures. After the artery clamp was removed, the thread was up to the middle cerebral artery (12.00-mm depth) through the internal carotid artery. Signals in the blood flow monitor were observed with an index of declining to ∼20%. After 1-h occlusion, the thread was taken out, followed by double ligation of the external carotid artery and internal carotid artery bifurcation suture entrance. The common carotid artery was untied. When the signal in the blood flow monitor returned to 100%, the skin was sutured along the incision. The mice in the sham group did not receive thread inserted into the artery. With the balance of the body temperature of mice being achieved during the surgery, the mice had ad libitum access to water and food after surgery. All procedures were in compliance with the Guide for Care and Use of Laboratory Animals, and the study protocol was approved by the Animal Ethics Committee of the Affiliated Hospital of Youjiang Medical University for Nationalities. The mice were divided into normal, sham, MCAO (WT MCAO mice), miR-378 mimic MCAO (MCAO mice infected with lentivirus expressing miR-378 mimic), miR-378+/− MCAO (miR-378 knockdown MCAO mice), and GRB2+/− MCAO (GRB2 knockdown MCAO mice) groups. The miR-378 mimic MCAO mice received tail vein injection shown as follows: the mice were fixed on a sterilized test table via fixator, followed by the tail being scrubbed repeatedly by using alcohol cotton balls until bilateral venous dilatation, and injected with miR-378 mimic and lentivirus (5 × 108 plaque-forming unit/100 μl) via tail vein. 3 days later, the mice were used for the establishment of MCAO models. The sequence of miR-378 mimic is 5´-CUCCUGACUCCAGUCCUGUGU-3´.
Evaluation of nerve function
Neurological deficit scores of MCAO model mice were evaluated separately by two investigators blinded to the group condition, using the Longa grade point standard at the 6th, 12th, and 24th h after the surgery. The scores were then averaged and recorded. The mice without any neurological deficit received a score of 0 points; mice showing left anterior extension disorder (mild neurological deficit) received a score of 1 point; mice turning to the left during crawling (moderate neurological deficit) received a score of 2 points; mice turning to hemiplegia side during walking (severe neurological deficit) received a score of 3 points; and mice unable to walk on their own (unconsciousness) received a score of 4 points (44).
Behavioral testing
The pole test was performed at the 1st and 3rd days after the surgery (45). The time mice taken to turn completely head downwards (Tturn) and the total time taken to descend down and reach the floor with their front paws (Ttotal) were recorded. Preoperative training including pole climbing, turning and lower pole was done for 3 days, with three trials per day. The average of the three trials was used for statistical analysis.
The foot fault test was performed at the 1st and 3rd days after the surgery (46). The numbers of misses on the injured side (right foot fault) and misses on the normal side (left foot fault) going through the overhead net were counted. The calculation formula was as follows: number of right foot fault/(number of right foot fault + number of left foot fault) × 100%. The mice were trained to be able to go through from the starting site of the overhead net to the ending site for 3 days before the test and three times (5 min each) every day. The mice in each group were tested three times to get the mean.
Triphenyltetrazolium chloride staining
The brain tissues of mice were prepared after the behavioral testing 3 days after the surgery, frozen at −20 °C for 20 min, cut into ∼1.5-mm coronal sections using blades, and cultured in 0.5% triphenyltetrazolium chloride + PBS at 37 °C for 20 min in the dark for even staining. The brain sections were put on a glass pane according to the anatomic order and scanned via a scanner. After the optimization and processing, the scanned images were analyzed for infarct size by using an Image-Pro Plus software. Infarct size calculation formula were shown as follows: brain edema rate (S) = (ΣLT − ΣRT)/(ΣLT + ΣRT) × 100%, where ΣLT represents the total volume of the left hemisphere, and ΣRT represents the total volume of the right hemisphere; volume percentage of infarct (A) = ΣSIN (1 − S)/(ΣLT + ΣRT)(1 − B) × 100%, where B represents total volume of noncoloring area in the sham group, and ΣSIN(1 − S) represents total infarct volume after removal of brain edema rate) (43, 47).
LCBF detection
The mice were injected with 1% pentobarbital sodium at 0.06 g/kg intraperitoneally and fixed on the stereotaxic apparatus, followed by local anesthesia in the skull using lidocaine. The skull was exposed, wiped by 3% H2O2 to remove tissues on the surface. We fixed a probe in the core area of arterial blood supply of brain to monitor LCBF baseline value and record the changes of LCBF. One-hour change of blood flow during MCAO and 20-min change during reperfusion were monitored and recorded. The baseline of LCBF was regarded as 100%, the ratio of LCBF during MCAO, and reperfusion to the baseline of LCBF indicated the change of LCBF.
Nissl staining
The right anterior cerebral cortex tissue of mice was isolated after the blood was removed using 0.9% NaCl solution and fixed in 4% paraformaldehyde overnight, followed by sucrose dehydration. After OCT embedding and freezing, the tissue was cut into 0.2-mm sections. Dried sections were washed with pure water for 10 s, replaced in tar violet working solution avoiding light for 1 h, and then immersed in pure water for a few seconds, followed by color separation in 70, 80, and 95% alcohol (each for 10 s). Afterward, the sections were immersed in the solution prepared with anhydrous alcohol, chloroform, and ether at the ratio of 1:1:1 for few seconds, immersed two times in anhydrous alcohol (5 min each), immersed two times in xylene (5 min each), mounted with neutral resin, and observed under a microscope.
Dual-Luciferase reporter assay
The binding site between miR-378 and GRB2 3´-UTR was analyzed on the TargetScan.org (http://www.targetscan.org/vert_72/) website and further tested by the Dual-Luciferase reporter assay. The target sequence in GRB2 3´-UTR was synthetized and inserted into the pMIR-reporter vector (Huayueyang Biotechnology Co., Ltd., Beijing, China) via SpeI and HindIII sites. The mutant (MUT) sequence complementary to seed regions in WT GRB2 (GRB2-WT) was designed. The target sequence was inserted into the pMIR-reporter vector by using T4 DNA ligase after restriction endonuclease cleavage, designated as GRB2-MUT. After that, the recombinant GRB2-WT and GRB2-MUT plasmids were co-transfected with miR-378 into human embryonic kidney (HEK)-293T cells (Shanghai Beinuo Biotech Ltd., Shanghai, China), respectively. After 48-h transfection, the cells were collected and lysed. The luciferase assay kit (K801-200, BioVision, Milpitas, CA, USA) and GloMax 20/20 luminometer (Promega, Madison, WI, USA) was used to detect luciferase activity. The experiment was repeated three times.
We predicted the binding relationship between MEG3 and miR-378 using the RNA22 database (RRID:SCR_016507) and further tested by the Dual-Luciferase reporter assay. The target sequence in miR-378 was synthetized and inserted into the pMIR-reporter vector (Huayueyang Biotechnology Co.) using the SpeI and HindIII sites. The complementary sequence mutation sites of seed sequences were designed on miR-378–WT. The target sequence was inserted into pMIR-reporter vector by T4 DNA ligase after restriction endonuclease digestion. After that, miR-378–WT and miR-378–Mut with correctly identified sequences were co-transfected with MEG3 or siRNA targeting MEG3 into HEK-293T cells (Shanghai Beinuo Biotech), respectively. After 48-h transfection, the cells were collected and lysed. The luciferase assay kit (K801-200, BioVision) and GloMax 20/20 luminometer (Promega) were used for the luciferase activity measurement. The experiment was repeated three times. MEG3 siRNA was synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, Guangdong, China), and its sequence was 5′-GATGAAGAGGACCGGAACTGA-3′.
Primary neuron extraction, culture, and treatment
Neonatal suckling mice of less than 24 h of age were purchased from SPF Biotechnology Co. The cerebral cortex was isolated from mouse brain with the meninges being removed, cut into small pieces, and detached with 5 ml of 0.25% trypsin at 37 °C for 15 min. The detached brain tissue was washed three times with Dulbecco's modified Eagle's medium (DMEM)–horse serum and triturated into cell suspension. The upper cell suspension was filtrated three times by cyto-screener. After that, the 8 × 105 cells were cultured with DMEM–horse serum in a 5% CO2 incubator at 37 °C for 4-6 h. Following DMEM–horse serum being discarded, the cells were added with culture medium. The culture medium was changed every 2–3 days. Serum-free neurobasal medium (Gibco) was employed for primary cortical neuron culture. The morphology of neurons was observed under a microscope.
Lentiviruses expressing siRNA were prepared for plasmid construction. Neurons cultured for 3 days were collected and cultured with 100 μl of lentivirus suspension, 500 μl of fresh medium and 5 µg/ml Polybrene for 24 h after the 3/4 medium was removed. The cells were then cultured in fresh medium for another 3 days for the establishment of OGD models. The neurons were grouped and treated as follows: control group (neurons without any treatment), blank group (nontransfected neurons exposed to OGD), OGD group (neurons transfected with scrambled sequence exposed to OGD), miR-378 inhibitor OGD group (neurons transfected with miR-378 inhibitor, a chemically synthesized single-stranded RNA that can specifically target and knockdown a single miR-378, and exposed to OGD), miR-378 mimic OGD group (neurons transfected with miR-378 mimic, a chemically synthesized sequence that can mimic the endogenous miR-378, and exposed to OGD), GRB2 siRNA OGD group (neurons transfected with siRNA targeting GRB2; 5′-GTGAGAACGATGAAATA-3′, and exposed to OGD), miR-378 inhibitor + GRB2 siRNA OGD group (neurons co-transfected with miR-378 inhibitor and siRNA targeting GRB2, and exposed to OGD). To conduct MEG3 intervention, the neurons were grouped and treated as follows: blank group (nontransfected neurons exposed to OGD), NC group (neurons transfected with scrambled sequence, and exposed to OGD), lncRNA MEG3 siRNA group (neurons transfected with siRNA targeting MEG3; 5´-GACUUAAACCAAUGCCCUA-3´, and exposed to OGD), pcDNA-MEG3 group (neurons transfected with pcDNA-MEG3, and exposed to OGD).
Establishment of OGD-induced neuron models
Neurons cultured for 7 days and in good growth condition were used to establish the model of ischemia injury in vitro and to develop a neuron model of OGD. The medium was replaced with sugar-free culture medium, and the neurons were cultured in an incubator with 5% CO2, 2% O2, and 93% N2 at 37 °C for 60 min. After that, the medium was replaced with high-glucose medium containing 10% fetal bovine serum, and the neurons were then cultured into a saturated normoxic medium with 5% CO2 incubator at 37 °C, with untreated neurons serving as control.
Monodansylcadaverine (MDC) staining
The neurons were cultured on a small and round slide coated with polylysine, treated with 20 μm MDC for incubation for 30 min, and then visualized using a fluorescence microscope. MDC fluorescence in the cytoplasm was changed from dispersion distribution to punctate distribution during the formation process of autophagosome.
LDH leakage rate
The Cytotox96 nonradioactive cytotoxicity kit (Promega) was employed to measure the LDH leakage rate. Neurons with a density of 105–106 were seeded in a coated 96-well plate, which was cultured in a 5% CO2 incubator at 37 °C until mature. After that, the mature neurons were performed with OGD for 1 h and cultured with 15 μl of lysate (9% (v/v) Triton® X-100 dissolved in water) in an incubator at 37 °C for 45 min. Cell supernatant (50 μl) was collected and added into a 96-well plate containing 50 μl of prepared substrate mixture for a 30-min reaction at room temperature. Each well was treated with 50 μl of stop buffer to terminate the enzymatic reaction. The OD value of each well was measured at 490 nm using a microplate reader.
Immunofluorescence
On the 7th day of neuron culture, the cell slides were washed three times with PBS (5 min each) and fixed in 4% paraformaldehyde for 30 min, followed by three PBS washes (5 min each). Subsequently, the cells were treated with 0.2% Triton X-100 for 15 min, blocked in 3% BSA at 4 °C for 30 min, and incubated with primary antibodies including rabbit anti-GRB2 (1:100, ab32037, Abcam, Cambridge, UK), mouse anti–microtubule-associated protein 2 (MAP-2, 1:200, ab11267, Abcam) and rabbit anti–GFAP (1:500, ab7260, Abcam) and mouse anti-GFAP (1:200, ab10062, Abcam) in a wet box at 4 °C overnight. Followed by three PBS washes (5 min each), the cells were incubated with secondary antibody, goat anti-rabbit IgG (1:500, ab150080, Alexa Fluor® 594) or goat anti-mouse IgG (1:200, ab150113, Alexa Fluor® 488) at room temperature for 2 h avoiding light and washed three times with PBS (5 min each). The cells were then treated with 4′,6-diamidino-2-phenylindole (1:100, ab104139, Abcam) avoiding light for 10 min at room temperature, washed three times with PBS (5 min each), sealed by mounting medium, and observed under an inverted fluorescence microscope.
In situ hybridization
The 10-μm-thick paraffin-embedded sections of mouse brain tissue were dewaxed with xylene and hydrated with gradient ethanol. The sections were detached with 40 μg/ml proteinase K for 20 min. Next, the sections were fixed in 4% paraformaldehyde for 10 min and treated with 20% acetic acid for 10 min, and pepsin was diluted with 3% sodium citrate for 2 min. The sections were then washed with 0.5 m TBS and diethyl pyrocarbonate (DEPC) water and incubated with 20 μl of prehybridization solution at 40 °C for 3 h. Afterward, the sections were hybridized with 20 μl of hybridization solution containing specific probes at 50 °C overnight. Then the sections were rinsed with three times in 2× SSC for 5 min each time and reacted with 20 μg/ml RNase A in 2× SSC for 30 min. The sections were rinsed with 2× SSC three times at 37 °C for 10 min each time, 0.5× SSC three times for 5 min each time, and 0.2× SSC three times for 5 min each time. Finally, the sections were blocked for 30 min at 37 °C and treated with biotinylated rat anti-digoxigenin for 1 h. After being rinsed in 0.5 m TBS four times for 5 min each time, the sections were stained with SABC-AP for 30 min and developed using 1-step TM NBT/BCIP (34041, Thermo Scientific, Rockford, IL, USA), followed by observation under an inverted fluorescence microscope.
Immunohistochemical staining
The paraffin-embedded sections of mouse brain tissue were heated at 60 °C for 1 h, dewaxed with xylene, and dehydrated with gradient alcohol. After antigen retrieval with 0.1 m sodium citrate, the sections were heated and boiled for 20 min. After being rinsed in 0.2 mol/liter PBS three times for 5 min each time, the activity of endogenous oxidase was blocked with 3% catalase for 15 min, followed by PBS washing. After being washed with 0.2 mol/liter PBS solution (pH 7.4), the sections were blocked with 5% BSA at 37 °C for 30 min and then incubated with rabbit anti-GRB2 (ab32037, 1:100, Abcam), p-Akt (ab38449, 1:250, Abcam), or p-S6 (ab59208, 1:100, Abcam) overnight at 4 °C, followed by PBS washing. The sections were incubated with the biotinylated goat anti-mouse IgG (ab6789, 1:2000, Abcam) 30 min and developed by diaminobenzidine (DA1010, Solarbio Science and Technology Ltd., Beijing, China). Hematoxylin was utilized for dyeing. After dehydration, clearing and sealing, the stained sections were visualized under the optical (XSP-36, BoShi Optical Instruments Co., Ltd., Shenzhen, China). The positive rate was scored proportionally, and the stained area was scored as follows: 0%, 0 point; 1–25%, 1 point; 26–50%, 2 points; 51–75%, 3 points; and 76–100%, 4 points). The staining intensity was scored as follows: negative, 0 point; weak positive, 1 point; moderate positive, 2 points; and strong positive, 3 points. The stained area was multiplied by staining intensity to produce the final scores. The scores were obtained independently from two pathologists.
RT-qPCR
TRIzol (16096020, Thermo Fisher Scientific) was used to extract the total RNA from cells. A total of 5 µg of RNA was used as a template for the reverse transcription reaction into cDNA in accordance with the manufacturer's protocol of the RT-qPCR kit (Applied Biosystems, Foster City, CA, USA). The PCR system was as follows: 300 ng of cDNA, 1× PCR buffer, 200 μmol/liter dNTPs, 80 pmol/liter forward primer, 80 pmol/liter reverse primer, 300 ng of cDNA, and 0.5 unit of Taq enzyme (S10118, Shanghai Yuanye Biotechnology Co., Ltd., Shanghai, China), with a total volume of 25 μl. The primers for miR-378, U6, GRB2, β-actin, and lncRNA MEG3 are shown in Table 1. The internal reference for miR-378 was U6, whereas the internal reference for GRB2 and MEG3 was β-actin. 2−ΔΔCt showed the multiple relationship between the target genes of experiment and control group, and the formula was as follow: ΔΔCT = ΔCt experiment group − ΔCtcontrol group, and ΔCt = Cttarget gene − Ctinternal reference. Each experiment was repeated three times.
Table 1.
Primer sequences of related genes for RT-qPCR
| Gene | Forward (5´–3´) | Reverse (5´–3´) |
|---|---|---|
| miR-378 | CCCGGGCTGAGGAATT | AAGCTTAAAAAAAGGCCTTC |
| U6 | GCATGACGTCTGCTTTGGA | CCACAATCATTCTGCCATCA |
| GRB2 | ATTCCTGCGGGACATAGAACA | GGTGACATAATTGCGGGGAAAC |
| β-Actin | CATCCGTAAAGACCTCTATGCCAAC | ATGGAGCCACCGATCCACA |
| MEG3 | GGGAGCAGCTATGGATCACC | ATAGCGCCCCCTATTCATGC |
Western blotting analysis
Brain tissues or cells were washed two times with PBS, treated with lysis buffer, shaken on a whirlpool instrument, and centrifuged at 12,000 rpm at 4 °C for 30 min to remove debris. The supernatant was collected for further use. Following the determination of total protein concentration by the bicinchoninic acid protein assay kit, 50 µg of total protein samples were dissolved in 2× SDS loading buffer, boiled for 5 min for denaturation, separated on 10% SDS-PAGE, and transferred onto a polyvinylidene fluoride membrane. Then the membrane was blocked in 5% skimmed milk at room temperature for 1 h and incubated with diluted primary antibodies to β-actin (1:1000, ab8227), GRB2 (1:5000, ab32037), Beclin I (1:500, ab62557), light chain 3 (LC3)–I/II (1:3000, ab51520), Akt (1:250, ab151279), phosphorylated Akt (p-Akt; 1:500, ab38449), mTOR (mTOR; 1:2000, ab2732), p-mTOR (1:2000, ab109268), S6 (1:10,000, ab32529) and phosphorylated S6 (p-S6, 1:1000, ab109393). All antibodies were purchased from Abcam. Later, the membrane was washed three times with TBS containing Tween 20 (TBST) and incubated with secondary antibody to horseradish peroxidase-labeled IgG for 1 h, followed by being rinsed in TBST. The membrane was placed on a clean glass plate. Equal amounts of solutions A and B in the enhanced chemiluminescence kit (ECL kit, BB-3501, Amersham Biosciences) were mixed in a dark room and added onto the membrane. Densitometric analysis of the bands was carried out using a gel imaging analysis system. The Gel Doc XR imager system (Bio-Rad) was used for imaging and Quantity One V4.6.2 for analysis. The average absorbance ratio of target protein band to internal reference (β-actin) band was considered to be relative protein expression. Each experiment was repeated three times.
Methylation-specific PCR
Specific primers of miR-378 methylation and nonmethylation (Table 2) were synthetized by TaKaRa (Dalian, Liaoning, China). The reaction system (20 μl) was as follows: 0.4 μm primer, 10 μm SYBR® Premix Ex TaqTM II, 20 ng of modified DNA, 0.4 μl of 50 × ROX (Takara Holdings Inc., Tokyo, Japan) and sterile distilled water. The reaction conditions were as follows: predenaturation at 95 °C for 30 s; and 40 cycles of denaturation at 95 °C for 5 s, annealing at 62 °C for 30 s and extension at 72 °C for 5 s; followed by a last extension at 72 °C for 30 s. We collected 5 μl of MS-PCR product for a 30-min electrophoresis on 20 g/liter Sepharose gel (110 V). The gel image analysis system was used for imaging to observe the methylation of target gene.
Table 2.
Primer sequences of miR-378 methylation and nonmethylation
| miR-378 primer | Forward (5´–3´) | Reverse (5´–3´) |
|---|---|---|
| Methylation | GGATGAGTTTTGAGTCGTTT | CCATACAAACCGCTCACTCC |
| Nonmethylation | TAGGATGAGTTTTGAGTTGT | ATAATCCCATACAAACCACT |
RNA-binding protein immunoprecipitation
The RIP kit (Millipore Corp., Billerica, MA, USA) was used to detect the binding of lncRNA MEG3 and AGO2. Neurons were washed with precooled PBS, with the supernatant removed, lysed by radio immunoprecipitation assay lysis buffer (P0013B, Beyotime Institute of Biotechnology, Shanghai, China) on an ice bath for 5 min, and centrifuged at 14000 rpm for 10 min at 4 °C. The supernatant was collected. A proportion of the cell lysate was used as input, and a proportion was cultured with antibody for co-precipitation. In each co-precipitation system, 50 μl of magnetic beads were resuspended in 100 μl of RIP wash buffer and cultured with 5 μg of antibody for binding. Magnetic bead–antibody complex was washed and resuspended in 900 μl of RIP wash buffer and cultured with 100 μl of cell lysate overnight at 4 °C. The samples were placed on the magnetic base to collect magnetic bead–antibody. The samples and input were detached by proteinase K, followed by RNA extraction for further PCR detection. The antibody used in RIP was rabbit anti-mouse AGO2 (1:50, ab186733, Abcam). Rabbit anti-mouse IgG (1:100, ab109489, Abcam) was used as the negative control.
Statistical analysis
The data were analyzed using SPSS 21.0 software (IBM Corp., Armonk, NY, USA). All data were tested for normality and homogeneity of variance. The measurement data obeying normal distribution and homogeneity of variance are expressed as means ± standard deviation, otherwise, the data are expressed by median ± interquartile range. Comparison between two groups was conducted by unpaired t test. Comparisons among multiple groups were assessed by one-way analysis of variance with Tukey's post hoc test. The skewed data between two groups were compared using nonparametric Wilcoxon-signed rank test, and those among multiple groups were compared using Kruskal–Wallis H test (n = 10 in animal experiments and n = 3 in cell experiments). p < 0.05 was considered to be statistically significant.
Data availability
All data are contained within the article.
Acknowledgments
We acknowledge and appreciate our colleagues for valuable efforts and comments on this article.
Author contributions—H.-C. L. and F.-G. H. conceptualization; H.-C. L. validation; H.-C. L., F.-G. H., and Y. W. investigation; H.-C. L., X.-P. L., and Q.-S. L. methodology; T-ZY software; T-ZY and X.-P. L. formal analysis; T-ZY, Y. W., and Q.-S. L. writing-original draft; F.-G. H. and X.-P. L. resources; F.-G. H. visualization; Y. W. and Q.-S. L. writing-review and editing; X.-P. L. data curation.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- miR
- microRNA
- RT-qPCR
- reverse transcription–quantitative PCR
- MCAO
- middle cerebral artery occluded
- OGD
- oxygen-glucose deprivation
- lncRNA
- long noncoding RNA
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- GFAP
- glial fibrillary acidic protein
- ISH
- in situ hybridization
- LCBF
- local cerebral blood flow
- MUT
- mutant
- DMEM
- Dulbecco's modified Eagle's medium
- MDC
- monodansylcadaverine
- LDH
- lactate dehydrogenase
- TBS
- Tris-buffered saline
- MS
- methylation-specific
- RIP
- RNA-binding protein immunoprecipitation
- NC
- negative control.
References
- 1. Santos H. N., Magedanz E. H., Guaragna J. C., Santos N. N., Albuquerque L. C., Goldani M. A., Petracco J. B., and Bodanese L. C. (2014) Predictors of stroke in patients undergoing cardiac surgery. Rev. Bras. Cir. Cardiovasc. 29, 140–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mozaffarian D., Benjamin E. J., Go A. S., Arnett D. K., Blaha M. J., Cushman M., Das S. R., de Ferranti S., Despres J. P., Fullerton H. J., Howard V. J., Huffman M. D., Isasi C. R., Jimenez M. C., Judd S. E., et al. (2016) Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation 133, 447–454 10.1161/CIR.0000000000000366 [DOI] [PubMed] [Google Scholar]
- 3. Kim A. S., and Johnston S. C. (2011) Global variation in the relative burden of stroke and ischemic heart disease. Circulation 124, 314–323 10.1161/CIRCULATIONAHA.111.018820 [DOI] [PubMed] [Google Scholar]
- 4. Mokdad A. H., Forouzanfar M. H., Daoud F., Mokdad A. A., El Bcheraoui C., Moradi-Lakeh M., Kyu H. H., Barber R. M., Wagner J., Cercy K., Kravitz H., Coggeshall M., Chew A., O'Rourke K. F., Steiner C., et al. (2016) Global burden of diseases, injuries, and risk factors for young people's health during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 387, 2383–2401 10.1016/S0140-6736(16)00648-6 [DOI] [PubMed] [Google Scholar]
- 5. Wang D., Hao Z., Tao W., Kong F., Zhang S., Wu B., Lin S., and Liu M. (2011) Acute ischemic stroke in the very elderly Chinese: risk factors, hospital management and one-year outcome. Clin. Neurol. Neurosurg. 113, 442–446 10.1016/j.clineuro.2011.01.005 [DOI] [PubMed] [Google Scholar]
- 6. Kato Y., Hayashi T., Tanahashi N., Kobayashi S., and Japan Standard Stroke Registry Study Group (2015) Cardioembolic stroke is the most serious problem in the aging society: Japan standard stroke registry study. J. Stroke Cerebrovasc. Dis. 24, 811–814 10.1016/j.jstrokecerebrovasdis.2014.11.019 [DOI] [PubMed] [Google Scholar]
- 7. Bushnell C. D., Reeves M. J., Zhao X., Pan W., Prvu-Bettger J., Zimmer L., Olson D., and Peterson E. (2014) Sex differences in quality of life after ischemic stroke. Neurology 82, 922–931 10.1212/WNL.0000000000000208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shu S., Li C. M., You Y. L., Qian X. L., Zhou S., and Ling C. Q. (2016) Electroacupuncture ameliorates cerebral ischemia–reperfusion injury by regulation of autophagy and apoptosis. Evid. Based Complement. Alternat. Med. 2016, 7297425 10.1155/2016/7297425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Backes C., Meder B., Hart M., Ludwig N., Leidinger P., Vogel B., Galata V., Roth P., Menegatti J., Grässer F., Ruprecht K., Kahraman M., Grossmann T., Haas J., Meese E., et al. (2016) Prioritizing and selecting likely novel miRNAs from NGS data. Nucleic Acids Res. 44, e53 10.1093/nar/gkv1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei C. W., Luo T., Zou S. S., and Wu A. S. (2017) Research progress on the roles of microRNAs in governing synaptic plasticity, learning and memory. Life Sci. 188, 118–122 10.1016/j.lfs.2017.08.033 [DOI] [PubMed] [Google Scholar]
- 11. Bhalala O. G., Srikanth M., and Kessler J. A. (2013) The emerging roles of microRNAs in CNS injuries. Nat. Rev. Neurol. 9, 328–339 10.1038/nrneurol.2013.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ouyang Y. B., Stary C. M., Yang G. Y., and Giffard R. (2013) microRNAs: innovative targets for cerebral ischemia and stroke. Curr. Drug Targets 14, 90–101 10.2174/138945013804806424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang N., Zhong J., Han S., Li Y., Yin Y., and Li J. (2016) MicroRNA-378 alleviates cerebral ischemic injury by negatively regulating apoptosis executioner caspase-3. Int. J. Mol. Sci. 17, 1427 10.3390/ijms17091427 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Sun F., Zhuang Y., Zhu H., Wu H., Li D., Zhan L., Yang W., Yuan Y., Xie Y., Yang S., Luo S., Jiang W., Zhang J., Pan Z., and Lu Y. (2019) lncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J. Mol. Cell. Cardiol. 133, 188–198 10.1016/j.yjmcc.2019.06.011 [DOI] [PubMed] [Google Scholar]
- 15. Wei H., Li Y., Han S., Liu S., Zhang N., Zhao L., Li S., and Li J. (2016) cPKCγ-modulated autophagy in neurons alleviates ischemic injury in brain of mice with ischemic stroke through Akt-mTOR pathway. Transl. Stroke Res. 7, 497–511 10.1007/s12975-016-0484-4 [DOI] [PubMed] [Google Scholar]
- 16. Liu X., Hou L., Huang W., Gao Y., Lv X., and Tang J. (2016) The mechanism of long non-coding RNA MEG3 for neurons apoptosis caused by hypoxia: mediated by miR-181b-12/15-LOX signaling pathway. Front. Cell. Neurosci. 10, 201 10.3389/fncel.2016.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murase S. (2015) Impaired focal adhesion kinase–Grb2 interaction during elevated activity in hippocampal neurons. Int. J. Mol. Sci. 16, 15659–15669 10.3390/ijms160715659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manu M. S., Rachana K. S., and Advirao G. M. (2017) Altered expression of IRS2 and GRB2 in demyelination of peripheral neurons: implications in diabetic neuropathy. Neuropeptides 62, 71–79 10.1016/j.npep.2016.12.004 [DOI] [PubMed] [Google Scholar]
- 19. Chen Y., Xie H. Q., Sha R., Xu T., Zhang S., Fu H., Xia Y., Liu Y., Xu L., and Zhao B. (2020) 2,3,7,8-Tetrachlorodibenzo-p-dioxin and up-regulation of neurofilament expression in neuronal cells: evaluation of AhR and MAPK pathways. Environ. Int. 134, 105193 10.1016/j.envint.2019.105193 [DOI] [PubMed] [Google Scholar]
- 20. Salama R. M., Abdel-Latif G. A., Abbas S. S., El Magdoub H. M., and Schaalan M. F. (2020) Neuroprotective effect of crocin against rotenone-induced Parkinson's disease in rats: Interplay between PI3K/Akt/mTOR signaling pathway and enhanced expression of miRNA-7 and miRNA-221. Neuropharmacology 164, 107900 10.1016/j.neuropharm.2019.107900 [DOI] [PubMed] [Google Scholar]
- 21. Tan M. C., Widagdo J., Chau Y. Q., Zhu T., Wong J. J., Cheung A., and Anggono V. (2017) The activity-induced long non-coding RNA Meg3 modulates AMPA receptor surface expression in primary cortical neurons. Front. Cell. Neurosci. 11, 124 10.3389/fncel.2017.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qu C., Jiang T., Li Y., Wang X., Cao H., Xu H., Qu J., and Chen J. G. (2013) Gene expression and IG-DMR hypomethylation of maternally expressed gene 3 in developing corticospinal neurons. Gene Exp. Patterns 13, 51–56 10.1016/j.gep.2012.11.003 [DOI] [PubMed] [Google Scholar]
- 23. Niaz S. (2018) The AGO proteins: an overview. Biol. Chem. 399, 525–547 10.1515/hsz-2017-0329 [DOI] [PubMed] [Google Scholar]
- 24. Ballantyne M. D., McDonald R. A., and Baker A. H. (2016) lncRNA/MicroRNA interactions in the vasculature. Clin. Pharmacol. Therap. 99, 494–501 10.1002/cpt.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Gao A., Feng D., Wang Y., Zhang L., Cui Y., Li B., Wang Z., and Chen G. (2014) Evaluation of the protective potential of brain microvascular endothelial cell autophagy on blood–brain barrier integrity during experimental cerebral ischemia–reperfusion injury. Transl. Stroke Res. 5, 618–626 10.1007/s12975-014-0354-x [DOI] [PubMed] [Google Scholar]
- 26. Zhang X., Yan H., Yuan Y., Gao J., Shen Z., Cheng Y., Shen Y., Wang R. R., Wang X., Hu W. W., Wang G., and Chen Z. (2013) Cerebral ischemia–reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9, 1321–1333 10.4161/auto.25132 [DOI] [PubMed] [Google Scholar]
- 27. Li H., Wang Z., Zhang W., Qian K., Liao G., Xu W., and Zhang S. (2015) VGLL4 inhibits EMT in part through suppressing Wnt/β-catenin signaling pathway in gastric cancer. Med. Oncol. 32, 83 10.1007/s12032-015-0539-5 [DOI] [PubMed] [Google Scholar]
- 28. Lu Y., Jian M. Y., Ouyang Y. B., and Han R. Q. (2015) Changes in rat brain microRNA expression profiles following sevoflurane and propofol anesthesia. Chinese Med. J. 128, 1510–1515 10.4103/0366-6999.157676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Costantino S., Paneni F., Lüscher T. F., and Cosentino F. (2016) MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur. Heart J. 37, 572–576 10.1093/eurheartj/ehv599 [DOI] [PubMed] [Google Scholar]
- 30. Ouseph M. M., Huang Y., Banerjee M., Joshi S., MacDonald L., Zhong Y., Liu H., Li X., Xiang B., Zhang G., Komatsu M., Komatsu M., Yue Z., Li Z., Storrie B., et al. (2015) Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 126, 1224–1233 10.1182/blood-2014-09-598722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hernaez B., Cabezas M., Munoz-Moreno R., Galindo I., Cuesta-Geijo M. A., and Alonso C. (2013) A179L, a new viral Bcl2 homolog targeting Beclin 1 autophagy related protein. Curr. Mol. Med. 13, 305–316 10.2174/156652413804810736 [DOI] [PubMed] [Google Scholar]
- 32. Zou M., Lu N., Hu C., Liu W., Sun Y., Wang X., You Q., Gu C., Xi T., and Guo Q. (2012) Beclin 1-mediated autophagy in hepatocellular carcinoma cells: implication in anticancer efficiency of oroxylin A via inhibition of mTOR signaling. Cell. Signal. 24, 1722–1732 10.1016/j.cellsig.2012.04.009 [DOI] [PubMed] [Google Scholar]
- 33. Oliván S., Calvo A. C., Gasco S., Munoz M. J., Zaragoza P., and Osta R. (2015) Time-point dependent activation of autophagy and the UPS in SOD1G93A mice skeletal muscle. PLoS One 10, e0134830 10.1371/journal.pone.0134830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen W., Sun Y., Liu K., and Sun X. (2014) Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen. Res. 9, 1210–1216 10.4103/1673-5374.135329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Le X. T., Nguyet Pham H. T., Van Nguyen T., Minh Nguyen K., Tanaka K., Fujiwara H., and Matsumoto K. (2015) Protective effects of Bacopa monnieri on ischemia-induced cognitive deficits in mice: the possible contribution of bacopaside I and underlying mechanism. J. Ethnopharmacol. 164, 37–45 10.1016/j.jep.2015.01.041 [DOI] [PubMed] [Google Scholar]
- 36. Rückerl D., Jenkins S. J., Laqtom N. N., Gallagher I. J., Sutherland T. E., Duncan S., Buck A. H., and Allen J. E. (2012) Induction of IL-4Rα–dependent microRNAs identifies PI3K/Akt signaling as essential for IL-4–driven murine macrophage proliferation in vivo. Blood 120, 2307–2316 10.1182/blood-2012-02-408252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Y., Shi Q. F., Ye Y. C., Tashiro S., Onodera S., and Ikejima T. (2012) Activated O2(*-) and H2O2-mediated cell survival in SU11274-treated non-small-cell lung cancer A549 cells via c-Met-PI3K-Akt and c-Met-Grb2/SOS-Ras-p38 pathways. J. Pharmacol. Sci. 119, 150–159 10.1254/jphs.12048fp [DOI] [PubMed] [Google Scholar]
- 38. Yin H., Li P., Hu F., Wang Y., Chai X., and Zhang Y. (2014) IL-33 attenuates cardiac remodeling following myocardial infarction via inhibition of the p38 MAPK and NF-κB pathways. Mol. Med. Rep. 9, 1834–1838 10.3892/mmr.2014.2051 [DOI] [PubMed] [Google Scholar]
- 39. Huang J., Peng J., and Guo L. (2015) Non-coding RNA: a new tool for the diagnosis, prognosis, and therapy of small cell lung cancer. J. Thorac. Oncol. 10, 28–37 10.1097/JTO.0000000000000394 [DOI] [PubMed] [Google Scholar]
- 40. Yu F., Yang J., Huang K., Pan X., Chen B., Dong P., and Zheng J. (2016) The epigenetically-regulated microRNA-378a targets TGF-β2 in TGF-β1–treated hepatic stellate cells. Cell. Physiol. Biochem. 40, 183–194 10.1159/000452536 [DOI] [PubMed] [Google Scholar]
- 41. Yan H., Yuan J., Gao L., Rao J., and Hu J. (2016) Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience 337, 191–199 10.1016/j.neuroscience.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 42. You D., and You H. (2019) Repression of long non-coding RNA MEG3 restores nerve growth and alleviates neurological impairment after cerebral ischemia–reperfusion injury in a rat model. Biomed. Pharmacother. 111, 1447–1457 10.1016/j.biopha.2018.12.067 [DOI] [PubMed] [Google Scholar]
- 43. Jiang Y., Yang S., Tao J., Lin Z., Ye X., You Y., Peng J., Hong Z., and Chen L. (2016) Opposing needling promotes behavior recovery and exerts neuroprotection via the cAMP/PKA/CREB signal transduction pathway in transient MCAO rats. Mol. Med. Rep. 13, 2060–2070 10.3892/mmr.2016.4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Longa E. Z., Weinstein P. R., Carlson S., and Cummins R. (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20, 84–91 10.1161/01.str.20.1.84 [DOI] [PubMed] [Google Scholar]
- 45. Balkaya M., Kröber J., Gertz K., Peruzzaro S., and Endres M. (2013) Characterization of long-term functional outcome in a murine model of mild brain ischemia. J. Neurosci. Methods 213, 179–187 10.1016/j.jneumeth.2012.12.021 [DOI] [PubMed] [Google Scholar]
- 46. Chen J., Zhang C., Jiang H., Li Y., Zhang L., Robin A., Katakowski M., Lu M., and Chopp M. (2005) Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J. Cereb. Blood Flow Metab. 25, 281–290 10.1038/sj.jcbfm.9600034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feng S., Li D., Li Y., Yang X., Han S., and Li J. (2013) Insight into hypoxic preconditioning and ischemic injury through determination of nPKCε-interacting proteins in mouse brain. Neurochem. Int. 63, 69–79 10.1016/j.neuint.2013.04.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.