

Abstract
Emerging base editing technology exploits CRISPR RNA-guided DNA modification effects for highly specific C > T conversion, which has been used to efficiently disrupt gene expression. These tools can enhance synthetic T cell immunity by restricting specificity, addressing histocompatibility leukocyte antigen (HLA) barriers, and promoting persistence. We report lentiviral delivery of a hepatitis B-virus (HBV)-specific recombinant T cell receptor (rTCR) and a linked CRISPR single-guide RNA for simultaneous disruption of endogenous TCRs (eTCRs) when combined with transient cytosine deamination. Discriminatory depletion of eTCR and coupled expression of rTCR resulted in enrichment of HBV-specific populations from 55% (SEM, ±2.4%) to 95% (SEM, ±0.5%). Intensity of rTCR expression increased 1.8- to 2.9-fold compared to that in cells retaining their competing eTCR, and increased cytokine production and killing of HBV antigen-expressing hepatoma cells in a 3D microfluidic model were exhibited. Molecular signatures confirmed that seamless conversion of C > T (G > A) had created a premature stop codon in TCR beta constant 1/2 loci, with no notable activity at predicted off-target sites. Thus, targeted disruption of eTCR by cytosine deamination and discriminatory enrichment of antigen-specific T cells offers the prospect of enhanced, more specific T cell therapies against HBV-associated hepatocellular carcinoma (HCC) as well as other viral and tumor antigens.
Keywords: CRISPR/Cas9Base editing, Recombinant TCR, Gene therapy, Cell therapy, Hepatocellular carcinoma (HCC), Hepatitis B virus (HBV)
Graphical Abstract
The authors deploy cytosine-deamination-mediated base editing to genetically disrupt endogenous T cell receptors (eTCRs), thereby reducing competition with recombinant TCRs (rTCRs) and allowing the enrichment of engineered T cells for immunotherapy against hepatitis B-driven hepatocellular carcinoma.
Introduction
T cells redirected with recombinant T cell receptors (rTCRs) are being investigated in early-phase human studies.1, 2, 3 Limitations include unpredictable “off-target effects” due to TCR cross-reactivity; for example, cardiac toxicity following therapy with MAGE-A3 rTCR4,5 and concerns that endogenous TCR α and β chains may mispair with rTCR chains and give rise to novel dimeric complexes with unpredictable specificities.6,7 These limitations have been partially mitigated by predictive modeling of rTCR cross-reactivity and by promoting exclusive rTCR pairing via additional disulfide bonds and other strategies.8, 9, 10, 11 Also of note is the importance of rTCR assembly on the cell surface as a multimeric complex with CD3 chains, as competition from the endogenous TCR (eTCR) for the shared components can limit cell-surface expression.12 Competition for such cellular components can be addressed either by overexpression of CD3, disruption of eTCR by RNA interference,13 or nuclease-mediated genetic disruption of eTCR chains. Previously, zinc-finger nucleases (ZFNs),14 transcription activator-like effector nucleases (TALENs),15 and clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) have all been used to disrupt one or both TCR α and β chains.16, 17, 18 These genome-editing approaches also reduce the likelihood of mispairing, but existing nuclease-based approaches all result in double-stranded DNA breaks and may create large insertions/deletions (indels), trigger translocation events, and increase activation of p53 pathways.19, 20, 21, 22, 23 Recently, a report of autologous anti-tumor therapy with T cells edited using Cas9 to disrupt both TCR and PD1 expression noted readily detectable chromosomal translocations in the infused products,24 and similar aberrations were found after TALEN editing of T cells modified to express anti-CD19 chimeric antigen receptors (CARs).25
Here, we report the application of emerging cytosine deaminase base-editing technology for efficient and seamless base conversion to introduce premature stop codons in homologous regions of TCR beta constant 1 and 2 (TRBC 1/2) chains.26,27 BE3 (third-generation base editor) is a CRISPR-guided nickase Cas9 (D10A), fused to a rat apolipoprotein B mRNA editing enzyme catalytic polypeptide (rAPOBEC1) deaminase at the N terminus, which operates within a 4- to 8-bp window distal to the protospacer adjacent motif (PAM) sequence. The inclusion of a C terminus fusion comprising a uracil-DNA glycosylase inhibitor (UGI) (derived from Bacillus subtilis bacteriophage PBS1) inhibits uracil-DNA glycosylase and blocks uracil excision, promoting conversion to thymidine as cells replicate. High levels of C > T conversion and low levels of indels have been reported for BE3.28, 29, 30 Here, we investigate a codon-optimized BE3 (coBE3) in the context of engineering T cells against HBV surface antigen, an important target in the treatment of hepatocellular carcinoma (HCC).31,32 HBV viral antigens are processed and presented by major histocompatibility complex (MHC) molecules on the surface of infected cells,33,34 and naturally occurring HBV-specific T cells can engage with peptides presented in the context of histocompatibility leukocyte antigen (HLA) to moderate viral and tumor burdens.35,36 Nevertheless, such HBV-specific T cell responses can become exhausted during chronic HBV infection,37, 38, 39 and synthetic HBV-specific T cells can be generated through the expression of rTCRs.40, 41, 42, 43, 44 The approach has already been tested clinically in HBV-associated HCC,45,46 with further studies planned.
Lentiviral vector delivery of a rTCR specific for HLA-A2/HBV peptide S183-91, incorporating murine constant regions, and coupled to a CRISPR single guide RNA (sgRNA) targeting TRBC1/2 loci resulted in high levels of targeted cytosine deamination after transient delivery of mRNA encoding coBE3. Thereafter, discriminatory removal of residual eTCR+ cells was achieved by magnetic-bead-mediated depletion using the anti-human TCRαβ monoclonal antibody. Consequently, rTCR expression was enriched, as the murine constant regions lack the specific epitope recognized by this antibody. Phenotypic and functional assessments, including migration and killing in a 3D microfluidic model, verified immunotherapeutic effects following genome editing, and molecular analysis of both DNA and RNA was performed to examine editor effects.
Results
Base Conversion Disrupts eTCR Expression and Allows Enrichment of T Cells Expressing rTCR
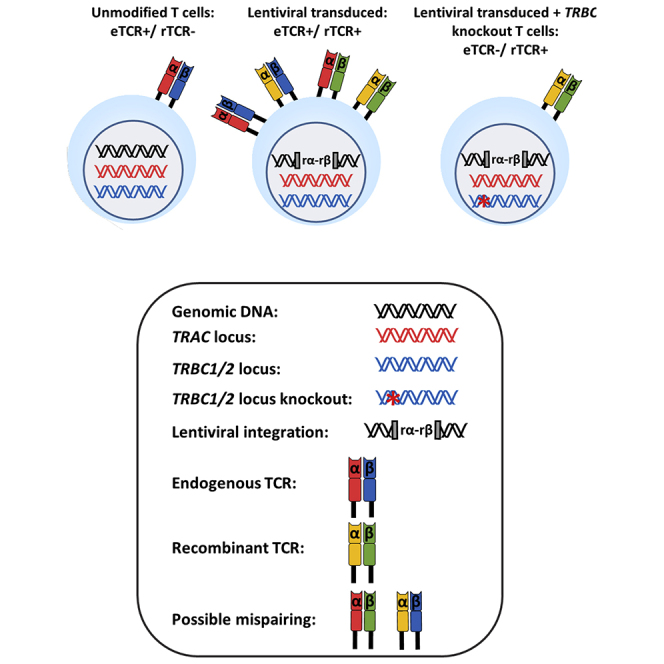
A third-generation self-inactivating (SIN) lentiviral vector was generated encoding an HLA-A0201 restricted rTCR (S183-91, FLLTRILTI) specific for HBV envelope protein47 and a linked sgRNA expression cassette targeting TRBC 1/2. The latter was embedded within a deleted unique (ΔU3) region of the 3′ long terminal repeat (LTR) under the transcriptional control of an RNA polymerase III human U6 promoter as previously described.48 This configuration is referred to as terminal-TRBC-S183-91 rTCR (TTRBC-S183-91 rTCR) (Figure 1). Upon electroporation of coBE3 mRNA, the sgRNA mediated highly targeted base conversion of two neighboring cytosine nucleotides within exon 1 of TRBC 1 /2 loci. Single or double base conversion produces a premature stop codon within a 4- to 8-bp window distal to the nCas9 (D10A) PAM sequence (Figure 2A). Consequently, disruption of endogenous TCR β chain expression eliminated eTCRαβ assembly, and the inclusion of murine constant regions within the rTCR further addressed any possibility of aberrant cross-pairing between residual recombinant and endogenous chains (Figure 2B). Following the timeline shown in Figure 2C, healthy T cells were readily activated and transduced resulting in 50%–60% rTCR expression (Figures 2D and 2Ei). Exposure to coBE3 led to disruption of eTCR expression and simultaneous emergence of rTCR+ populations, increasing in proportion to approximately 60%–65% of the cultures (Figures 2D and 2Ei). Furthermore, because eTCR was amenable to detection by anti-TCRαβ monoclonal antibody, magnetic bead-mediated depletion of residual eTCRαβ-expressing cells was possible. Notably, rTCR (constructed with murine C domains) was not susceptible to these reagents; thus, at the end of production, cells could be enriched for endogenous TCR−/recombinant TCR+ (eTCR−/rTCR+), resulting in a highly homogeneous product (>99% eTCR−/95.9% rTCR+) (Figures 2D and 2Ei). There was also a significant increase in the mean florescence intensity (MFI) of rTCR in eTCR−/rTCR+ cells compared to eTCR+/rTCR± , suggesting enhanced cell-surface expression of rTCR in the absence of eTCR, which may otherwise have competed for CD3 chains during assembly (one-way ANOVA, p < 0.02) (Figure 2Eii).
Figure 1.

Terminal-CRISPR Lentiviral Vector Configuration Coupling HBV rTCR and CRISPR TRBC1/2 sgRNA Delivery
Lentiviral plasmid configuration, coupling the expression of a recombinant T cell receptor (rTCR) against the hepatitis B virus (HBV) envelope surface antigen 183-91 (S183-91) and a T cell receptor beta constant (TRBC)-specific single guide RNA (sgRNA). The S183-91 rTCR is placed under the transcriptional control of an internal human phosphoglycerate kinase 1 (hPGK) promoter, while TRBC1/2 sgRNA is expressed via a human U6 promoter. The rTCR is expressed as a single transcript with the rTCR α chain first, followed by the rTCR β chain separated by a porcine teschovirus-1 2A (P2A) self-cleavage sequence. These recombinant chains are composed of the T cell receptor α variable 34 (TRAV34) and the T cell receptor β variable 28 (TRBV28) domains, as well as either murine TRAC (muTRAC) or murine TRBC 1 (muTRBC1). The rTCR chains contained an additional cysteine-cysteine disulfide bond between murine constant regions. CMV, cytomegalovirus; cPPT, central polypurine tract; WPRE, woodchuck post-transcriptional regulatory element; LTR, long terminal repeat; ΔU3, deleted unique 3′; R, repeat; U5, unique 5′; ψ, psi; D, diversity region; J, joining region.
Figure 2.

Generation of eTCR−/rTCR+ T cells Using Coupled Cytosine Deaminase Base Editing
(A) Schematic representation of base editor 3 (BE3) targeting exon 1 of the TRBC 1/2 loci. Editing window (blue) of the BE3 ranged from 4 to 8 bp distal to the PAM (red) with the conversion of tryptophan (Trp) codons to create premature stop codons. (B) Theoretical TCR chain pairing when introducing a rTCR with or without knockout of the endogenous TCR β chain. Incorporation of murine constant regions with an additional disulfide bridge in the recombinant α and β (rα-rβ) chains reduced the potential for the mispairing indicated in the middle panel, and disruption of eTCR further reduces the likelihood of mispairing. (C) Schema of cell production. Human peripheral blood lymphocytes were isolated and activated with TransAct (anti-CD3/CD28) (day 0) before transduction (day 1) and electroporation with codon-optimized (co)BE3 mRNA (day 4). After overnight hypothermic culture at 30°C, cells were expanded in G-Rex10 flasks for 7 days. Discriminatory depletion of residual endogenous TCR (eTCR)-expressing cells was carried out (day 11) before cryopreservation on day 14. (D) Representative flow cytometry phenotyping of unmodified and terminal TRBC-S183-91 rTCR (TTRBC-S183-91 rTCR)-transduced cells. Delivery of coBE3 mRNA by electroporation caused reduction of eTCR expression (38.1%) and emergence of eTCR−/ rTCR+ cells (red boxes). Magnetic-bead-mediated depletion of residual eTCR+ T cells enriched eTCR− populations, resulting in >99% eTCR−/95.9% rTCR+ (gated on CD45+). (E) Expression of S183-91 rTCR in three healthy donors. (i) Histogram of rTCR (183-91) expression exhibiting transduction ranging from 59.8% to 63.9% in cells exposed to both vector and BE3, which, following TCRαβ bead-mediated depletion, resulted in the enrichment of genome-edited cells, with rTCR levels increased to 93.9%–96.1%. The three colors represent different donors. (ii) Levels of cell-surface rTCR expression measured by mean fluorescence intensity (MFI) (n = 3) showed increased eTCR−/rTCR+ compared to eTCR+/rTCR± cells (gated on CD45+ > rTCR+ population). One-way ANOVA with Tukey’s multiple comparison test, p < 0.02. Error bars represent ±1 standard error of the mean (SEM). nCas9, nickase CRISPR-associated protein 9; UGI, uracil DNA glycosylase inhibitor; rAPOBEC1, rat apolipoprotein B mRNA editing enzyme catalytic polypeptide 1.
Hepatitis B Antigen-Specific Responses of eTCR−/rTCR+ T Cells
Three different in vitro assessments of antigen-specific function were undertaken. First, production of cytokines including interferon-γ (IFNγ), tumor necrosis factor α (TNF-α), interleukin-2 (IL-2), and C-C motif chemokine ligand 4 (CCL4) was determined by flow cytometry in T cells responding to HepG2 cells pulsed with the irrelevant control peptide (HBV core C18-27, FLPSDFFPSV) or a gradient of Hepatitis B target surface envelope peptide (S183-91, FLLTRILTI) concentrations. In all three donors tested, cytokine production was higher in eTCR−/rTCR+ T cells in response to target S183-91 peptide (Figure 3A, i and ii), with absent response to control C18-27 peptide and no-peptide control (Figure S1). Next, we investigated effector function at different effector:target (E:T) ratios in a previously described xCELLigence impedance assay and calculated the relevant normalized cell indices over 72 h after the addition of effector T cells.49 An increased index indicated HepG2 target cell proliferation, whereas cell death or apoptosis resulted in a reduced index, signifying higher levels of effector T cell activity (Figure 3B). Control groups included target cells alone (HepG2 alone), and non-transduced effectors (eTCR+/rTCR−), where, as expected, there was a progressive increase and plateau in index. In contrast, both effector groups exhibited a transient rise and then decline in index, with more rapid reductions mediated by eTCR−/rTCR+ cells compared to eTCR+/rTCR± T cells at all E:T ratios (Figure 3Ci). Overall effector function was calculated by area under the curve, as shown in Figure 3Cii, reflecting the increased cytotoxicity by enriched eTCR−/rTCR+ effector cells compared to their unedited, non-eTCR-depleted counterparts (eTCR+/rTCR±).
Figure 3.

Anti-HBV Responsiveness of eTCR+/rTCR±, compared to Base-Edited eTCR−/rTCR+ Effector T Cells
(A) Cytokine responses of effector T cells to the HepG2 cell line pulsed with target HBV surface peptide (S183-91, FLLTRILTI). n = 3. (i) Histograms of tumor necrosis factor α (TNF-α) responses to HepG2 target cells pulsed with 1 μM target peptide. Both the eTCR+/rTCR± and eTCR−/rTCR+ effector groups are gated on CD45+ > CD3+ > rTCR+ > CD8+, whereas unmodifed eTCR+/rTCR− effectors are gated on CD45+ > CD3+ > rTCR− > CD8+. The three different colors represent results from three donors. (ii) Cytokine responsiveness at different concentrations of target peptide (S183-91). HepG2 target cells were pulsed with 1 μM control peptide (C18-27) to ensure specificity of response, showing cytokine responsiveness comparable to that of the no-peptide control. Effector groups eTCR+/rTCR± and eTCR−/rTCR+ are gated on CD45+ > CD3+ > rTCR+ > CD8+, whereas unmodified cells are gated on CD45+ > CD3+ > rTCR− > CD8+. Error bars represent ±1 SEM. (B) Schematic depiction of the xCELLigence impedance assay showing cancer cells (green) seeded in wells with micro-electrode array (yellow) in the presence of effector T cells (blue). Where T cells recognize cancer cells, this leads to cell death (brown) and reduced impedance, resulting in lower cell index values and area under the curve (AUC). (C) xCELLigence data across different effector:target (E:T) ratios (1:1, 1:2, and 1:4). (i) Visualization of normalized cell index (NCI) over time; all donors showed an increased NCI with decreased E:T ratio. Both HepG2 alone (red) and eTCR+/rTCR− (orange) show steadily increasing NCIs over time, whereas the eTCR+/rTCR± (purple) and eTCR−/ rTCR+ (green) groups show an initially increased NCI followed by a marked decline. Normalized to time point prior to effector T cell addition. (ii) Summary data of AUC. Increased AUC values were observed at the lower E:T ratios, with eTCR−/ rTCR+ consistently presenting with the lowest AUC values. Error bars represent ±1 SEM. IFNγ, interferon γ; IL-2, interleukin-2; CCL4, C-C motif chemokine ligand 4.
Finally, migration and target cell killing by engineered T cells was determined in a 3D microfluidics device. The system captured the migration of effector T cells from a fluidics channel to a collagen gel embedded with target PreS1-GFP-HepG2 cells. Phenotyping of effector T cells confirmed rTCR expression (Figure 4A) and minimal cytokine expression in the absence of stimulation after thawing. Comparable numbers of T cells were observed migrating into the gel between the effector groups (Figure S2) before cytokine expression profiles were compared between cells recovered from inside or outside the gel area (Figure 4B). Both eTCR+/rTCR± and eTCR−/rTCR+ effector groups presented higher levels of IL-2, IFNγ, and TNF-α expression within the gel. Killing of PreS1-GFP-HepG2 cells by eTCR−/rTCR+ cells was confirmed within 24 h, whereas eTCR+/rTCR± cells at this time point were comparable to control eTCR+/rTCR− indices and the control PreS1-GFP-HepG2-alone groups (Figure 4C). Direct visualization revealed greater clearance of HepG2 cells after co-culture with eTCR−/rTCR+ T cells (Figure 4D).
Figure 4.

Effector T Cell Cytokine Responsiveness and Target Cell Killing in a 3D Microfluidics Device
(A) Flow-cytometry-based phenotyping of effector T cells used in 3D killing assay post-cryopreservation (gated on CD45+). (B) Histogram depicting cytokine responsiveness of effector T cells isolated from either outside the collagen gel (gray) or inside the collagen gel (blue) after 24 h (cells pooled from 3 replicate devices, gated on rTCR+CD8+; unmodified cells gated on rTCR−CD8+). (C) Normalized killing of target PreS1-GFP-HepG2 cells in response to effector T cell groups presented as violin plot with median (solid black line) and 25th/75th quartiles (dotted black lines). PreS1-GFP-HepG2 alone (orange) and 20% DMSO (red) were used as negative and positive controls, respectively. Increased cytotoxicity was observed with eTCR−/rTCR+ (purple) effectors, compared to PreS1-GFP-HepG2 alone (p < 0.0001), eTCR+/rTCR− (green, p < 0.002), and eTCR+/rTCR± (blue, p = 0.0001). Each point represents a section of a 3D microfluidics device from n = 3 technical replicates (3 sections analyzed per device). One-way ANOVA with Tukey’s multiple comparison test. (D) Visualization of a region within the collagen gel. Addition of 20% DMSO resulted in cell death (red), while PreS1-GFP-HepG2 target cells alone resulted in high viability (green). Addition of effector T cells (blue) resulted in different degrees of target cell killing between different effector groups. Scale bars, 100 μm.
Molecular Characterization of Base Editor Effects
The application of novel genome-editing tools necessitated further investigation of anticipated and unexpected molecular consequences of T cell engineering. There is an established experience of lentiviral mediated effects, including their propensity to integrate into transcriptionally active genes,50, 51, 52 and we did not re-examine these aspects. However, base conversion effects of coBE3 were characterized in depth, extending comparisons to the effects of SpCas9 disruption in similar experiments disrupting eTCR expression in T cells engineered to express a CAR against CD19. Both modalities had mediated high levels of TCRαβ disruption (means ± SEM: coBE3, 40.4% ± 5.4%; SpCas9, 52.7% ± 6%) (n = 4; Figure S3), but as anticipated, we found reduced indel frequencies following electroporation of coBE3 (11.9% ± 1.8%) compared to SpCas9 delivery (48.7% ± 6%).
In the context of rTCR delivery, direct sequencing of TRBC 1/2 in TCRαβ-depleted eTCR-/rTCR+ T cells was undertaken and analyzed using EditR, with cytosines at positions 5 and 6 distal to the PAM of particular interest (Figure 5A, i and ii). High levels of C > T conversion (G > A sense strand) were captured at these positions (37.3 ± 3.9% and 24.3 ± 2.2% at C5 and C6, respectively), with little activity at other nearby C residues (C1, 5 ± 1.2%; C2, 2.3 ± 1%; and C3, 4.3 ± 1.8%). Next-generation sequencing (NGS) revealed similar levels of C > T conversion at both positions C5 (40 ± 2.9%) and C6 (32.3 ± 3%) (Figure S4). Although mostly seamless, a minority of reads exhibited small (<10 bp; 8.4 ± 1.4%) or large (10–100 bp; 8.2 ± 0.7%) indel signatures (Figure 5Aiii), as others have noted previously.28, 29, 30
Figure 5.

Molecular Analysis of On-/Off-Target DNA Editing and Fidelity of CDR3α and β Regions within rTCR mRNA Transcripts
(A) Sanger sequencing of on-target editing at TRBC 1/2 loci in eTCR−/rTCR+ cells. (i) Representative EditR analysis with wild-type sequence (top) and four possible bases (side) shown at each position. Target G > A conversions (red box) generate a premature stop codon (Trp > ∗). (ii) Summary of EditR data for three donors at cytosine positions 5 and 6 distal to the PAM, presented as C > T changes (black), non-C > T changes (gray), and no editing (white). Error bars represent ±1 SEM. (iii) NGS analysis of on-target editing of TRBC 1/2 loci and quantification and characterization of indels after BE3 editing found only low levels of small (<10 bp, black) or large (10–100 bp, gray) indels, with the majority of reads presenting with no indels (white). Error bars represent ± 1 SEM. (B) Boxplots showing off-target editing detected by NGS analysis at the top 6 in silico predicted off-target sites for the TRBC 1/2 sgRNA, with comparison of unedited eTCR+/rTCR− and edited eTCR−/rTCR+ groups (n = 3). Larger dots represent outliers; in all cases, ⩽1.3% conversion. Two-tailed independent t test between unmodified (eTCR+/rTCR−) and edited (eTCR−/rTCR+) samples shown for donor 1 (p > 0.5), donor 2 (p = 0.001), and donor 3 (p > 0.1). (C) Serial examination of RNA from rTCR HBs183-91 for 8 days post-coBE3 mRNA delivery (days 5–12 post-activation) found no evidence of promiscuous deamination, with fidelity of CDR3α and CDR3β regions maintained. Amplicon positions are marked above for C residues, and schematic highlights hypervariable CDR3α and CDR3β regions that confer HLA-peptide specificity. CDR3 regions were mapped as a heatmap in R using the gplots library for C > T conversion rates at the marked sites (TCR Clone software: TCRmodel).53
In silico analysis of sgRNA binding and possible off-target activity was undertaken using the Benchling software platform and presented no exonic off-targets with <3 mismatches. Six genomic loci with the highest scores for off-target activity, all of which contained cytosine bases within the BE3 editing window, were interrogated directly by NGS in three different donors (Figure 5B). We found very low levels (<1%) of conversion activity at these sites, and only one intronic site exhibited C > T changes higher than in its respective non-edited control sample. Recent reports in cell lines have also suggested that promiscuous rAPOBEC1 RNA deamination (including by BE3) can arise following plasmid-mediated expression of base editors.54, 55, 56, 57 In the T cell context, and with coBE3 transiently expressed by mRNA electroporation, we investigated whether regions directing antigen receptor specificity might be affected. Analysis of RNA from T cells exposed to coBE3 focused on high-throughput interrogation of TCR hypervariable regions (TCRVα and TCRVβ CDR3 regions). Analysis of samples collected at serial time points, from 1–8 days after BE3 mRNA delivery, found no obvious evidence of aberrant deamination compared to controls (99%–100% cysteines unmodified), and intact sequence integrity of HBs183-91 rTCR was verified (Figure 5C). In addition, transcriptomic analysis on these samples detected anticipated effects of T cell activation and transduction over time (Figure S5). Thus, the first principal component (PC1) accounted for 76% of variance when comparing day 5 and day 12. As the second principal component (PC2) accounted for only 13% of variance, no major transcriptional changes between edited and non-edited cells were noted. In silico analysis had identified a further 24 unique sites of possible off-target BE activity in exonic regions. However, these were all found to have low transcriptional activity (averaged <100 reads) in both edited and non-edited T cells and, therefore, unlikely to be of importance.
Thus, while on-target deamination and creation of TCR-stop codons were highly efficient, there was no notable activity at sites of potential interest at either the DNA or RNA level for coBE3.
Discussion
T cell immunotherapy against conventional tumor-associated targets such as NY-ESO-1 are being widely investigated, and recent reports indicate that autologous T cells with additional CRISPR-Cas9 modifications designed to improve persistence and efficacy can be safely infused.24,58 Emerging base editor technologies offer the prospect of highly specific C > T (G > A) base conversion that can be harnessed to create seamless premature stop codons or modify splice sites to disrupt gene expression for advanced T cell engineering.
We previously reported the first therapeutic use of autologous T cells modified to express HBsAg-specific T cell receptors in a subject with chemoresistant, extrahepatic, metastatic disease. In that case, HBV antigens were detectable in HCC metastases but not in donor-derived liver (following cadaveric liver transplantation), thereby reducing the risk of T cell-mediated hepatitis. Gene-modified T cells survived, expanded, and mediated a reduction in HBsAg levels, and while efficacy was not established, there was no significant on- or off-target toxicity.2 A small number of additional subjects have been treated subsequently, although the approach remains highly patient tailored, and extending to larger numbers of patients is logistically challenging and costly. Similar hurdles are being addressed in the arena of hematological malignancies through the generation of “universal” T cells expressing CARs from non-HLA-matched healthy donors. As such, depletion of endogenous TCR and other antigens by genome editing has allowed HLA barriers to be circumvented, and ongoing trials suggest that such universal CAR T cells can expand and persist sufficiently to induce molecular remission.25 The editing tools applied in clinic have included TALENS and CRISPR-Cas9, and they rely on targeted DNA cleavage and repair by non-homologous end joining (NHEJ), which results in the creation of indels leading to gene disruption. Application of CRISPR-guided base conversion to create stop codons or alter the critical splice site to disrupt gene expression offers the possibility of seamless gene disruption with greatly reduced likelihood of translocations or toxicity. We report the application of APOBEC deaminase technology for the generation of engineered T cells, which are then rendered devoid of endogenous TCRs and uniformly express rTCR specific for an epitope of HBsAg. The resulting product was homogeneous and exhibited enhanced rTCR intensity, greater levels of cytokine production, and antigen-specific functional integrity in models of HCC elimination. An ability to discriminate, selectively process, and deplete eTCR T cells while rTCR populations are untouched provides critical advantages, especially for strategies when allogeneic donor cells bearing potentially alloreactive eTCRs can be eliminated. Non-human protein sequences within constructs have the potential to be immunogenic, although murine TCR constant regions are considered unlikely determinants in the generation of human anti-mouse antibodies.59 Likewise, the BE configurations use bacterial and rodent-derived elements, but expression is transient during ex vivo culture and unlikely to be problematic in vivo.
The rapid development of tools enabling highly targeted base conversion through deamination effects promises tantalizing opportunities, although the in-depth characterization of desirable and unwanted effects in subsequent therapeutic applications has to be mapped. Existing CRISPR-Cas base editors using rAPOBEC1 (including coBE3) are known to mediate off-target DNA edits and transcriptome-wide RNA deamination in both protein-coding and non-coding regions.54, 55, 56 While these could be problematic, newer variants with more precise DNA restricted editing are already in development and should continue to evolve as ever more efficient, specific, and non-toxic editing tools. Our analysis of possible off-target sgRNA activity in three donors found minimal base conversion effects at predicted DNA sites. Importantly, examination of RNA detected no major differences in gene expression levels between base edited and non-edited T cells, with only very minor perturbations and C > U conversions of the CDR3 variable regions, no greater than in control cells. Such changes could otherwise redirect the specificity of the introduced TCR and would risk causing autoimmunity or off-target T cell effects.
Conclusions
Removal of eTCR enhances the expression of introduced rTCR, reduces the risk of aberrant cross-pairing, and allows discriminatory enrichment of engineered T cells. The strategy also opens the door to generating “universal” allogeneic T cells from healthy HLA-mismatched donors by reducing the risk of graft versus host disease. In the case of the rTCR specific for HBs183-91, blood from healthy HLA-A201 donors could readily be further edited to disrupt mismatched HLA molecules, creating immunologically stealthy cells. Additional multiplexed editing of T cell exhaustion markers may promote enhanced persistence and anti-tumor effects. Ultimately, pre-manufactured banks of eTCR−/rTCR+ T cells specific for groups of dominant HLA/peptide combinations could provide treatment options for large numbers of subjects.
Materials and Methods
CRISPR Guide RNA
Guide sequences compatible with coBE3 targeting homologous sequences in TRBC1 and -2 were designed using the CRISPR design tool Benchling (https://www.benchling.com) and provided an on-target editing score for predicted activity at each cytosine around the editing window;28 TRBC1/2: C0.8C11C5.7AC21.9 C21.4AGCUCAGCUCCACG (anti-sense, numbers indicate predicted editing scores for the specific cytosine base). Predicted exonic off-target binding required at least 3 mismatches within the protospacer.
Lentiviral Construct for rTCR and sgRNA Delivery
Lentiviral design for coupled transgene and guide RNA expression has been previously described.48 Briefly, rTCR HLA-A0201/HBs183-91 was cloned under the control of an internal human phosphoglycerate kinase 1 (hPGK) promoter, and a CRISPR guide expression cassette was embedded in the lentiviral 3′ LTR. This comprised a 5′ RNA polymerase III promoter (U6) and a sgRNA specific for TCRB1/2 with a 5′ G for improved transcription. Vector stocks were produced in 293T cells by transient transfection with third-generation packaging plasmids and concentrated by ultracentrifugation prior to storage at −80°C.
Primary Human Lymphocyte Culture and Modification
Blood was obtained from consented healthy volunteers compliat with ethic (REC number 0141/001). Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient and subsequently activated with TransAct Reagent (130-111-160, Miltenyi Biotec) at 10 μL/mL. TexMACS medium (130-097-196, Miltenyi Biotec) with 3% human AB serum (GEM-100-512-HI, Seralabs) and 100 U/mL IL-2 was used for all lymphocyte cell culture. Transduction with lentiviral vector was performed 24 h post-activation at a multiplicity of infection (MOI) of 5. Electroporation of coBE3 mRNA was performed at day 4 post-activation, after which cells were cultured in a G-Rex 10 (P/N 80040S, Wilsonwolf). Lymphocytes were cultured for 11 days post-activation and magnetically depleted using anti-TCR α/β-biotin (130-098-219, Miltenyi Biotec) followed by incubation with Anti-Biotin Microbeads Ultrapure (130-105-637, Miltenyi Biotec) and separation through LD Columns (130-042-901, Miltenyi Biotec). Cells were rested overnight before flow-cytometry-based phenotyping and cryopreservation.
Phenotyping Flow Cytometry
Flow cytometry was performed on a 4-laser BD LSRII (BD Biosciences), with subsequent analysis executed using FlowJo v.10 (TreeStar). Cells were stained according to the manufacturer’s instructions with Mouse TCR β constant-APC (clone H57-597, BioLegend, catalog no. 109211), Human TCRα/β PerCP-Vio 700 (clone REA652, Miltenyi Biotec, catalog no. 130-113-540), PD1-PE (clone PD1.3.1.3, Miltenyi Biotec, catalog no. 130-117-384), CD4-VioBlue (clone REA623, Miltenyi Biotec, catalog no. 130-114-534), and CD45-VioGreen (clone REA747, Miltenyi Biotec, catalog no. 130-110-638).
Antigen-Specific Responses
Target HepG2 cells were pulsed with HBV surface envelope peptide S183-91 (FLLTRILTI, JPT Peptide Technologies) and irrelevant control HBV core peptide C18-27 (FLPSDFFPSV, JPT Peptide Technologies) at gradient concentrations for 1 h at 37°C. Cryopreserved effector T cells (eTCR+/rTCR−, eTCR+/rTCR±, and eTCR-/rTCR+) were thawed and cultured at an E:T ratio of 1:1, and 0.1 μg/mL Brefeldin A (Sigma) was added before overnight co-culture. A BD LSR Fortessa X20 flow cytometer (BD Biosciences) was used for cell acquisition, with FlowJo v10 (TreeStar) used to analyze phenotype and function of effector T cell groups. Phenotyping included intracellular staining with TNF-α FITC (clone MAb11, BD Biosciences, catalog no. 502906), MIP-1b PE (clone D21-1351, BD Biosciences, catalog no. 550078), IL-2 PerCP-eFluor 710 (clone MQ1-17H12, eBioscience, catalog no. 46-7029-42), GranzymeB AF700 (clone GB11, BD Biosciences, catalog no. 560213), IFNγ V450 (clone B27, BD Biosciences, catalog no. 560371), and surface staining with CD3 BUV395 (clone UCHT1, BD Biosciences, catalog no. 563546) and mouse TCR β constant-APC (clone H57-597, BioLegend, catalog no. 109211).
Electroporation of Base Editor mRNA
The BE3 amino-acid sequence was sourced from previously published work containing a single C terminus nuclear localization signal.28 Additionally, the DNA sequence has been codon optimized by GeneArt (Thermo Fisher Scientific). coBE3 mRNA was produced by TriLink and was clean-capped (Cap 1), polyadenylated, and purified by high-performance liquid chromatography (HPLC). Electroporation used a 100-μL tip kit and the Neon Transfection System (Thermo Fisher Scientific). Cells were electroporated at 20 × 106 cells per milliliter in buffer T, using protocol 24 (1,600 V, 10 ms, 3 pulses) with 50 μg/mL coBE3 mRNA.
Following electroporation, T cells were incubated overnight at 30°C before restoration to 37°C.
Molecular Characterization of On-Target DNA Editing
Genomic DNA extraction was performed using the DNeasy Blood and Tissue Kit (69504, QIAGEN) and PCR sequencing was undertaken using primers for TRBC1/2 loci: TRBC forward, 5′-AGGTCGCTGTGTTTGAGC-3′; and TRBC reverse, 5′-CTATCCTGGGTCCACTCGTC-3′. Sanger sequencing data (Eurofins Genomics) was analyzed using EditR (https://moriaritylab.shinyapps.io/editr_v10/).60 In addition, amplified products were library prepped for NGS using a Nextera XT Kit (Illumina, Cambridge, UK). After the library preparation, individually barcoded samples were pooled and run in MiSeq using a 500-V2 nano-cartridge. Demultiplexed fastq files were uploaded to Galaxy61 for trimming and alignment. NHEJ signatures were analyzed using Pindel,62 and haplotypes were analyzed using Freebayes.63 Figures were created in R.
Molecular Characterization of Off-Target DNA Editing
The online software Benchling was used to predict off-targets for the TRBC guide. Libraries were prepared on the top six off-targets using the same methodology as described earlier (NGS for on-target DNA editing) and combinations of target-specific primers (Supplemental Materials and Methods).
Characterization and Analysis of the Transcriptome
Sequential RNA samples from engineered T cells were prepared for RNA sequencing (RNA-seq) using the KAPA mRNA HyperPrep Kit (Roche) at UCL Genomics. Initial analysis was performed on a customized Galaxy workflow followed by transcriptomics analysis on iDEP 9.1 (workflow using R packages). The online software tool CRISPR RGEN “Cas-OFFinder” predicted 1,071 off-target sites for the TRBC guide, with parameters set for up to 3 mismatches and a 1-nt bulge). A pipeline was developed for further investigation of these sites in RNA-seq data (Supplemental Materials and Methods).
Screening for rTCR RNA Editing Effects
Total RNA was extracted using a QIAamp RNA Blood Mini Kit (QIAGEN, 52304) for TCR library preparation and sequencing as previously described.64,65 rTCR RNA was reverse transcribed using a murine TRBC-specific primer (5′-TGGACTTCTTTGCCGTTGAC-3′). Following ligation of an oligonucleotide containing the Illumina SP2 primer and unique molecular identifiers, products were amplified using primers specific to the murine constant α and β chains (5′-CGTTGATCTGGCTGTCGAAG-3′ and 5′-TTGACCCACCAAGACAGCTC-3′, respectively). Finally, libraries were built in two further steps of amplification during which the SP1 sequencing primer, indices, and Illumina adaptors were added. Part of the primers used in these were also specific for the constant regions (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNGCCAATGCACGTTGATCTGGCTGTCGAAG-3′ and 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNGCCAATCCGTTGACCCACCAAGACAGCTC-3′). The final purified libraries were verified using TapeStation (Agilent Technologies) and Qubit (Thermo Fisher Scientific) and were multiplexed and sequenced on a MiSeq system (Illumina) using 500-V2 cartridges (Illumina). Fastq files were demultiplexed using Demultiplexor (https://github.com/innate2adaptive/Decombinator).66 Using Galaxy tools,61 the demultiplexed fastq files were trimmed (Trim Galore and Trimmomatic) and aligned (Bowtie2) to the relative TCR HBV gene map. Aligned files were interrogated for the frequency of the reference sequence per base around the complementarity-determining region 3 (CDR3) (100-bp total window).
Data Availability
All fastq files will be available on the NCBI Sequence Read Archive upon publication of this article (BioProject: PRJNA637371).
xCELLigence Impedance Assay
Target HepG2 cells were seeded (1 × 105 per well) in the dedicated device (E-Plate VIEW 16, ACEA Biosciences) and cultured for 24 h. Impedance measurement was acquired with an interval of 15 min by an array of electrodes located at the bottom of the plate. Different T cell preparations and E:T ratios were added in the well after 24 h, and the impedance signal was recorded for the subsequent 72 h. Three different donors were tested in triplicate conditions.
3D Microfluidics Device
Briefly, dissociated PreS1-GFP-HepG2 target cells were mixed with Collagen Type I gel (Rat Tail, Corning) and injected into the dedicated region of the 3D cell-culture chip (DAX-1, AIM Biotech), before gel polymerization, following a previously developed protocol.38,43,49 R10 media with 3 μM DRAQ7 (BioLegend) cell-impermeable nuclear dye was then added to the media channels to hydrate the gel, and chips were incubated at 37°C. T cells were stained with 3 μM CellTracker Violet BMQC (Thermo Fisher Scientific) and were injected into one of two media channels flanking the gel region before overnight incubation. 3D confocal images were acquired daily with a high-content imaging system (Phenix, PerkinElmer). T cells from the liquid channel were collected by manual pipetting; then, collagenase solution was injected into the device to retrieve the immune cells migrating in the hydrogel region for flow cytometry analysis on a 4-laser BD LSRII (BD Biosciences).
Statistics
Statistical analysis was performed using GraphPad Prism software, v.8.0.0.
Author Contributions
W.Q. developed the concept, supervised the project, designed experiments, and reviewed the manuscript. R.P. designed and performed experiments, analysed data, and wrote the manuscript. A.P., Z.M., J.Y.J., S.A.G., and K.A.S. designed and performed experiments, analysing data, and reviewed the manuscript. C.G. was involved in experimental design, and reviewing the manuscript. A.P., A.B., and M.K.M. supervised scientist performing experiments and reviewed the manuscript.
Conflicts of Interests
W.Q. holds interests unrelated to this project in Autolus. W.Q. received unrelated research funding from Cellectis, Servier, Miltenyi Biotec, and Bellicum. The other authors declare no competing interests.
Acknowledgments
This work is supported by the NIHR Blood and Transplant Research Units (RP-2014-05-007), the Great Ormond Street Biomedical Research Centre (IS-BRC-1215-20012), Wellcome Trust (215619/Z/19/Z), and the National Research Foundation Singapore (NRF-CRP17-2017-06).
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, Wellcome Trust, or the Department of Health.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.09.002.
Supplemental Information
References
- 1.Morgan R.A., Dudley M.E., Wunderlich J.R., Hughes M.S., Yang J.C., Sherry R.M., Royal R.E., Topalian S.L., Kammula U.S., Restifo N.P. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qasim W., Brunetto M., Gehring A.J., Xue S.A., Schurich A., Khakpoor A., Zhan H., Ciccorossi P., Gilmour K., Cavallone D. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015;62:486–491. doi: 10.1016/j.jhep.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Chapuis A.G., Egan D.N., Bar M., Schmitt T.M., McAfee M.S., Paulson K.G., Voillet V., Gottardo R., Ragnarsson G.B., Bleakley M. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019;25:1064–1072. doi: 10.1038/s41591-019-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron B.J., Gerry A.B., Dukes J., Harper J.V., Kannan V., Bianchi F.C., Grand F., Brewer J.E., Gupta M., Plesa G. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendle G.M., Linnemann C., Hooijkaas A.I., Bies L., de Witte M.A., Jorritsma A., Kaiser A.D.M., Pouw N., Debets R., Kieback E. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010;16:565–570. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 7.van Loenen M.M., de Boer R., Amir A.L., Hagedoorn R.S., Volbeda G.L., Willemze R., van Rood J.J., Falkenburg J.H., Heemskerk M.H. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA. 2010;107:10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen C.J., Zhao Y., Zheng Z., Rosenberg S.A., Morgan R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y., Moysey R., Molloy P.E., Vuidepot A.L., Mahon T., Baston E., Dunn S., Liddy N., Jacob J., Jakobsen B.K., Boulter J.M. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 10.Kuball J., Dossett M.L., Wolfl M., Ho W.Y., Voss R.H., Fowler C., Greenberg P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bentzen A.K., Such L., Jensen K.K., Marquard A.M., Jessen L.E., Miller N.J., Church C.D., Lyngaa R., Koelle D.M., Becker J.C. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4303. Published online November 19, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadi M., King J.W., Xue S.A., Voisine C., Holler A., Wright G.P., Waxman J., Morris E., Stauss H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood. 2011;118:3528–3537. doi: 10.1182/blood-2011-04-346338. [DOI] [PubMed] [Google Scholar]
- 13.Bunse M., Bendle G.M., Linnemann C., Bies L., Schulz S., Schumacher T.N., Uckert W. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol. Ther. 2014;22:1983–1991. doi: 10.1038/mt.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provasi E., Genovese P., Lombardo A., Magnani Z., Liu P.Q., Reik A., Chu V., Paschon D.E., Zhang L., Kuball J. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berdien B., Mock U., Atanackovic D., Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014;21:539–548. doi: 10.1038/gt.2014.26. [DOI] [PubMed] [Google Scholar]
- 16.Legut M., Dolton G., Mian A.A., Ottmann O.G., Sewell A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. doi: 10.1182/blood-2017-05-787598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth T.L., Puig-Saus C., Yu R., Shifrut E., Carnevale J., Li P.J., Hiatt J., Saco J., Krystofinski P., Li H. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559:405–409. doi: 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schober K., Müller T.R., Gökmen F., Grassmann S., Effenberger M., Poltorak M., Stemberger C., Schumann K., Roth T.L., Marson A., Busch D.H. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019;3:974–984. doi: 10.1038/s41551-019-0409-0. [DOI] [PubMed] [Google Scholar]
- 19.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 20.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 21.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 22.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 23.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 26.Billon P., Bryant E.E., Joseph S.A., Nambiar T.S., Hayward S.B., Rothstein R., Ciccia A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell. 2017;67:1068–1079 e1064. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuscu C., Parlak M., Tufan T., Yang J., Szlachta K., Wei X., Mammadov R., Adli M. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods. 2017;14:710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 28.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komor A.C., Zhao K.T., Packer M.S., Gaudelli N.M., Waterbury A.L., Koblan L.W., Kim Y.B., Badran A.H., Liu D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017;3:eaao4774. doi: 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webber B.R., Lonetree C.L., Kluesner M.G., Johnson M.J., Pomeroy E.J., Diers M.D., Lahr W.S., Draper G.M., Slipek N.J., Smeester B.A. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019;10:5222. doi: 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sung W.K., Zheng H., Li S., Chen R., Liu X., Li Y., Lee N.P., Lee W.H., Ariyaratne P.N., Tennakoon C. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 32.Amaddeo G., Cao Q., Ladeiro Y., Imbeaud S., Nault J.C., Jaoui D., Gaston Mathe Y., Laurent C., Laurent A., Bioulac-Sage P. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820–829. doi: 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brechot C., Pourcel C., Louise A., Rain B., Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–535. doi: 10.1038/286533a0. [DOI] [PubMed] [Google Scholar]
- 34.Edman J.C., Gray P., Valenzuela P., Rall L.B., Rutter W.J. Integration of hepatitis B virus sequences and their expression in a human hepatoma cell. Nature. 1980;286:535–538. doi: 10.1038/286535a0. [DOI] [PubMed] [Google Scholar]
- 35.El-Serag H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 36.Chen X.P., Long X., Jia W.L., Wu H.J., Zhao J., Liang H.F., Laurence A., Zhu J., Dong D., Chen Y. Viral integration drives multifocal HCC during the occult HBV infection. J. Exp. Clin. Cancer Res. 2019;38:261. doi: 10.1186/s13046-019-1273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye B., Liu X., Li X., Kong H., Tian L., Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;6:e1694. doi: 10.1038/cddis.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otano I., Escors D., Schurich A., Singh H., Robertson F., Davidson B.R., Fusai G., Vargas F.A., Tan Z.M.D., Aw J.Y.J. Molecular Recalibration of PD-1+ Antigen-Specific T Cells from Blood and Liver. Mol. Ther. 2018;26:2553–2566. doi: 10.1016/j.ymthe.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuch A., Salimi Alizei E., Heim K., Wieland D., Kiraithe M.M., Kemming J., Llewellyn-Lacey S., Sogukpinar Ö., Ni Y., Urban S. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut. 2019;68:905–915. doi: 10.1136/gutjnl-2018-316641. [DOI] [PubMed] [Google Scholar]
- 40.Sastry K.S., Too C.T., Kaur K., Gehring A.J., Low L., Javiad A., Pollicino T., Li L., Kennedy P.T., Lopatin U. Targeting hepatitis B virus-infected cells with a T-cell receptor-like antibody. J. Virol. 2011;85:1935–1942. doi: 10.1128/JVI.01990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koh S., Shimasaki N., Suwanarusk R., Ho Z.Z., Chia A., Banu N., Howland S.W., Ong A.S., Gehring A.J., Stauss H. A practical approach to immunotherapy of hepatocellular carcinoma using T cells redirected against hepatitis B virus. Mol. Ther. Nucleic Acids. 2013;2:e114. doi: 10.1038/mtna.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertoletti A., Brunetto M., Maini M.K., Bonino F., Qasim W., Stauss H. T cell receptor-therapy in HBV-related hepatocellularcarcinoma. OncoImmunology. 2015;4:e1008354. doi: 10.1080/2162402X.2015.1008354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavesi A., Tan A.T., Koh S., Chia A., Colombo M., Antonecchia E., Miccolis C., Ceccarello E., Adriani G., Raimondi M.T. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight. 2017;2:e89762. doi: 10.1172/jci.insight.89762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kah J., Koh S., Volz T., Ceccarello E., Allweiss L., Lütgehetmann M., Bertoletti A., Dandri M. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J. Clin. Invest. 2017;127:3177–3188. doi: 10.1172/JCI93024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qasim W., Amrolia P.J., Samarasinghe S., Ghorashian S., Zhan H., Stafford S., Butler K., Ahsan G., Gilmour K., Adams S. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood. 2015;126(23):2046. [Google Scholar]
- 46.Tan A.T., Yang N., Krishnamoorthy T.L., Oei V., Chua A., Zhao X., Tan H.S., Chia A., Le Bert N., Low D. Use of Expression Profiles of HBV-DNA Integrated Into Genomes of Hepatocellular Carcinoma Cells to Select T Cells for Immunotherapy. Gastroenterology. 2019;156:1862–1876.e9. doi: 10.1053/j.gastro.2019.01.251. [DOI] [PubMed] [Google Scholar]
- 47.Gehring A.J., Xue S.A., Ho Z.Z., Teoh D., Ruedl C., Chia A., Koh S., Lim S.G., Maini M.K., Stauss H., Bertoletti A. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011;55:103–110. doi: 10.1016/j.jhep.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 48.Georgiadis C., Preece R., Nickolay L., Etuk A., Petrova A., Ladon D., Danyi A., Humphryes-Kirilov N., Ajetunmobi A., Kim D. Long Terminal Repeat CRISPR-CAR-Coupled “Universal” T Cells Mediate Potent Anti-leukemic Effects. Mol. Ther. 2018;26:1215–1227. doi: 10.1016/j.ymthe.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee H., Kim J.S. Unexpected CRISPR on-target effects. Nat. Biotechnol. 2018;36:703–704. doi: 10.1038/nbt.4207. [DOI] [PubMed] [Google Scholar]
- 50.Bushman F., Lewinski M., Ciuffi A., Barr S., Leipzig J., Hannenhalli S., Hoffmann C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005;3:848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- 51.Wang G.P., Ciuffi A., Leipzig J., Berry C.C., Bushman F.D. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cattoglio C., Maruggi G., Bartholomae C., Malani N., Pellin D., Cocchiarella F., Magnani Z., Ciceri F., Ambrosi A., von Kalle C. High-definition mapping of retroviral integration sites defines the fate of allogeneic T cells after donor lymphocyte infusion. PLoS ONE. 2010;5:e15688. doi: 10.1371/journal.pone.0015688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gowthaman R., Pierce B.G. TCRmodel: high resolution modeling of T cell receptors from sequence. Nucleic Acids Res. 2018;46(W1):W396–W401. doi: 10.1093/nar/gky432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grünewald J., Zhou R., Iyer S., Lareau C.A., Garcia S.P., Aryee M.J., Joung J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019;37:1041–1048. doi: 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 57.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 58.Lu Y., Xue J., Deng T., Zhou X., Yu K., Deng L., Huang M., Yi X., Liang M., Wang Y. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020;26:732–740. doi: 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 59.Davis J.L., Theoret M.R., Zheng Z., Lamers C.H., Rosenberg S.A., Morgan R.A. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin. Cancer Res. 2010;16:5852–5861. doi: 10.1158/1078-0432.CCR-10-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012 arXiv:1207.3907. [Google Scholar]
- 64.Oakes T., Heather J.M., Best K., Byng-Maddick R., Husovsky C., Ismail M., Joshi K., Maxwell G., Noursadeghi M., Riddell N. Quantitative Characterization of the T Cell Receptor Repertoire of Naïve and Memory Subsets Using an Integrated Experimental and Computational Pipeline Which Is Robust, Economical, and Versatile. Front. Immunol. 2017;8:1267. doi: 10.3389/fimmu.2017.01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gkazi A.S., Margetts B.K., Attenborough T., Mhaldien L., Standing J.F., Oakes T., Heather J.M., Booth J., Pasquet M., Chiesa R. Clinical T Cell Receptor Repertoire Deep Sequencing and Analysis: An Application to Monitor Immune Reconstitution Following Cord Blood Transplantation. Front. Immunol. 2018;9:2547. doi: 10.3389/fimmu.2018.02547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thomas N., Heather J., Ndifon W., Shawe-Taylor J., Chain B. Decombinator: a tool for fast, efficient gene assignment in T-cell receptor sequences using a finite state machine. Bioinformatics. 2013;29:542–550. doi: 10.1093/bioinformatics/btt004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All fastq files will be available on the NCBI Sequence Read Archive upon publication of this article (BioProject: PRJNA637371).