

Abstract
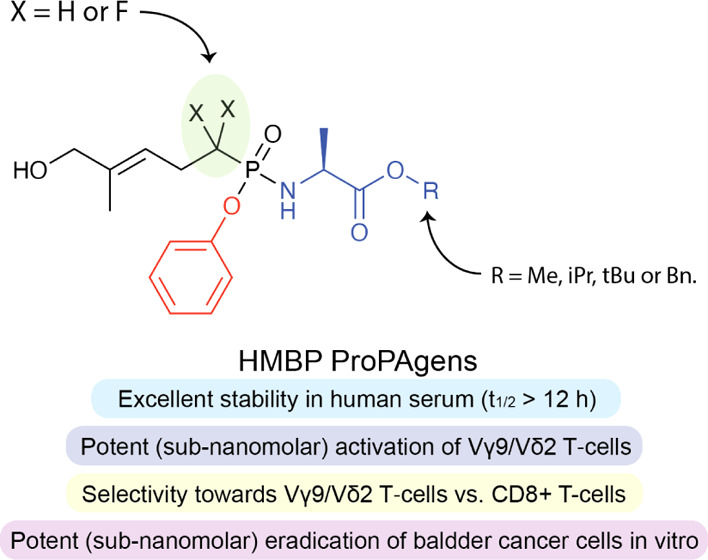
Vγ9/Vδ2 T-cells are activated by pyrophosphate-containing small molecules known as phosphoantigens (PAgs). The presence of the pyrophosphate group in these PAgs has limited their drug-like properties because of its instability and polar nature. In this work, we report a novel and short Grubbs olefin metathesis-mediated synthesis of methylene and difluoromethylene monophosphonate derivatives of the PAg (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBP) as well as their aryloxy diester phosphonamidate prodrugs, termed ProPAgens. These prodrugs showed excellent stability in human serum (t1/2 > 12 h) and potent activation of Vγ9/Vδ2 T-cells (EC50 ranging from 5 fM to 73 nM), which translated into sub-nanomolar γδ T-cell-mediated eradication of bladder cancer cells in vitro. Additionally, a combination of in silico and in vitro enzymatic assays demonstrated the metabolism of these phosphonamidates to release the unmasked PAg monophosphonate species. Collectively, this work establishes HMBP monophosphonate ProPAgens as ideal candidates for further investigation as novel cancer immunotherapeutic agents.
Introduction
Vγ9/Vδ2 T-cells are the dominant subtype of human γδ T-cells in adult peripheral blood.1 They are involved in immune responses to many diseases such as tuberculosis, leprosy, typhoid, malaria, and toxoplasmosis.1 Studies in primate models have also implicated Vγ9/Vδ2 T-cells in immunity to Mycobacterium tuberculosis.2 These cells have been shown to target and lyse a diverse range of cancer cells in vitro.1 Together, these observations have made the Vγ9/Vδ2 subset a major focus of investigation for the therapeutic exploitation of γδ T-cells.3
To date, a handful of small molecule activators of Vγ9/Vδ2 T-cells have been reported. Among these are the two aminobisphosphonate drugs, risedronate and zoledronate (Figure 1), which are currently used clinically to treat osteoporosis and some types of cancer.4−6 These agents inhibit isopentenyl pyrophosphate (IPP) catabolism via farnesyl diphosphate (FPP) synthase, leading to intracellular IPP accumulation, which ultimately results in Vγ9/Vδ2 T-cell activation.7−9 Beyond these two compounds, the microbially derived phosphoantigen (PAg) (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP) and the host-derived PAg IPP itself are both established Vγ9/Vδ2 T-cell activators (Figure 1).10,11 These are known to activate Vγ9/Vδ2 T-cells by binding the type-1 transmembrane protein butyrophilin 3A1 (BTN3A1) that is expressed on target cells.12 Although the binding site for these PAgs initially remained unclear amid conflicting reports as to whether they bind the extracellular or intracellular domains of this transmembrane protein, there is now compelling evidence to support the notion that HMBPP binds the intracellular B30.2 domain of butyrophilin 3A1, triggering a conformational change.12−16 Although the molecular events subsequent to this are unclear, PAg-mediated activation is dependent upon target cells co-expressing BTN3A1 alongside a second family member, butyrophilin 2A1 (BTN2A1), with which it associates and which is able to directly ligate the Vγ9Vδ2 T-cell receptor (TCR).17,18 One plausible sensing mechanism involves PAg exposure regulating BTN3A1-mediated recruitment of an additional molecule18 that in combination with BTN2A1, forms a “composite ligand” for the Vγ9Vδ2 TCR complex, thereby triggering activation.18
Figure 1.

Chemical structure of reported small molecule Vγ9/Vδ2 T-cell activators; HMBPP (EC50: 60–500 pM),19−21 IPP (EC50: 1–10 μM),22,23 risedronate (EC50: 0.08–5 μM),24,25 and zoledronate (EC50: 0.003–0.5 μM) [this work and Davey et al.(20)]
Encouraged by the potency of the PAg HMBPP in activating Vγ9/Vδ2 T-cells, EC50 = 60–500 pM,19−21 and given our interest in developing phosphate- and phosphonate-containing drugs,26−28 we previously reported the application of McGuigan’s phosphoramidate prodrug technology29,30 to the monophosphate derivative of HMBPP, that is, (E)-4-hydroxybut-2-enyl phosphate (HMBP), as a means of improving its drug-like properties.29 In this phosphoramidate prodrug approach, the monophosphate or monophosphonate groups are masked by an aryl group and an amino acid ester (Figure 2), which are both enzymatically cleaved off inside cells to release the monophosphate or monophosphonate species.29 As these are prodrugs of PAgs, we previously20 termed them ProPAgens to distinguish them from ProTides,29prodrugs of nucleotides. Although the HMBP ProPAgens that we reported previously20 exhibited potent activation of Vγ9/Vδ2 T-cells (EC50 = 0.45–10.62 nM), their stability was poor (t1/2 < 30 min).20 The observed instability was shown to be because of the cleavage of the −P–O– bond of these compounds.20 To circumvent this problem, we herein report our application of McGuigan’s phosphoramidate prodrug technology to HMBP methylene and difluoromethylene monophosphonates to generate their aryloxy diester phosphonamidate prodrugs (Figure 2).
Figure 2.
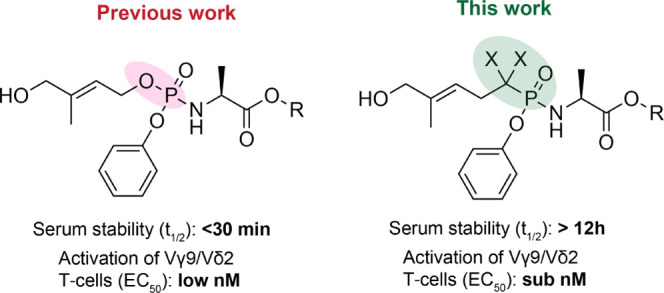
Chemical structures of HMBP ProPAgens (previous work) and the ProPAgens of their phosphonate derivatives (this work) as well as a general indication of their stability and Vγ9/Vδ2 T-cell activation.
Results and Discussion
Design and Synthesis of HMBP Monophosphonate ProPAgens
In the design of HMBP monophosphonates, we switched the liable −O–PO(OH)2 to a more stable −CH2–PO(OH)2 or −CF2–PO(OH)2 bond. The introduction of a phosphonate bond (−CH2–PO(OH)2) instead of a phosphate one (−O–PO(OH)2) is quite common in drug discovery and has been used with success in addressing stability issues observed with phosphate-containing compounds.31 A prime example of this are the antiviral drugs cidofovir and adefovir.31 Although the change from a −O–PO(OH)2 bond to a −CH2–PO(OH)2 bond achieves better stability, the pKa value of the second deprotonation of the phosphonate group (pKa = 7.49) is significantly different from that of the phosphate group (pKa = 6.31) (Figure 3).32 As a result, this affects their full ionization under physiological pH (<7.4) and, hence, their binding to the target protein, which optimally requires the full ionization of the phosphate group to bind a positively charged pocket (arginine-rich) on the BTN3A1 intracellular B30.2 domain.16 To address this, switching the phosphate (−O–PO(OH)2) bond to a difluoromethylene phosphonate (−CF2–PO(OH)2) has emerged as a better substitution because these have excellent stability in physiological environments coupled with a pKa value for the second deprotonation (pKa = 5.5) as acidic as that of the native phosphate (Figure 3).32 Therefore, under physiological pH, the −CF2–PO(OH)2 will be fully deprotonated akin to natural phosphate groups −O–PO(OH)2, and this ensures similar binding to that achieved by the natural phosphate group.
Figure 3.

pKa values of phosphate and different phosphonate groups.
With this in mind, we synthesized the ProPAgens of both HMBP methylene and difluoromethylene monophosphonates (4a–d and 9a–d, Scheme 1). Previously, the synthesis of HMBP methylene monophosphonate prodrugs was achieved in five steps starting from dimethyl homoprenylphosphonate,33 which itself has to be prepared in two steps from the commercially available 3-methylbut-2-en-1-ol.19 Some of these reactions are low yielding such as the oxidation of the side chain to introduce HMBP’s side chain, the hydroxyl group.33 Notably, to date, no synthesis of HMBP difluoromethylene monophosphonate prodrugs has been reported.
Scheme 1.
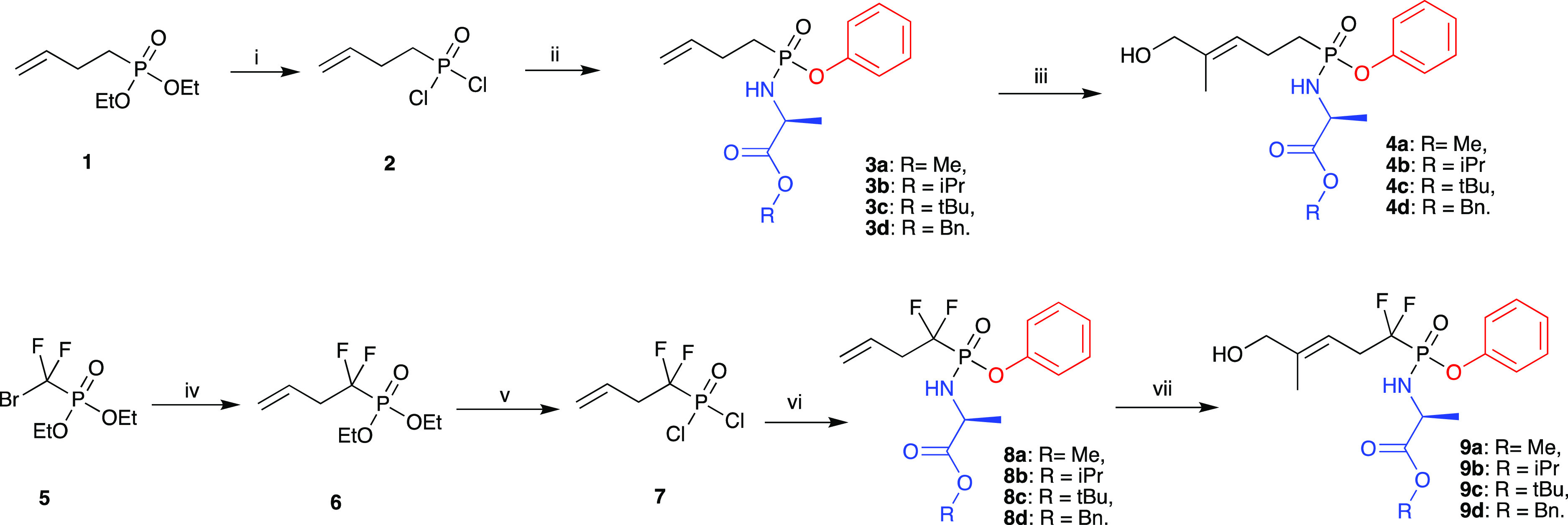
Reagents and conditions: (i) TMSBr, DCM, rt, 2 h then (COCl)2, DMF cat, DCM, rt, 18 h; (ii) a. Phenol, Et3N, DCM, −78 °C for 30 min then rt, 3 h; b. Substituted l-alanine ester hydrochloride, Et3N, DCM, rt, 12 h, yields: 38–61%; (iii) 2-methyl-2-propenol, 1,4-benzoquinone, Hoveyda–Grubbs catalyst 2nd generation, DCM, rt, yields: 57–64%; (iv) diethyl (bromodifluoromethyl)phosphonate, DMF, zinc powder, rt, N2, 3 h then CuBr, allyl bromide, rt, 40 h; (v) (i) TMSBr, DCM, rt, 2 h then (COCl)2, DMF cat, DCM, rt, 18 h; (ii) a. Phenol, Et3N, DCM, −78 °C for 30 min then rt, 3 h; b. Substituted l-alanine ester hydrochloride, Et3N, DCM, rt, 12 h, yields: 24–46%; and (iii) 2-methyl-2-propenol, 1,4-benzoquinone, Hoveyda–Grubbs Catalyst 2nd generation, DCM, rt, yields: 58–69%.
In order to accomplish the synthesis of HMBP methylene and difluoromethylene monophosphonate prodrugs (4a–d and 9a–d, Scheme 1), we employed the Grubbs olefin metathesis34 in the construction of these prodrugs. Using commercially available starting materials, our new synthetic approach allowed the rapid and efficient synthesis of the PAg ProPAgens in three steps for HMBP methylene monophosphonate prodrugs (4a–d) and four steps for HMBP difluoromethylene monophosphonate prodrugs (9a–d) as shown in Scheme 1.
The synthesis of ProPAgens 4a–d started by treating the commercially available diethyl 3-butenylphosphonate (1) with bromotrimethylsilane (TMSBr) at room temperature to remove the ethoxy groups and generate the phosphonic acid derivative.35 This was followed by a chlorination reaction using oxalyl chloride in the presence of a catalytic amount of DMF. The product of this reaction, 2, was subsequently treated with phenol in the presence of triethylamine and then with the appropriate amino acid ester to yield phosphonamidates 3a–d in moderate to good yields (38–61%). Next, these compounds underwent Grubbs olefin metathesis with 2-methyl-2-propenol employing the Hoveyda–Grubbs second generation catalyst in the presence of 1,4-benzoquinone to prevent alkene isomerization.36 This gave ProPAgens 4a–d in good yields (57–64%).
For the synthesis of ProPAgens 9a–d, the commercially available diethyl (bromodifluoromethyl)phosphonate 5 was reacted with allyl bromide in the presence of zinc and copper bromide in DMF as reported.37 The generated compound, 6, was subsequently chlorinated and then treated with phenol and the appropriate amino acid ester to yield phosphonamidates 8a–d in moderate yields (24–46%) as described for the synthesis of compounds 3a–d above. Subsequently, these phosphonamidates were treated with 2-methyl-2-propenol in the presence of Hoveyda–Grubbs second generation catalyst and catalytic amounts of 1,4-benzoquinone.36 The final ProPAgens 9a–d were generated in good yields (58–69%).
In terms of the design of the prodrugs, l-alanine was used as the amino acid of choice in the synthesis of these ProPAgens because it has historically shown optimum biological activity,30 while the phenol motif was chosen as it has been used successfully in the discovery of two FDA-approved drugs: sofosbuvir and tenofovir alafenamide.29 Four different ester motifs were chosen in the synthesis of ProPAgens 4a–d and 9a–d, methyl (Me), isopropyl (iPr), tert-butyl (tBu), and benzyl (Bn) because they show varying biological activities that range from low (tBu) to high (Bn).20,26,29
Serum Stability of HMBP Monophosphonate ProPAgens
Because the motive for synthesizing the HMBP monophosphonate ProPAgens was to address the poor stability observed with their parent HMBP monophosphate ProPAgens,20 upon the completion of their synthesis, we first studied their stability in human serum. As an example, we incubated the difluoromethylene ProPAgen 9a with human serum at 37 °C for 12 h and monitored the sample by 31P NMR, as we reported previously.26 As shown in Figure 4, the 31P NMR spectra of ProPAgen 9a showed six phosphorous peaks because of the two diastereoisomers that arise from the chiral phosphorous center, which is typical of these prodrugs, and the phosphorous coupling with the fluorine atoms. Following the addition of human serum and monitoring of the sample by 31P NMR, these original 9a31P NMR peaks remained present through the 12 h of the study, and no new 31P NMR peaks were observed. A similar stability profile was observed with the methylene ProPAgen 4d (Figure S1).
Figure 4.

Stability of HMBP monophosphonate ProPAgen 9a in human serum at 37 °C for 12 h as monitored by 31P NMR.
Collectively, these data indicated the superior stability of these compounds relative to that previously observed with HMBP monophosphate ProPAgens.20 The observed −CH2–PO(OH)2 and −CF2–PO(OH)2 groups’ stability profile is in-line with what has been observed for difluoromethyl and methyl phosphonates.31
Metabolism of HMBP Monophosphonate ProPAgens
The breakdown of aryloxy triester phosphoramidate and aryloxy diester phosphonamidate prodrugs is known to be mediated by two enzymes: carboxypeptidase Y and a phosphoramidase-type enzyme, for example, Hint-1 (Figure 5A).29 Indeed, upon cell entry, the breakdown of these phosphonate masking groups is initiated by hydrolysis of the ester motif by carboxypeptidase Y, and this generates a carboxylate group (A), which performs a nucleophilic attack onto the phosphonate group. This leads to the loss of the aryl group and the formation of a highly unstable five-membered heterocyclic ring (B), which is rapidly opened up by a water molecule to generate the amidate metabolite C. Finally, a phosphoramidase-type enzyme, for example, Hint-1 cleaves the P–N bond and releases the monophosphonate species. As this postulated mechanism of McGuigan’s phosphoramidate prodrugs has been generated through extensive studies on nucleoside monophosphate aryloxy triester phosphoramidate prodrugs, we investigated whether this was the case for non-nucleosides, PAg aryloxy diester phosphonamidate prodrugs in this case. Thus, ProPAgen 9b was incubated in vitro with recombinant carboxypeptidase Y at 37 °C, and the reaction was then monitored by 31P NMR. As shown in Figure 5B, at t = 0 h, the 31P NMR scan showed the presence of six phosphorous peaks, δP = 9.25, 9.52, 9.71, 10.08, 10.19, and 10.51 ppm, as expected. However, 30 min after the addition of recombinant carboxypeptidase Y and incubation at 37 °C, the phosphorous peaks corresponding to ProPAgen 9b were almost consumed and three prominent phosphorous peaks, δP = 6.50, 6.90, and 7.20 ppm, appeared. These peaks remained present throughout the 15 h period of this study. The disappearance of the six phosphorous peaks corresponding to ProPAgen 9b indicates that the carboxypeptidase Y cleaved off the ester motif and triggered the breakdown of the aryloxy diester phosphoramidate moiety as depicted in Figure 5A. Given that the sample lacks Hint-1, the anticipated product from this in vitro study is metabolite C (Figure 5A). This notion was supported by the fact that the loss of the ester and phenyl groups from the parent ProPAgen and the generation of the corresponding metabolite C leads to the loss of the chirality at the phosphorous center. Hence, the 31P NMR would show only a triplet because of the phosphorous–fluorine coupling, and such a splitting pattern was observed in our study (Figure 5B). To confirm whether the product generated after 15 h was metabolite C, the sample was analyzed by LC–MS, and indeed, a new major peak was detected by high-performance liquid chromatography (HPLC) and this had a mass that corresponded to metabolite C (Figure S2).
Figure 5.

In vitro carboxypeptidase-mediated breakdown of HMBP phosphonate ProPAgen 9b. (a) Postulated mechanism of aryloxy diester prodrugs metabolism. (b) 31P NMR spectrum of ProPAgen 9b alone and at different time points, following incubation with recombinant carboxypeptidase Y at 37 °C for 15 h.
In order to probe whether metabolite C could be processed by Hint-1 to cleave the P–N bond and release the monophosphonate species,38−40 we performed in silico docking of metabolite C into the cocrystal structure of Hint-1 with AMP (Figure S3). Analysis of the different predicted poses indicated that the amidate moiety of metabolite C was positioned in a pocket that included the key catalytic Hint-1 amino acid residues (serine 107, histidines 112 and 114) (Figure S3). Such a position suggests that metabolite C is likely to be processed by Hint-1 to release the unmasked monophosphonate, in agreement with previous conclusions.27
Together, the in vitro carboxypeptidase Y assay and the Hint-1 in silico docking support the metabolism of HMBP phosphonate ProPAgens to release the unmasked monophosphonate species.
Vγ9/Vδ2 T-cell Activation and Cancer Cells Lysis by HMBP Monophosphonate ProPAgens
Once the synthesis, stability, and metabolism of HMBP monophosphonate ProPAgens were established, they were subsequently studied for their ability to activate Vγ9/Vδ2 T-cells and lyse cancer cells in vitro. For this, peripheral blood mononuclear cells (PBMCs) containing Vγ9/Vδ2 T-cells derived from healthy donors were incubated with increasing concentrations of HMBPP, zoledronate, or HMBP ProPAgens 4a–d and 9a–d (up to 100 μM) (Figure 6). Peripheral blood γδ T-cells lack appreciable levels of surface CD69 or CD25 under steady-state conditions, but TCR stimulation upregulates both T-cell activation markers.41 PAg-responsive Vγ9/Vδ2 T-cells were then assessed for the upregulation of CD69 and CD25.
Figure 6.
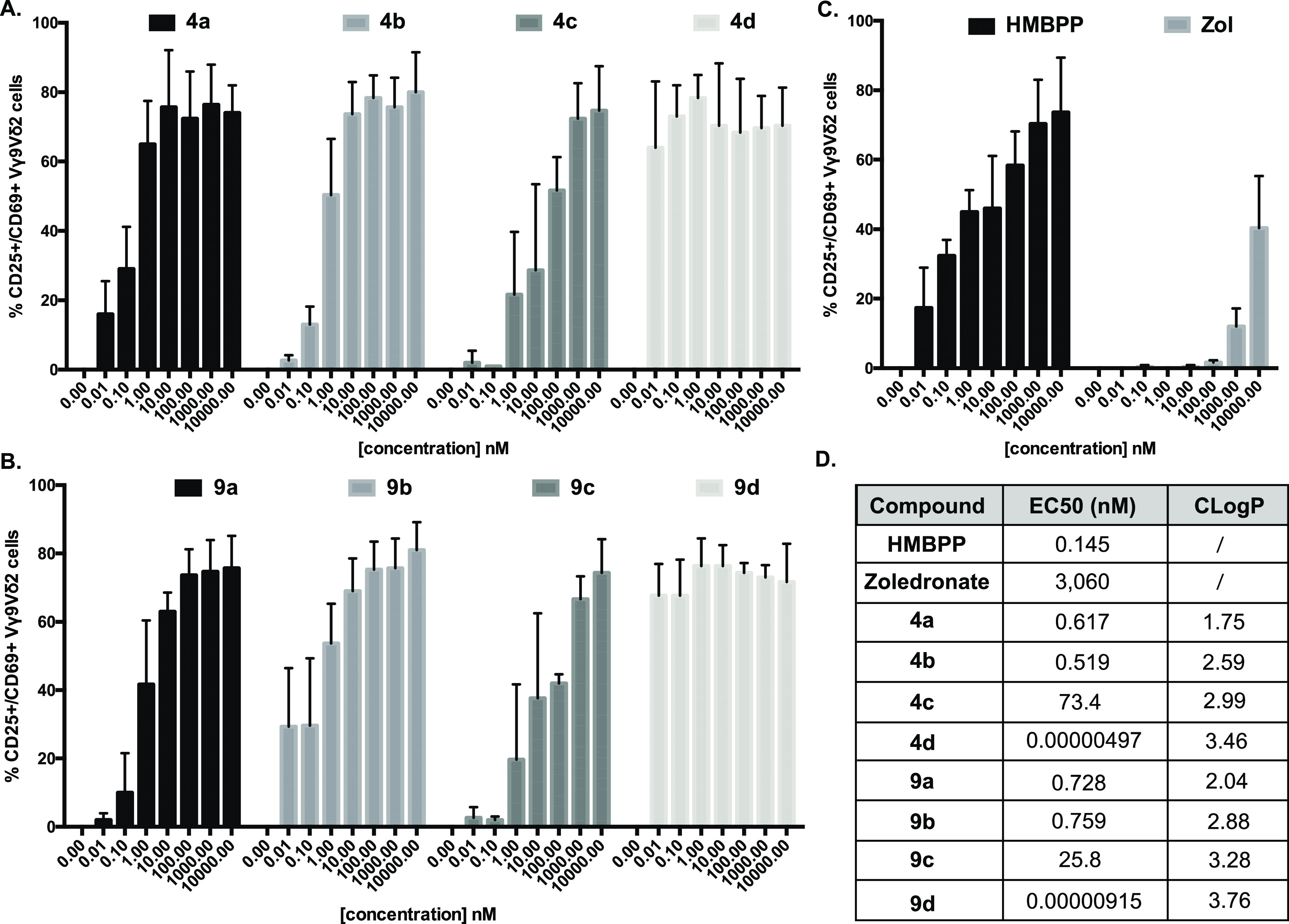
In vitro ProPAgen-mediated activation of Vγ9/Vδ2 T-cells, following overnight incubation with HMBPP, zoledronate, and HMBPP ProPAgens. Levels of activation are represented as % of Vγ9/Vδ2 T-cells that are CD69+ CD25+. Data is shown as mean ± SE (n = 4). (A) Activation of Vγ9/Vδ2 stimulated by ProPAgens 4a–d. (B) Activation of Vγ9/Vδ2 stimulated by ProPAgens 9a–d. (C) Activation of Vγ9Vδ2 stimulated by HMBPP and zoledronate. (D) EC50 values calculated based on the results of the activation assay. CLogP values were calculated using ChemDraw.
The results showed that the natural PAg HMBPP exhibited significant activation of Vγ9/Vδ2 T-cells, EC50 = 0.145 nM (Figure 6C), comparable to its reported potency.19,21 Additionally, zoledronate showed a much weaker activation, EC50 ≈ 3,060 nM as expected (Figure 6C). Initially, we tested the activation of Vγ9/Vδ2 T-cells using all of the eight HMBP phosphonate ProPAgens (4a–d and 9a–d) and employing concentrations ranging from 10 pM to 100 μM (Figures 6A,B and S4). For simplicity, the data presented in Figure 6A–C only show the activation up to 10 μM, while a graphical representation of the data up to 100 μM is provided in Figure S4. All of the ProPAgens showed potent activation of Vγ9/Vδ2 T-cells with potencies (EC50) comparable to the natural PAg HMBPP. Interestingly, two ProPAgens 4d and 9d, which are the benzyl ester prodrugs, exhibited far more potent activation of Vγ9/Vδ2 T-cells: even at 10 nM, these two prodrugs achieved >50% activation of Vγ9/Vδ2 T-cells. To determine the EC50 values of these compounds, we repeated the Vγ9/Vδ2 T-cells activation assay using sub-nanomolar concentrations (from 1 attomolar [aM] to 100 μM) (Figure S5). The data showed that ProPAgen 4d had an EC50 = 9.15 fM, whereas 9d had an EC50 = 4.97 fM (Figure 6D), demonstrating that these ProPAgens are 15,000–30,000-fold more potent than the PAg HMBPP in the same assay. To the best of our knowledge, this makes ProPAgens 4d and 9d the most potent small molecule Vγ9/Vδ2 T-cells activators reported to date.
Notably, at the highest concentration studied (100 μM), the activation of Vγ9/Vδ2 T-cells by ProPAgens 4d and 9d was less than that achieved with 10 μM (Figure S4). This could be explained by negative feedback mechanisms induced by antigen overstimulation that lead to downregulation of the TCR, subsequently resulting in lower expression of the CD25 activation marker.42
Although the potencies of compounds 4d and 9d are significantly higher than the other HMBP ProPAgens studied in this work (Figure 6D), this is consistent with other studies of this type of prodrug when used as anticancer agents. An example of this is the aryloxy triester phosphoramidate prodrugs of the anticancer agent gemcitabine, where the benzyl ester phosphoramidates exhibit potent sub-micromolar potencies across four cancer cell lines compared to the isopropyl ester prodrugs, which had an IC50 > 5 μM.43 Additionally, HMBP methylene phosphoramidate prodrugs using glycine as an amino acid have been shown to exhibit sub-nanomolar activation of Vγ9/Vδ2 T-cells,33 and this was the same with mixed aryl prodrugs of HMBP.44,45 Notably, glycine phosphoramidates often generate less potent prodrugs compared to the l-alanine counterparts.29 Thus, the sub-nanomolar potencies of Vγ9/Vδ2 T-cell activation by HMBP prodrugs seems to be consistent and correlates with the lipophilicity of the prodrugs.
Considering the structure–activity relationship (SAR) of our HMBP ProPAgens, we did not observe significant differences between the methyl and difluoromethyl phosphonate ProPAgens, while the Vγ9/Vδ2 T-cell activation potency varied with the ester motif used. Indeed, ProPAgens with the benzyl ester groups (4d and 9d) exhibited the most potent activation of Vγ9/Vδ2 T-cells, followed by those with methyl and isopropyl ester groups, and the tBu-bearing ProPAgens exhibited the least potency in activating Vγ9/Vδ2 T-cells within the series presented here.
Such an influence of the ester group on the biological activity of this type of phosphonamidate prodrugs is consistent with the established SAR of these prodrugs.29 The difference in biological activity in such prodrugs is because of the differential lipophilicity between these compounds and the efficiency of the esterases in hydrolyzing the ester groups. For instance, it is now well-established that phosphonamidates with a benzyl ester often show the best biological activity because they often have the highest lipophilicity, which facilitates their efficient passive uptake into cells as well as rapid hydrolysis of this ester group by esterases.29
In contrast to the benzyl moieties, the tBu esters of phosphonamidates are known to be metabolized relatively slowly by esterases, which explains their relatively poor biological activity as compared to their benzyl ester phosphonamidates. This may also explain the superior Vγ9/Vδ2 T-cell activation by the benzyl ester ProPAgens 4d and 9d compared to the other studied ProPAgens—the 20 h activation assay described here is often sufficient for the benzyl ester prodrugs to be metabolized, while it may be of too short duration for the other nonbenzyl ester ProPAgens to be metabolized to levels that generate intracellular concentrations of the monophosphonate species sufficient to induce potent Vγ9/Vδ2 T-cell activation. Indeed, classically, the potency of aryloxy triester phosphoramidates is determined after 48 or 72 h, and in such studies, the nonbenzyl ester aryloxy triester phosphoramidates (apart from the tBu esters) show potent pharmacological activity.43,46
Given the high potency of HMBPP ProPAgen presented in this work, we subsequently studied their specificity toward the activation of Vγ9/Vδ2 T-cells. For this, we repeated the same Vγ9/Vδ2 T-cell activation assay using ProPAgens 4d and 9d (1 aM and 100 μM concentrations) and also, as a specificity control, we assessed the activation of CD8+ αβ T-cells, which are activated by peptides.47 As expected, our data confirmed that these prodrugs did not induce any activation of αβ T-cells (Figure S6). The specificity of these HMBP prodrugs is, therefore, in line with previous observations.20
Encouraged by the potency and specificity of our HMBP ProPAgens, especially 4d and 9d, we subsequently studied their ability to sensitize the urinary bladder carcinoma cell line T24 for targeted killing by in vitro expanded Vγ9/Vδ2 T-cells. In brief, T24 cells were retrovirally transduced to express GFP. These GFP + T24 cells were incubated in media containing 10 pM HMBPP or the indicated HMBPP ProPAgens for a period of 4 h. Media-only treated controls (no drug) were also included. The cells were then washed and cocultured with ex vivo expanded Vγ9/Vδ2 T-cells overnight at different effector/target ratios, and the viability of GFP + T24 cells was then measured on the following day. In the absence of effectors (Vγ9/Vδ2 T-cells), ProPAgen-pulsed T24 cells were poorly targeted, but upon pulsing for 4 h with Vγ9/Vδ2 T-cells, T24 cells were specifically lysed with increasing effector/target ratio, particularly with ProPAgens 4d and 9d (Figure 7A). In addition, the sensitizing effects of ProPAgens 4d and 9d are evidently much more potent in comparison to HMBPP and the tBu ester ProPAgens 4c and 9c (Figure 7B,C). Notably, ProPAgen 9d showed more significant sensitizing effects compared to HMBPP and ProPAgen 9c at 1:1 effector/target ratio (Figure 7B). Collectively, these results are consistent with our previous20 and current findings that benzyl ester ProPAgens are the most potent activators of Vγ9/Vδ2 T-cells based on the data generated by in vitro activation assays, while, alternatively, the tBu ester ProPAgens are comparatively less potent.
Figure 7.

Cytotoxicity of Vγ9/Vδ2 T-cells toward ProPAgen-treated T24 cells. % killing represents % GFP + eFluor780+ cells (i.e. dead T24 cells). (A) Cytotoxicity of Vγ9/Vδ2 T-cells toward T24 cells treated with ProPAgens 4c, 4d, 9c, and 9d at different effector/target ratios. Ratio 0:1 refers to the basal effect of ProPAgens on T24 cells in the absence of effectors. (B,C) Cytotoxicity of ProPAgens 4c, 4d, 9c, and 9d (used at 10 pM) compared to the 10 pM HMBPP-induced effect after subtracting the basal cytotoxic effect of Vγ9/Vδ2 on untreated T24 cells. Data is shown as mean ± SE (n = 3). Statistical analysis was performed using one-way ANOVA and Tukey’s multiple comparisons test. *p < 0.05.
Conclusions
A series of HMBP methylene and difluoromethylene monophosphonate ProPAgens were synthesized using the Grubbs olefin metathesis, the first report of this approach in synthesizing the HMBP phosphonate core. These ProPAgens exhibited superior serum stability compared to their phosphate ProPAgen derivatives, and which we have previously reported.20 Critically, these prodrugs were specific and potent activators of Vγ9/Vδ2 T-cells (femtomolar EC50) in PBMC assays and this translated into potent sensitization of the urinary bladder carcinoma cell line T24 to Vγ9/Vδ2 T-cell-mediated lysis. In PBMC assays, although it is unclear which cells take up the ProPAgens, it has previously been reported that aminobisphosphonates such as zoledronate, as well as the PAg HMBPP, are taken up and “presented” to Vγ9Vδ2 T-cells by monocytes and dendritic cells (DCs).48,49 While this may also be the case for ProPAgens, it is likely they are taken up and processed by a wider range of cells, as they are more lipophilic than zoledronate and HMBPP, allowing intracellular access through diverse cell membranes likely via passive diffusion. Enhanced and potentially broadened uptake in PBMC could be responsible for their higher potency compared to HMBPP and zoledronate. Regarding their ability to directly sensitize cancer cells for Vγ9/Vδ2 T-cell-mediated lysis, in the future, it will be beneficial to explore the potency of these ProPAgens against a wider range of cancer targets, as well as assessing the tumor selectivity in comparison to healthy cells. Of relevance to future efforts, substantial levels of Vγ9/Vδ2 T-cell-mediated killing were observed in T24 killing assays in the absence of ProPAgen sensitization. This is likely to be affected by several factors, including the activation state of the γδ T-cells, the levels of endogenous PAg in target cells (e.g., isopentenyl pyrophosphate50), and the presence on target cells of ligands for natural killer cell receptors expressed on Vγ9/Vδ2 T-cells such as the Natural Killer Group 2D (NKG2D) and DNAX Accessory Molecule-1 (DNAM1).51 These factors in combination will likely exert an important influence on Vγ9/Vδ2 T-cell effector responses, but have substantial potential to synergize with ProPAgen-mediated sensitization.
Collectively, the stability and potency profiles of the new HMBP phosphonate ProPAgens presented in this work make them highly promising candidates for in vivo safety and efficacy studies, and future development as new immunotherapeutics for treating challenging cancers and infections that can be targeted by Vγ9/Vδ2 T-cell responses. Regarding their clinical application, these ProPAgens could conceivably either be employed as part or subsequent to a clinical regimen to expand γδ T-cells in vivo or, alternatively, they could be administered to patients receiving adoptive cell therapy with ex vivo-expanded γδ T-cells, to directly augment Vγ9/Vδ2 T-cell-mediated antitumor activity.
Experimental Section
General Information
All reagents and solvents were of general purpose or analytical grade and were purchased from Sigma-Aldrich Ltd., Fisher Scientific, Fluorochem, or Acros. 31P, 1H, and 13C NMR data were recorded on a Bruker AVANCE DPX500 spectrometer operating at 202, 500, and 125 MHz, respectively. Chemical shifts (δ) are quoted in ppm, and J values are quoted in Hz. In reporting spectral data, the following abbreviations were used: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), td (triplet of doublets), and m (multiplet). All of the reactions were carried out under a nitrogen atmosphere and were monitored using analytical thin layer chromatography on precoated silica plates (kiesel gel 60 F254, BDH). Compounds were visualized by illumination under UV light (254 nm) or by the use of KMnO4 stain followed by heating. Flash column chromatography was performed with silica gel 60 (230–400 mesh) (Merck). HPLC was carried out on a SHIMADZU Prominence-i quaternary low-pressure gradient pump with a Prominence-i UV detector (190 to 700 nm). All solvents for HPLC were HPLC grade purchased from Fisher Scientific. HPLC data analysis was performed using the SHIMADZU Lab solutions software package. The purity of the tested ProPAgens was determined by HPLC, and they were all of ≥95% purity.
But-3-en-1-ylphosphonic Dichloride (2)
Trimethylsilylbromide (13.72 mL, 104.06 mmol, 10 equiv) was slowly added over 30 min to diethylbut-3-en-1-yl phosphonate 1 (2 g, 10.40 mmol, 1 equiv) in DCM (50 mL) under nitrogen at room temperature. The mixture was stirred for 2 h followed by the removal of volatiles under reduced pressure to obtain a yellow liquid δP NMR (202 MHz, CDCl3): 24.70. This was then dissolved in 50 mL of DCM, two drops of dry DMF were added, the mixture was cooled to 0 °C. Oxalyl chloride (2.68 mL, 31.20 mmol, 3 equiv) was then added dropwise, and the reaction mixture was allowed to warm to room temperature and stirred for 18 h. The volatiles were evaporated, and additional DCM (10 mL) was evaporated three more times to give the crude product (1.79 g, 100%) as a brown liquid which was used in the next step without further purification. δP NMR (202 MHz, CDCl3): 49.66.
General Procedure 1. Synthesis of Allylphosphonoamidates 3a–d
The crude product but-3-en-1-ylphosphonic dichloride (2) was dissolved in 5 mL of DCM and added dropwise to a solution of phenol (1 equiv), dry Et3N (2 equiv), and DCM (10 mL) at −78 °C. After stirring at −78 °C for 30 min, the reaction mixture was allowed to warm to room temperature, and the stirring was continued for another 3 h. Once the reaction is complete as indicated by 31P NMR [δP NMR (202 MHz, CDCl3): ∼39.93], the mixture was filtered, and the volatiles were removed under reduced pressure and washed twice with Et2O, which was subsequently removed under reduced pressure to give a crude oil. This product was then dissolved in DCM (10 mL) and was added dropwise over 15 min to a stirring mixture of l-alanine ester hydrogen chloride (1 equiv) and dry Et3N (2 equiv) in dry DCM (10 mL) under nitrogen at −78 °C. After stirring at −78 °C for 30 min, the reaction was allowed to warm to room temperature and was left stirring overnight. The solvents were removed under reduced pressure, and the mixture was filtered and washed with Et2O, which was then removed under reduced pressure to give a crude oil. The final products were then purified by column chromatography (6:4 Hex/EtOAc) as colorless oils.
Methyl (but-3-en-1-yl(phenoxy)phosphoryl)-l-alaninate (3a)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine methyl ester hydrogen chloride (0.373 g, 2.23 mmol, 1 equiv) to give product 3a (0.248 g, 38%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.88, 31.22. δH NMR (500 MHz, CDCl3): 7.30 (m, 2H, Ar), 7.10 (m, 3H, Ar),5.82 (m, 1H, CH2=CH), 5.00 (m, 2H, CH2=CH), 4.11–3.86 (m, 1H,CH–NH), 3.60 (d, J = 6.6 Hz, 3H, OCH3), 3.18 (m, 1H,NH), 2.48–2.27 (m, 2H,=CH–CH2), 1.92 (m, 2H, CH2–P), 1.21 (2 d, J = 7.1 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 174.68 (d, J = 6.3 Hz, C=O), 174.38 (d, J = 5.1 Hz, C=O), 150.72 (d, J = 9.1 Hz), 150.51 (d, J = 9.4 Hz), 129.78, 124.78 (d, J = 5.5 Hz, CH=CH2), 120.86 (d, J = 4.6 Hz, C–Ar), 120.71 (d, J = 4.7 Hz), 115.56, 52.52 (d, J = 3.1 Hz, CH3–O), 49.58 (d, J = 14.7 Hz, CH–NH), 27.88 (d, J = 130.9 Hz, CH2–P) 27.60 (d, J = 131.6 Hz, CH2–P), 26.59 (d, J = 4.1 Hz, CH2–CH2–P), 21.68 (2 d, J = 4.3 Hz, CHCH3).
Isopropyl (but-3-en-1-yl(phenoxy)phosphoryl)-l-alaninate (3b)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine isopropyl ester hydrogen chloride (0.373 g, 2.23 mmol, 1 equiv) to give product 3b (0.348 g, 48%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.93, 31.24. δH NMR (500 MHz, CDCl3): 7.30 (m, 2H, Ar), 7.21 (m, 3H, Ar), 5.81 (m, 1H, CH2=CH), 5.11 (m, 2H, CH2=CH), 4.96 (m, 1H, CH–iPr), 4.04–3.94 (m, 1H, CH–NH), 3.21 (m, 1H, NH), 2.45 (m, 2H, =CH–CH2), 2.02–1.88 (m, 2H, CH2–P), 1.29–1.18 (m, 9H, CH3–CH–NH, CH–iPr). δC NMR (126 MHz, CDCl3): 173.67 (d, J = 6.3 Hz, C=O), 150.74 (d, J = 9.1 Hz), 129.76 (d, J = 6.7 Hz), 124.73 (d, J = 5.0 Hz), 120.86 (d, J = 4.6 Hz), 120.69 (d, J = 4.6 Hz), 115.50, 69.23 (d, J = 5.6 Hz, CH–iPr), 49.75 (d, J = 9.5 Hz, CH–NH), 27.89 (d, J = 130.9 Hz, CH2–P), 27.54 (d, J = 131.4 Hz, CH2–P), 26.74 (d, J = 4.3 Hz, CH2–CH2–P), 26.57 (d, J = 4.0 Hz, CH2–CH2–P), 21.58 (2 d, J = 4.4 Hz, CHCH3).
tert-Butyl (but-3-en-1-yl(phenoxy)phosphoryl)-l-alaninate (3c)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine tert-butyl ester hydrogen chloride (0.405 g, 2.23 mmol, 1 equiv) to give product 3c (0.461 g, 61%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.95, 31.21. δH NMR (500 MHz, CDCl3): 7.32 (m, 2H, Ar), 7.20 (m, 3H, Ar), 5.87 (m, 1H, CH2=CH), 5.12 (m, 2H, CH2=CH), 4.00–3.86 (m, 1H, CH–NH), 3.26 (m, 1H, NH), 2.45 (m, 2H,=CH–CH2), 2.04–1.85 (m, 2H, P–CH2), 1.42 (s, 9H, tBu-H), 1.22 (2 d, J = 7.2 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 173.13 (d, J = 5.5 Hz, C=O), 150.77 (d, J = 9.2 Hz), 137.43 (d, J = 7.1 Hz), 137.32 (d, J = 7.6 Hz), 129.76 (d, J = 6.2 Hz), 124.71 (d, J = 4.0 Hz), 120.88 (d, J = 4.6 Hz), 120.72 (d, J = 4.7 Hz), 115.47, 81.99 (d, J = 8.2 Hz), 60.54, 50.18 (d, J = 4.0 Hz, CH–NH), 28.05, 27.91 (d, J = 131.6 Hz, CH2–P), 27.20 (d, J = 130.2 Hz, CH2–P), 21.82 (2 d, J = 4.2 Hz, CHCH3).
Benzyl (but-3-en-1-yl(phenoxy)phosphoryl)-l-alaninate (3d)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine benzyl ester hydrogen chloride (0.405 g, 2.23 mmol, 1 equiv) to give product 3d (0.461 g, 55%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.85, 31.21. δH NMR (500 MHz, CDCl3): 7.38–7.27 (m, 7H, Ar), 7.21–7.18 (m, 2H, Ar), 7.16–7.09 (m, 1H, Ar), 5.85 (m, 1H, CH2=CH), 5.04 (m, 4H, CH2=CH, −OCH2), 4.28–4.03 (m, 1H, CH–NH), 3.24 (m, 1H, NH), 2.56–2.34 (m, 2H, =CH–CH2), 2.08–1.82 (m, 2H, P–CH2), 1.24 (2 d, J = 7.1 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 173.74 (d, J = 5.2 Hz, C=O), 150.70 (d, J = 9.1 Hz), 137.35, 135.40 (d, J = 6.7 Hz), 129.79 (d, J = 6.1 Hz), 128.79 (d, J = 2.8 Hz), 128.64 (d, J = 6.4 Hz), 128.33, 120.86 (d, J = 4.6 Hz), 120.68 (d, J = 4.7 Hz), 115.55, 67.31 (d, J = 3.0 Hz, CH2–O), 49.72 (d, J = 11.1 Hz, CH–NH), 27.88 (d, J = 130.7 Hz, CH2–P), 27.57(d, J = 131.3 Hz, CH2–P), 26.74 (d, J = 4.3 Hz, CH2–CH2–P), 26.57 (d, J = 4.1 Hz, CH2–CH2–P), 21.70 (2 d, J = 4.4 Hz, CHCH3).
General Procedure for the Synthesis of Phosphonoamidates through Hoveyda–Grubbs Cross Metathesis
To a solution of allylphosphonoamidates 3a–d (1 equiv) and 2-methyl-2-propen-1-ol (85 μL, 1 mmol, 2 equiv), 1,4-benzoquinone (5.40 mg, 10 mol %) in dry DCM (10 mL) was added to Hoveyda–Grubbs catalyst 2nd generation (23.5 mg, 0.038 mmol, 7.5 mol %). The catalyst was added in three equal portions of 2.5 mol % (7.8 mg, 0.013 mmol) at t = 0, 2, and 4 h over the course of the reaction. The solution was then heated to reflux at 45 °C under a nitrogen atmosphere for 18 h. After cooling to room temperature, a scoop of activated carbon was added, and the mixture stirred for another 2 h and then filtered through a Celite pad. Volatiles were evaporated, and the residue was purified by extensive silica gel column chromatography (hexane/EtOAc, 7:3 to 0:10) to give 4a–d as colorless oils.
Methyl (((E)-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (4a)
Synthesized following general procedure 2 using 3a (150 mg, 0.5 mmol, 1 equiv) to give 4a (91 mg, 57%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.89, 31.31. δH NMR (500 MHz, CDCl3): 7.31 (m, 2H, Ar), 7.18 (m, 3H, Ar), 5.48 (m,1H, =CH), 4.24–3.98 (m, 3H, CH2OH, CH–NH), 3.68 (d, J = 7.9 Hz, 3H, OCH3), 3.39–3.18 (m, 1H, NH), 2.55–2.41 (m, 2H, =CH–CH2), 2.12–1.87 (m, 2H, CH2–P), 1.71 (d, J = 6.6 Hz, 3H, CH3(CH2)C=), 1.27 (2 × d, J = 7.1 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 176.71 (d, J = 5.9 Hz, C=O), 150.72, 129.81 (d, J = 6.9 Hz), 124.81 (d, J = 8.6 Hz, CH=CH2), 124.17, 124.04, 120.94, 120.69 (d, J = 4.6 Hz), 68.56 (d, J = 9.1 Hz, CH2–OH), 52.64 (d, J = 3.5 Hz, CH3–O), 49.51 (d, J = 4.1 Hz, CH–NH), 28.33 (d, J = 129.6 Hz CH2–P), 28.06 (d, J = 130.2 Hz, CH2–P), 21.86 (2 d, J = 4.9 Hz, CHCH3), 21.00 (d, J = 4.8 Hz, CH2–CH2–P), 20.89 (d, J = 4.4 Hz, CH2–CH2–P),13.87. HRMS (ES+, m/z): calcd for (M + Na)+ C16H24NO5NaP, 364.1290; found, 364.1293. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, tRt = 5.36 min (96.5%).
Isopropyl (((E)-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (4b)
Synthesized following general procedure 2 using 3b (0.150 g, 0.46 mmol, 1 equiv) to give product 4b (97 mg, 59%) as a colorless oil. δP NMR (202 MHz, CDCl3): 31.11, 31.49. δH NMR (500 MHz, CDCl3): 7.28 (m, 2H, Ar), 7.20 (m, 3H, Ar), 5.46 (m, 1H, =CH), 4.95 (m, 1H, CH–iPr), 4.08–3.79 (m, 3H, CH2OH, CH–NH), 3.47–3.25 (m, 1H, NH), 2.51–2.35 (m, 2H,=CH–CH2), 2.02–1.85 (m, 2H, CH2–P), 1.73–1.56 (d, J = 6.0 Hz, 3H, CH3(CH2)C=), 1.35–1.08 (m, 9H, CH3–CH–NH, CH–iPr). δC NMR (126 MHz, CDCl3): 173.62 (d, J = 5.8 Hz, C=O), 150.74 (d, J = 9.0 Hz),136.76, 124.75, 123.95 (d, J = 5.4 Hz), 123.84 (d, J = 6.9 Hz), 120.90 (d, J = 4.5 Hz), 120.67 (d, J = 4.6 Hz),119.83, 115.58, 69.41 (d, J = 4.7 Hz, CH–iPr) 68.39 (d, J = 7.0 Hz, CH2–OH), 49.65, 28.36 (d, J = 129.7 Hz, CH2–P), 27.92 (d, J = 131.1 Hz, CH2–P), 21.83 (2 d, J = 6.2 Hz, CHCH3), 20.90 (d, J = 4.4 Hz, CH2–CH2–P), 20.84 (d, J = 4.4 Hz, CH2–CH2–P),13.84.HRMS (ES+, m/z): calcd for (M + Na)+ C18H28NO5NaP, 392.1603; found, 392.1613. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 5.23 min (98%).
tert-Butyl (((E)-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (4c)
Synthesized following general procedure 2 using 3c (0.150 g, 0.44 mmol, 1 equiv) to give product 4c (108 mg, 64%) as a colorless oil. δP NMR (202 MHz, CDCl3): 31.05, 31.42. δH NMR (500 MHz, CDCl3): 7.30 (m, 2H, Ar), 7.17 (m, 3H,Ar), 5.46 (m, 1H, =CH), 4.17–3.78 (m, 3H, CH2OH, CH–NH), 3.41–3.18 (m, 1H, NH), 2.60–2.30 (m, 2H, =CH–CH2), 2.01–1.87 (m, 2H, CH2–P), 1.69 (d, J = 6.8 Hz, 3H, CH3(CH2)C=), 1.42 (s, 9H, tBu), 1.27 (2 d, J = 7.9 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 173.34 (d, J = 6.0 Hz, C=O), 150.79 (d, J = 9.0 Hz), 136.81 (d, J = 9.6 Hz), 115.59, 82.22 (d, J = 5.6 Hz), 68.45 (d, J = 10.4 Hz, CH2–OH), 50.07 (d, J = 4.5 Hz, CH–NH), 28.01 (d, J = 129.7 Hz, CH2–P), 27.26 (d, J = 130.4 Hz, CH2–P), 21.98 (d, J = 3.8 Hz, CHCH3), 20.97 (d, J = 4.7 Hz, CH2–CH2–P), 20.88 (d, J = 4.4 Hz, CH2–CH2–P), 13.86 (CH3). HRMS (ES+, m/z): calcd for (M + Na)+ C19H30NO5NaP, 406.1759; found, 406.1762. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 5.41 min (97%).
Benzyl (((E)-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (4d)
Synthesized following general procedure 2 using 3d (0.150 g, 0.4 mmol, 1 equiv) to give product 4d (100 mg, 59%) as a colorless oil. δP NMR (202 MHz, CDCl3): 30.90, 31.32, δH NMR (500 MHz, CDCl3): 7.39–7.28 (m, 7H, Ar), 7.22–7.17 (m, 2H,Ar), 7.12 (m, 1H, Ar), 5.44 (m, 1H, =CH), 5.09 (m, 2H, OCH2), 4.22–3.86 (m, 3H, m, 3H, CH2OH, CH–NH), 3.49–3.17 (m, 1H, NH), 2.64–2.36 (m, 2H, =CH–CH2), 2.04–1.80 (m, 2H, CH2–P), 1.69 (d, J = 6.8 Hz, 3H, CH3(CH2)C=), 1.33 (2 d, J = 7.0 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 176.76, 129.81 (d, J = 7.4 Hz), 128.75 (d, J = 14.9 Hz), 128.39, 124.77, 124.15 (d, J = 15.0 Hz), 120.93–120.84, 120.67 (d, J = 4.7 Hz), 68.57 (d, J = 9.6 Hz), 67.42 (d, J = 2.9 Hz), 49.65, 28.05 (d, J = 129.9 Hz), 21.77 (d, J = 4.1 Hz), 20.87 (d, J = 4.4 Hz), 13.87. HRMS (ES+, m/z): calcd for (M + Na)+ C22H28NO5NaP, 440.1603; found, 440.1609. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 5.18 min (95%).
Diethyl (1,1-difluorobut-3-en-1-yl)phosphonate (6)37
Anhydrous DMF (20 mL) was added to a 250 mL round bottom flask containing activated zinc powder (2.50 g, 38.23 mmol, 1 equiv) under nitrogen. This was followed by slow dropwise addition of diethyl (bromodifluoromethyl)phosphonate (6.80 mL, 38.23 mmol, 1 equiv), and the mixture was stirred for 3 h at room temperature. CuBr (5.48 g, 38.23 mmol, 1 equiv) was added followed by slow dropwise addition of allyl bromide (3.96 mL, 45.87 mmol, 1.2 equiv) to prevent exothermic reaction. After stirring for 40 h, the mixture was filtered and then partitioned between DCM and 10% aqueous NH4Cl. The aqueous phase was extracted three times with DCM. The combined organic phases were dried over anhydrous MgSO4 and concentrated under reduced pressure, and the residue obtained was purified by column chromatography using 20% EtOAc in hexane to give 6 (5.41 g, 62%) as a pale-yellow oil. δP NMR (202 MHz, CDCl3): 6.93 (t, J = 107.4 Hz). δH NMR (500 MHz, CDCl3): 5.84 (m, 1H, =CH), 5.26 (m, 2H, CH2=), 4.32–4.21 (m, 4H, 2 × OCH2CH3), 2.82 (m, 2H, =CH–CH2), 1.37 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3).
(1,1-Difluorobut-3-en-1-yl)phosphonic Dichloride (7)
Synthesized as described for 2 using 6 (2.5 g, 10.95 mmol, 1 equiv) to give the crude product 7 (2.28 g, 100%) as a brown liquid, which was used in the next step without further purification. δP NMR (202 MHz, CDCl3): 31.56 (t, J = 138.8 Hz).
Methyl ((1,1-difluorobut-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (8a)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine methyl ester hydrogen chloride (0.261 g, 1.87 mmol, 1 equiv) to give product 8a (0.150 g, 24%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.05 (dd, J = 101.1, 49.9), 8.41 (dd, J = 100.0, 51.1 Hz). δH NMR (500 MHz, CDCl3): 7.35 (m, 2H, Ar), 7.21 (m, 3H, Ar), 5.89 (m, 1H, CH2=CH), 5.29 (m, CH2=CH), 4.14 (m, 1H, CH–NH), 3.69 (d, J = 6.6 Hz, 3H, OCH3),3.64 (m, 1H, NH), 3.13–2.84 (m, 2H, =CH–CH2), 1.38 (2 × d, 7.1 Hz, 3H, CH–CH). δC NMR (126 MHz, CDCl3): 173.76 (d, J = 4.1 Hz, C=O), 130.02 (d, J = 4.1 Hz), 127.40–127.07 (m), 125.64, 121.54 (d, J = 9.3 Hz), 120.54 (t, J = 4.8 Hz), 52.69, 50.09 (d, J = 6.6 Hz), 39.39–37.22 (m), 21.74(2d, J = 3.5 Hz, CH3).
Isopropyl ((1,1-difluorobut-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (8b)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine isopropyl ester hydrogen chloride (0.314 g, 1.87 mmol, 1 equiv) to give product 8b (0.165 g, 25%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.17 (dd, J = 101.1, 30.3 Hz), 8.46 (dd, J = 99.9, 38.0 Hz). δH NMR (500 MHz, CDCl3). 7.34 (m, 2H, Ar), 7.20 (m, 3H, Ar), 5.89 (m, 1H, CH2=CH), 5.29 (m, 2H, CH2=CH), 4.99 (m, 1H, CH–iPr), 4.14–3.99 (m, 1H, CH–NH), 3.68 (m, 1H, NH), 3.00–2.89 (m, 2H, =CH–CH2), 1.35–1.17 (m, 9H, CH3–CH–NH, CH–iPr). δC NMR (126 MHz, CDCl3): 172.68 (d, J = 5.9 Hz, C=O), 149.46, 129.88 (d, J = 4.2 Hz), 127.11 (d, J = 5.4 Hz), 125.46, 121.36 (d, J = 8.9 Hz), 120.40 (t, J = 4.9 Hz), 69.39 (d, J = 2.8 Hz, CH–iPr), 50.15, 38.63–37.99 (m), 21.58 (d, J = 1.6 Hz), 21.44 (2 d, J = 3.2 Hz, CHCH3).
tert-Butyl ((1,1-difluorobut-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (8c)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine tert-butyl ester hydrogen chloride (0.340 g, 1.87 mmol, 1 equiv) to give product 8c (0.320 g, 46%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.23 (dd, J = 101.0, 34.4 Hz), 8.52 (dd, J = 99.6, 36.0 Hz). δH NMR (500 MHz, CDCl3): 7.34 (m, 2H, Ar),7.21 (m, 3H, Ar), 5.89 (m, 1H, CH2=CH), 5.28 (m, 2H, CH2=CH), 4.08–3.98 (m, 1H, CH–NH), 3.67 (m, 1H, NH), 3.01–2.84 (m, 2H, =CH–CH2), 1.42 (d, J = 5.2 Hz,9H, tBu), 1.30 (d, J = 7.2 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 172.48 (d, J = 4.5 Hz, C=O), 149.72, 129.99 (d, J = 2.1 Hz), 128.59–126.95 (m), 125.54, 121.46 (d, J = 9.2 Hz), 120.54 (t, J = 4.8 Hz), 82.32 (d, J = 5.8 Hz), 50.69 (d, J = 2.8 Hz, CH–NH), 38.80–38.12 (m), 27.99, 21.85 (2d, J = 3.1 Hz, CHCH3).
Benzyl ((1,1-difluorobut-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (8d)
Synthesized following general procedure 1 using phenol (0.210 g, 2.23 mmol, 1 equiv) and l-alanine benzyl ester hydrogen chloride (0.403 g, 1.87 mmol, 1 equiv) to give product 8d (0.350 g, 45% yield) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.08 (dd, J = 101.2, 45.4 Hz), 8.39 (dd, J = 100.1, 46.5 Hz). δH NMR (500 MHz, CDCl3): 7.41–7.29 (m, 7H, Ar), 7.25–7.17 (m, 3H, Ar), 5.87 (m, 1H, CH2=CH), 5.27 (m, 2H, CH2=CH), 5.11 (m, 2H, −OCH2), 4.33–4.09 (m, 1H, CH–NH), 3.68 (m, 1H, NH), 2.93 (m, 2H, P–CH2), 1.36 (d, J = 7.2 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 173.15 (d, J = 3.2 Hz, C=O), 149.68, 135.26 (d, J = 6.4 Hz), 130.01 (d, J = 4.1 Hz), 128.87–128.52 (m), 128.35 (d, J = 1.1 Hz), 127.38–127.03 (m), 125.62, 122.24–120.38 (m), 120.51, 67.49, 50.20, 38.74–38.07 (m), 21.96 (2 d, J = 3.5 Hz, CHCH3).
Methyl (((E)-1,1-difluoro-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (9a)
Synthesized following general procedure 2 using 8a (0.150 g, 0.45 mmol, 1 equiv) to give product 9a (97 mg, 58%) as a colorless oil. δP NMR (202 MHz, CDCl3) δ 9.27 (dd, J = 112.1, 18.4 Hz), 8.44 (dd, J = 126.4, 20.2 Hz). δH NMR (500 MHz, CDCl3): 7.35 (m, 2H, Ar), 7.21 (m, 3H, Ar), 5.56 (m, 1H, =CH), 4.18 (m, 1H, CH–NH), 4.07 (m, 2H, =CH–CH2), 3.75–3.46 (m, 4H, NH, OCH3), 3.10–2.79 (m, 2H, =CH–CH2), 1.83 (m, 1H, OH), 1.76 (s, 3H, CH3(CH2)C=CH),1.37 (2 × d, J = 7.1 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 176.76, 141.38 (d, J = 7.6 Hz), 130.04 (d, J = 3.2 Hz), 125.65 (d, J = 4.0 Hz), 120.56 (d, J = 4.6 Hz), 120.47 (d, J = 4.7 Hz), 68.25 (d, J = 1.9 Hz), 52.80 (CH3–O), 50.09 (d, J = 4.1 Hz, CH–NH), 32.82, 21.82 (2 d, J = 2.6 Hz, CHCH3), 14.15 (CH3). HRMS (ES+, m/z): calcd for (M + Na)+ C16H22F2NO5NaP, 400.1101; found, 400.1109. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 5.45 min (99%).
Isopropyl (((E)-1,1-difluoro-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (9b)
Synthesized following general procedure 2 using 8b (0.150 g, 0.41 mmol, 1 equiv) to give product 9b (117 mg, 69%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.30 (dd, J = 109.1, 34.4 Hz), 8.42 (dd, J = 111.1, 38.4 Hz). δH NMR (500 MHz, CDCl3): 7.35 (m, 2H, Ar), 7.21 (m, 3H, Ar), 5.56 (m, 1H, =CH), 5.00 (m, 1H, CH–iPr), 4.17–3.91 (m, 3H, CH–NH, =CH–CH2), 3.67 (m, 1H, NH), 3.08–2.75 (m, 2H, =CH–CH2), 1.86 (m, 1H, OH), 1.70 (s, 3H, CH3(CH2)C=CH), 1.34–1.19 (m, 9H, CH3–CH–NH, CH–iPr). δC NMR (126 MHz, CDCl3): 173.09 (d, J = 6.8 Hz, C=O), 141.37 (d, J = 7.9 Hz), 130.03 (d, J = 2.6 Hz), 125.60 (d, J = 4.4 Hz), 120.50 (dd, J = 10.6, 4.7 Hz), 113.79–113.43 (m), 69.71 (d, J = 4.5 Hz), 68.25, 50.29 (d, J = 9.6 Hz), 30.88–28.72 (m), 21.73 (d, J = 8.2 Hz), 21.84 (2d, J = 4.1 Hz, CHCH3), 14.18 (CH3). HRMS (ES+, m/z): calcd. for (M + Na)+ C18H26F2NO5NaP, 428.1414; found, 428.1414. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 5.46 min (97.5%).
tert-Butyl (((E)-1,1-difluoro-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (9c)
Synthesized following general procedure 2 using 8c (0.150 g, 0.39 mmol, 1 equiv) to give product 9c (105 mg, 63%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.36 (dd, J = 109.1, 34.4 Hz), 8.48 (dd, J = 111.1, 36.4 Hz). δH NMR (500 MHz, CDCl3): 7.36 (m, 2H, Ar), 7.22 (m, 3H, Ar), 5.56 (m, 1H, =CH), 4.16–3.93 (m, 3H, CH–NH, =CH–CH2), 3.63 (m, 1H, NH), 3.08–2.77 (m, 2H, =CH–CH2), 1.90 (m, 1H, OH), 1.71 (s, 3H, CH3(CH2)C=CH), 1.43 (d, J = 5.2 Hz, 9H, tBu), 1.30 (2 × d, J = 7.1 Hz, 3H, CH–CH3). δC NMR (126 MHz, CDCl3): 172.79 (d, J = 6.8 Hz, C=O), 130.02, 125.56 (d, J = 5.8 Hz), 120.55 (d, J = 4.6 Hz), 120.47 (d, J = 4.4 Hz), 82.59, 68.26, 50.72 (d, J = 7.4 Hz, CH–NH), 32.97–32.80 (m), 28.02 (d, J = 1.1 Hz, 3 × CH3), 21.98 (2 d, J = 3.8 Hz, CHCH3), 14.16. HRMS (ES+, m/z) calcd. for (M + Na)+ C19H28F2NO5NaP: 442.1571; found: 442.1578. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 80:20 in 12 min, λ = 254 nm, Rt = 4.87 min (96%).
Benzyl (((E)-1,1-difluoro-5-hydroxy-4-methylpent-3-en-1-yl)(phenoxy)phosphoryl)-l-alaninate (9d)
Synthesized following general procedure 2 using 8d (0.150 g, 0.36 mmol, 1 equiv) to give product 9d (101 mg, 61%) as a colorless oil. δP NMR (202 MHz, CDCl3): 9.21 (dd, J = 113.2, 22.3 Hz), 8.37 (dd, J = 111.1, 24.3 Hz). δH NMR (500 MHz, CDCl3): 7.39–7.28 (m, 7H, Ar), 7.24–7.16 (m, 3H, Ar), 5.55 (m,1H, =CH), 5.13 (m, 2H, OCH2), 4.27–4.14 (m, 1H, CH–NH), 4.06 (m, 2H, =CH–CH2), 3.68 (m, 1H, NH), 3.05–2.73 (m, 2H, =CH–CH2), 1.80 (m, 1H, OH), 1.69 (s, 3H, CH3(CH2)C=CH), 1.35 (2 × d, J = 7.1 Hz, 3H, CH–CH3). δC (126 MHz, CDCl3): 173.38 (d, J = 7.9 Hz, C=O), 149.71, 141.37 (d, J = 8.3 Hz), 130.04 (d, J = 3.1 Hz), 128.76 (d, J = 12.8 Hz), 128.35 (s), 125.63 (d, J = 5.5 Hz), 120.49 (dd, J = 13.7, 4.6 Hz), 113.75–113.34 (m), 68.25 (d, J = 2.4 Hz), 67.61, 50.22 (d, J = 8.0 Hz), 34.32–31.18 (m), 21.84 (d, J = 3.8 Hz, CHCH3), 14.15 (CH3). HRMS (ES+,m/z): calcd for (M + Na)+ C22H26F2NO5NaP, 476.1414; found, 476.1421. HPLC (reverse-phase) 0.5 mL/min MeOH/H2O 70:30 in 12 min, λ = 254 nm, Rt = 6.58 min (96%).
In Silico Docking
Computational studies were performed using the OpenEye molecular modelling software suite provided by the OpenEye Scientific Software (http://www.eyesopen.com). An iMac with Intel Core i5/2.9 GHz microprocessor, 8 GB RAM, and running macOS operating system 10.14.1 was used to execute the docking studies. The three-dimensional cocrystal structure of AMP with the human Hint-1 was retrieved from the Protein Data Bank (PDB code 1KPF), and the site of AMP binding was identified as the docking site.52 Omega2 was used to generate multiple conformers for metabolite C using the default settings.53 FRED (fast rigid exhaustive docking) implements a rigid docking approach to fit these conformers into the predefined binding site and rank the poses by scoring functions.54,55 The VIDA module was then used to visualize and inspect the docked poses and identify key interactions between metabolite C and Hint-1.
Serum Stability
The experiment was carried out according to a published procedure,43 ProPAgens 4d or 9d (5.0 mg) in DMSO (0.050 mL) and D2O (0.15 mL). After recording the control 31P NMR at 37 °C, 0.30 mL of a freshly defrosted human serum aliquot (code: H4522, Sigma, U.K.) was added to the sample, which was next submitted to the 31P NMR experiments at 37 °C. The spectra were recorded every 30 min over 12 h. 31P NMR recorded data were processed and analyzed using the Bruker TopSpin 2.1 program.
In Vitro Carboxypeptidase Y Assay
The experiment was carried out according to the published protocol.43 This involved dissolving ProPAgens 94 (3.0 mg) in acetone-d6 (0.15 mL), followed by addition of 0.30 mL of T TRIZMA buffer (pH 7.6). After recording the control 31P NMR at 25 °C, a previously defrosted carboxypeptidase Y aliquot (0.1 mg dissolved in 0.15 mL of TRIZMA) was added to the sample, which was then immediately submitted to the 31P NMR experiments (at 25 °C). The spectra were recorded after 10 min, and every 1 h for 12 h.
Isolation of PBMCs from Healthy Donors
All healthy donors participating in this study were aged over 21 and were recruited from the Institute of Immunology and Immunotherapy, University of Birmingham. Written informed consent was obtained from all donors prior to any blood withdrawal, and ethical approval was obtained from the NRES Committee West Midlands ethical board (REC reference 14/WM/1254). Approximately 50 mL of peripheral blood (PB) was collected, and PB mononuclear cells (PBMCs) were extracted via a ficoll gradient method using Lymphoprep reagent. A volume of 30–35 mL of undiluted whole blood was carefully layered over 15 mL of Lymphoprep, and the cells were then centrifuged at 1200g for 20 min without brake. PBMCs were harvested, washed twice in PBS at 1900 rpm for 5 min, and counted manually using a hemocytometer. The required number of cells was taken for experiments, and any excess was frozen down with freezing media consisting of FCS +10% v/v DMSO. The Vγ9Vδ2 T-cells were identified in all assays by flow cytometric staining as Vγ9+ Vδ2+ population within the live (Zombie Aqua viability dye) CD3+ gate.
In Vitro Vγ9Vδ2 T-Cell Activation Assays
PBMCs were seeded out into round-bottom 96-well plates at 5 × 105 cells in a total volume of 200 μL per well, and stimulated with zoledronate and HMBPP (concentrations range 1 pM to 100 μM) and HMBPP ProPAgens, initially at a concentration range of 100 μM to 10 pM, and then, compounds 4d and 9d were selected and tested at 1 aM to 100 μM in a separate experiment. The cells were incubated at 37 °C/5% CO2 overnight and were stained by flow cytometry to assess Vγ9Vδ2 T-cell activation on the following day. The staining panel included viability dye (Zombie Aqua; 1:400), CD3 (BV421; 1:100), Vγ9 (PEcy5; 1:400), Vδ2 (APC; 1:400), CD8 (BV650; 1:100), CD69 (PE; 1:25), and CD25 (FITC; 1:100). All samples were acquired using a LSRFortessa X20 (BD Biosciences), and all data were analyzed via FlowJo v10 and GraphPad Prism software.
Retroviral Transduction of T24 Cells with GFP
For production of retrovirus encoding green fluorescent protein (GFP), Phoenix cells were transiently transfected with GFP-retroviral vector (kindly provided by Prof. C. Baum, Hannover Medical School, Hannover, Germany) and pCl ampho using Fugene 6 (Promega). The retrovirus particles were collected after 48 h of transfection and were then immobilized to a fibronectin (RetroNectin, Takara)-coated nontissue culture-treated plate (Corning). Subsequently, T24 bladder carcinoma cells (ATCC HTB4) were incubated with the immobilized retrovirus for 72 h at 37 °C. Cells were then acquired on a BD LSRFortessa X20 (BD Biosciences), and GFP expression data were analyzed with FlowJo V10.1.
Cytotoxicity Assay
Prior to carrying out the assay, Vγ9Vδ2 T-cells were expanded ex vivo from healthy donor PBMCs with 5 μM of zoledronate and the addition of 100 U/mL IL2 every 2–3 days over a period of 14 days. On the final day (day 14), GFP + T24 bladder carcinoma cells were cultured for 4 h in the presence of 10 pM ProPAgens or 10 pM HMBPP in round-bottom nontissue culture-treated plates. After washing the plates three times with RPMI medium, expanded Vγ9Vδ2 T-cells were then added, and the cocultures were incubated for 18 h at 37 °C. Cells were then labelled with eFluor 780 fixable viability dye (Invitrogen). Samples were subsequently acquired on a BD LSRFortessa X20 (BD Biosciences), and the data were analyzed with FlowJo V10.1 and Graphpad PRISM 7 (Graphpad Software Inc). The percentage of killing was determined by calculating the number of fixable viability dye + GFP + T24 (dead cells) of the total number of GFP + T24 cells. This assay was performed with n = 3 donors.
Acknowledgments
The work was supported by a Wellcome Trust ISSF grant awarded to Y.M. (Grant code: 514079), a Wellcome Trust Investigator award funding to B.E.W. (Grant code: 099266/Z/12/Z), and a Wellcome Trust Pathfinder award funding to B.E.W. (Grant code: 200983/Z/16/Z).
Glossary
Abbreviations
- Bn
benzyl
- DCM
dichloromethane
- BTN2A1
butyrophilin 2A1
- BTN3A1
butyrophilin 3A1
- Et3N
triethylamine
- HMBP
(E)-4-hydroxy-3-methylbut-2-enyl monophosphate
- HMBPP
(E)-4-hydroxy-3-methylbut-2-enyl pyrophosphate
- IP
isopentenyl pyrophosphate
- iPr
isopropyl
- Me
methyl
- PAg
phosphoantigen
- ProPAgen
prodrug of a phosphoantigen
- TCR
T-cell receptor
- tBu
tert-butyl
- TCR
T-cell receptor
- TMSBr
trimethylsilyl bromide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01232.
Accession Codes
PDB code 1KPF.
Author Present Address
⊥ (H.K.) School of Chemistry, South Road, Durham University, Durham DH1 3LE, U.K.
Author Contributions
H.K., T.E.T., Q.X., and M.S. contributed equally. H.K. and Q.X. synthesized the compounds reported in this work and carried out the in vitro metabolism assays. T.E.T. and M.S. conducted the biological evaluation of the compounds. E.A. contributed to the initial biological assay validation. R.T.B., B.E.W., and Y.M. designed the experiments and supervized the work. The manuscript was written through contributions of all authors, and all of the authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Y.M. and B.E.W. are named inventors on a patent application filed by Cardiff University (GB1810965.2), which covers the compounds discussed in this work.
Supplementary Material
References
- Morita C. T.; Jin C.; Sarikonda G.; Wang H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vγ2Vδ2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 2007, 215, 59–76. 10.1111/j.1600-065x.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Zhou D.; Qiu L.; Lai X.; Simon M.; Shen L.; Kou Z.; Wang Q.; Jiang L.; Estep J.; Hunt R.; Clagett M.; Sehgal P. K.; Li Y.; Zeng X.; Morita C. T.; Brenner M. B.; Letvin N. L.; Chen Z. W. Adaptive immune response of Vγ2Vδ2+ T cells during mycobacterial infections. Science 2002, 295, 2255–2258. 10.1126/science.1068819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. P.; Heuijerjans J.; Yan M.; Gustafsson K.; Anderson J. γδ T cells for cancer immunotherapy. Oncoimmunology 2014, 3, e27572 10.4161/onci.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraka S.; Kennel K. A. Bisphosphonates for the prevention and treatment of osteoporosis. Br. Med. J. 2015, 351, h3783. 10.1136/bmj.h3783. [DOI] [PubMed] [Google Scholar]
- Neville-Webbe H. L.; Holen I.; Coleman R. E. The anti-tumour activity of bisphosphonates. Canc. Treat Rev. 2002, 28, 305–319. 10.1016/s0305-7372(02)00095-6. [DOI] [PubMed] [Google Scholar]
- Kunzmann V.; Bauer E.; Feurle J.; Tony F. W. H.-P.; Wilhelm M.; Wilhelm M. Stimulation of γδ T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. 10.1182/blood.v96.2.384. [DOI] [PubMed] [Google Scholar]
- Gober H.-J.; Kistowska M.; Angman L.; Jenö P.; Mori L.; De Libero G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. 10.1084/jem.20021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt R. E.; Lissina A.; Green A. E.; Slay E. S.; Price D. A.; Sewell A. K. The bisphosphonate acute phase response: rapid and copious production of proinflammatory cytokines by peripheral blood γδ T cells in response to aminobisphosphonates is inhibited by statins. Clin. Exp. Immunol. 2005, 139, 101–111. 10.1111/j.1365-2249.2005.02665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson K.; Rojas-Navea J.; Rogers M. J. Alkylamines cause Vγ9Vδ2 T-cell activation and proliferation by inhibiting the mevalonate pathway. Blood 2006, 107, 651–654. 10.1182/blood-2005-03-1025. [DOI] [PubMed] [Google Scholar]
- Hintz M.; Reichenberg A.; Altincicek B.; Bahr U.; Gschwind R. M.; Kollas A.-K.; Beck E.; Wiesner J.; Eberl M.; Jomaa H. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human γδ T cells in Escherichia coli. FEBS Lett. 2001, 509, 317–322. 10.1016/s0014-5793(01)03191-x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Morita C. T.; Tanaka Y.; Nieves E.; Brenner M. B.; Bloom B. R. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature 1995, 375, 155–158. 10.1038/375155a0. [DOI] [PubMed] [Google Scholar]
- Vavassori S.; Kumar A.; Wan G. S.; Ramanjaneyulu G. S.; Cavallari M.; El Daker S.; Beddoe T.; Theodossis A.; Williams N. K.; Gostick E.; Price D. A.; Soudamini D. U.; Voon K. K.; Olivo M.; Rossjohn J.; Mori L.; De Libero G. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 2013, 14, 908–916. 10.1038/ni.2665. [DOI] [PubMed] [Google Scholar]
- Rhodes D. A.; Chen H.-C.; Price A. J.; Keeble A. H.; Davey M. S.; James L. C.; Eberl M.; Trowsdale J. Activation of human γδ T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. 10.4049/jimmunol.1401064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom A.; Peigné C.-M.; Léger A.; Crooks J. E.; Konczak F.; Gesnel M.-C.; Breathnach R.; Bonneville M.; Scotet E.; Adams E. J. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vγ9Vδ2 T cells. Immunity 2014, 40, 490–500. 10.1016/j.immuni.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Morita C. T. Sensor function for butyrophilin 3A1 in prenyl pyrophosphate stimulation of human Vγ2Vδ2 T cells. J. Immunol. 2015, 195, 4583–4594. 10.4049/jimmunol.1500314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim M.; Knowles T. J.; Baker A. T.; Davey M. S.; Jeeves M.; Sridhar P.; Wilkie J.; Willcox C. R.; Kadri H.; Taher T. E.; Vantourout P.; Hayday A.; Mehellou Y.; Mohammed F.; Willcox B. E. BTN3A1 discriminates γδ T cell phosphoantigens from nonantigenic small molecules via a conformational sensor in its B30.2 domain. ACS Chem. Biol. 2017, 12, 2631–2643. 10.1021/acschembio.7b00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigau M.; Ostrouska S.; Fulford T. S.; Johnson D. N.; Woods K.; Ruan Z.; McWilliam H. E. G.; Hudson C.; Tutuka C.; Wheatley A. K.; Kent S. J.; Villadangos J. A.; Pal B.; Kurts C.; Simmonds J.; Pelzing M.; Nash A. D.; Hammet A.; Verhagen A. M.; Vairo G.; Maraskovsky E.; Panousis C.; Gherardin N. A.; Cebon J.; Godfrey D. I.; Behren A.; Uldrich A. P. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020, 367, eaay5516 10.1126/science.aay5516. [DOI] [PubMed] [Google Scholar]
- Karunakaran M. M.; Willcox C. R.; Salim M.; Paletta D.; Fichtner A. S.; Noll A.; Starick L.; Nöhren A.; Begley C. R.; Berwick K. A.; Chaleil R. A. G.; Pitard V.; Déchanet-Merville J.; Bates P. A.; Kimmel B.; Knowles T. J.; Kunzmann V.; Walter L.; Jeeves M.; Mohammed F.; Willcox B. E.; Herrmann T. Butyrophilin-2A1 directly binds germline-encoded regions of the Vγ9Vδ2 TCR and is essential for phosphoantigen sensing. Immunity 2020, 52, 487–498. 10.1016/j.immuni.2020.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao C.-H. C.; Lin X.; Barney R. J.; Shippy R. R.; Li J.; Vinogradova O.; Wiemer D. F.; Wiemer A. J. Synthesis of a phosphoantigen prodrug that potently activates Vγ9Vδ2 T-lymphocytes. Chem. Biol. 2014, 21, 945–954. 10.1016/j.chembiol.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Davey M. S.; Malde R.; Mykura R. C.; Baker A. T.; Taher T. E.; Le Duff C. S.; Willcox B. E.; Mehellou Y. Synthesis and biological evaluation of (E)-4-hydroxy-3-methylbut-2-enyl phosphate (HMBP) aryloxy triester phosphoramidate prodrugs as activators of Vγ9/Vδ2 T-cell immune responses. J. Med. Chem. 2018, 61, 2111–2117. 10.1021/acs.jmedchem.7b01824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl M.; Hintz M.; Reichenberg A.; Kollas A.-K.; Wiesner J.; Jomaa H. Microbial isoprenoid biosynthesis and human γδ T cell activation. FEBS Lett. 2003, 544, 4–10. 10.1016/s0014-5793(03)00483-6. [DOI] [PubMed] [Google Scholar]
- Reichenberg A.; Hintz M.; Kletschek Y.; Kuhl T.; Haug C.; Engel R.; Moll J.; Ostrovsky D. N.; Jomaa H.; Eberl M. Replacing the pyrophosphate group of HMB-PP by a diphosphonate function abrogates its potential to activate human γδ T cells but does not lead to competitive antagonism. Bioorg. Med. Chem. Lett. 2003, 13, 1257–1260. 10.1016/s0960-894x(03)00138-0. [DOI] [PubMed] [Google Scholar]
- Gossman W.; Oldfield E. Quantitative structure--activity relations for γδ T cell activation by phosphoantigens. J. Med. Chem. 2002, 45, 4868–4874. 10.1021/jm020224n. [DOI] [PubMed] [Google Scholar]
- van Beek E.; Pieterman E.; Cohen L.; Löwik C.; Papapoulos S. Nitrogen-containing bisphosphonates inhibit isopentenyl pyrophosphate isomerase/farnesyl pyrophosphate synthase activity with relative potencies corresponding to their antiresorptive potencies in vitro and in vivo. Biochem. Biophys. Res. Commun. 1999, 255, 491–494. 10.1006/bbrc.1999.0224. [DOI] [PubMed] [Google Scholar]
- Sanders J. M.; Song Y.; Chan J. M. W.; Zhang Y.; Jennings S.; Kosztowski T.; Odeh S.; Flessner R.; Schwerdtfeger C.; Kotsikorou E.; Meints G. A.; Gómez A. O.; González-Pacanowska D.; Raker A. M.; Wang H.; van Beek E. R.; Papapoulos S. E.; Morita C. T.; Oldfield E. Pyridinium-1-yl bisphosphonates are potent inhibitors of farnesyl diphosphate synthase and bone resorption. J. Med. Chem. 2005, 48, 2957–2963. 10.1021/jm040209d. [DOI] [PubMed] [Google Scholar]
- Osgerby L.; Lai Y.-C.; Thornton P. J.; Amalfitano J.; Le Duff C. S.; Jabeen I.; Kadri H.; Miccoli A.; Tucker J. H. R.; Muqit M. M. K.; Mehellou Y. Kinetin riboside and its ProTides activate the Parkinson’s disease associated PTEN-induced putative kinase 1 (PINK1) independent of mitochondrial depolarization. J. Med. Chem. 2017, 60, 3518–3524. 10.1021/acs.jmedchem.6b01897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y.; Valente R.; Mottram H.; Walsby E.; Mills K. I.; Balzarini J.; McGuigan C. Phosphoramidates of 2′-β-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem. 2010, 18, 2439–2446. 10.1016/j.bmc.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. An investigation into the anti-HIV activity of 2′,3′-didehydro-2′,3′-dideoxyuridine (d4U) and 2′,3′-dideoxyuridine (ddU) phosphoramidate “ProTide” derivatives. Org. Biomol. Chem. 2009, 7, 2548–2553. 10.1039/b904276h. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Rattan H. S.; Balzarini J. The ProTide prodrug technology: from the concept to the clinic. J. Med. Chem. 2018, 61, 2211–2226. 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y. The ProTides boom. ChemMedChem 2016, 11, 1114–1116. 10.1002/cmdc.201600156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clercq E. D.; Holý A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discovery 2005, 4, 928–940. 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- Elliott T. S.; Slowey A.; Ye Y.; Conway S. J. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. MedChemComm 2012, 3, 735–751. 10.1039/c2md20079a. [DOI] [Google Scholar]
- Lentini N. A.; Foust B. J.; Hsiao C.-H. C.; Wiemer A. J.; Wiemer D. F. Phosphonamidate prodrugs of a butyrophilin ligand display plasma stability and potent Vγ9Vδ2 T cell stimulation. J. Med. Chem. 2018, 61, 8658–8669. 10.1021/acs.jmedchem.8b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vougioukalakis G. C.; Grubbs R. H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746–1787. 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Higa M. T.; Cheung N. H.; McKenna M.-C. The facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 1977, 18, 155–158. 10.1016/s0040-4039(01)92575-4. [DOI] [Google Scholar]
- Bessières M.; Sari O.; Roy V.; Warszycki D.; Bojarski A. J.; Nolan S. P.; Snoeck R.; Andrei G.; Schinazi R. F.; Agrofoglio L. A. Sonication-assisted synthesis of (E)-2-methyl-but-2-enyl nucleoside phosphonate prodrugs. ChemistrySelect 2016, 1, 3108–3113. 10.1002/slct.201600879. [DOI] [Google Scholar]
- Martin B. P.; Vasilieva E.; Dupureur C. M.; Spilling C. D. Synthesis and comparison of the biological activity of monocyclic phosphonate, difluorophosphonate and phosphate analogs of the natural AChE inhibitor cyclophostin. Bioorg. Med. Chem. 2015, 23, 7529–7534. 10.1016/j.bmc.2015.10.044. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Zhou X.; Chou T.-F.; Ghosh B.; Liu B.; Wagner C. R. Identification of the amino acid-AZT-phosphoramidase by affinity T7 phage display selection. Bioorg. Med. Chem. Lett. 2009, 19, 6379–6381. 10.1016/j.bmcl.2009.09.067. [DOI] [PubMed] [Google Scholar]
- Murakami E.; Tolstykh T.; Bao H.; Niu C.; Steuer H. M. M.; Bao D.; Chang W.; Espiritu C.; Bansal S.; Lam A. M.; Otto M. J.; Sofia M. J.; Furman P. A. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. 10.1074/jbc.m110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congiatu C.; Brancale A.; McGuigan C. Molecular modelling studies on the binding of some protides to the putative human phosphoramidase Hint1. Nucleosides, Nucleotides Nucleic Acids 2007, 26, 1121–1124. 10.1080/15257770701521656. [DOI] [PubMed] [Google Scholar]
- Davey M. S.; Lin C.-Y.; Roberts G. W.; Heuston S.; Brown A. C.; Chess J. A.; Toleman M. A.; Gahan C. G. M.; Hill C.; Parish T.; Williams J. D.; Davies S. J.; Johnson D. W.; Topley N.; Moser B.; Eberl M. Human neutrophil clearance of bacterial pathogens triggers anti-microbial γδ T cell responses in early infection. PLoS Pathog. 2011, 7, e1002040 10.1371/journal.ppat.1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran J. R.; Cameron T. O.; Stern L. J. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity 2000, 12, 241–250. 10.1016/s1074-7613(00)80177-6. [DOI] [PubMed] [Google Scholar]
- Slusarczyk M.; Lopez M. H.; Balzarini J.; Mason M.; Jiang W. G.; Blagden S.; Thompson E.; Ghazaly E.; McGuigan C. Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
- Foust B. J.; Poe M. M.; Lentini N. A.; Hsiao C.-H. C.; Wiemer A. J.; Wiemer D. F. Mixed aryl phosphonate prodrugs of a butyrophilin ligand. ACS Med. Chem. Lett. 2017, 8, 914–918. 10.1021/acsmedchemlett.7b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust B. J.; Li J.; Hsiao C. H. C.; Wiemer D. F.; Wiemer A. J. Stability and efficiency of mixed aryl phosphonate prodrugs. ChemMedChem 2019, 14, 1597–1603. 10.1002/cmdc.201900344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Murziani P.; Slusarczyk M.; Gonczy B.; Vande Voorde J.; Liekens S.; Balzarini J. Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem. 2011, 54, 7247–7258. 10.1021/jm200815w. [DOI] [PubMed] [Google Scholar]
- Townsend A.; Rothbard J.; Gotch F. M.; Bahadur G.; Wraith D.; McMichael A. J. The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 1986, 44, 959–968. 10.1016/0092-8674(86)90019-x. [DOI] [PubMed] [Google Scholar]
- Wei H.; Huang D.; Lai X.; Chen M.; Zhong W.; Wang R.; Chen Z. W. Definition of APC presentation of phosphoantigen (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate to Vγ2Vδ2 TCR. J. Immunol. 2008, 181, 4798–4806. 10.4049/jimmunol.181.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler D. W.; Copier J.; Dalgleish A. G.; Bodman-Smith M. D. Zoledronic acid causes γδ T cells to target monocytes and down-modulate inflammatory homing. Immunology 2014, 143, 539–549. 10.1111/imm.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertner-Dardenne J.; Castellano R.; Mamessier E.; Garbit S.; Kochbati E.; Etienne A.; Charbonnier A.; Collette Y.; Vey N.; Olive D. Human Vγ9Vδ2 T cells specifically recognize and kill acute myeloid leukemic blasts. J. Immunol. 2012, 188, 4701–4708. 10.4049/jimmunol.1103710. [DOI] [PubMed] [Google Scholar]
- Rincon-Orozco B.; Kunzmann V.; Wrobel P.; Kabelitz D.; Steinle A.; Herrmann T. Activation of Vγ9Vδ2 T cells by NKG2D. J. Immunol. 2005, 175, 2144–2151. 10.4049/jimmunol.175.4.2144. [DOI] [PubMed] [Google Scholar]
- Lima C. D.; Klein M. G.; Hendrickson W. A. Structure-based analysis of catalysis and substrate definition in the HIT protein family. Science 1997, 278, 286–290. 10.1126/science.278.5336.286. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. 10.1021/ci300314k. [DOI] [PubMed] [Google Scholar]
- McGann M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- McGann M. R.; Almond H. R.; Nicholls A.; Grant J. A.; Brown F. K. Gaussian docking functions. Biopolymers 2003, 68, 76–90. 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.