

Abstract
The identification and lead optimization of a series of pyrazolo[3,4-d]pyridazinone derivatives are described as a novel class of potent irreversible BTK inhibitors, resulting in the discovery of compound 8. Compound 8 exhibited high potency against BTK kinase and acceptable PK profile. Furthermore, compound 8 demonstrated significant in vivo efficacy in a mouse-collagen-induced arthritis (CIA) model.
Keywords: BTK inhibitor, irreversible, pyridazinone, enzymatic potency, rheumatoid arthritis
Bruton’s tyrosine kinase (BTK) is an important member of the Tec family of non-receptor tyrosine kinases mostly expressed in B cells, mast cells, and macrophages and plays a central role in B-cell survival, activation, proliferation, differentiation, and maturation through multiple signaling pathways downstream of the B cell receptor (BCR) and Fcγ receptor (FcR).1−4 Therefore, BTK has been considered as a promising and attractive target for the treatment of B cell related diseases, such as hematological malignancies and rheumatoid arthritis (RA).5,6
In fact, many research groups have been focusing their efforts on the development of small molecular BTK inhibitors. On the basis of the binding modes with the BTK catalytic domains, the reported small molecular BTK inhibitors are generally divided into irreversible inhibitors and reversible inhibitors.7−12 The irreversible BTK inhibitors can achieve a strong binding to the relatively unique Cys481 residue of BTK enzyme and exhibit a powerful advantage.5 Among them, ibrutinib is the most successful example and has been approved by the U.S. FDA as the first-in-class irreversible BTK inhibitor for the treatment of mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), and Waldenström’s macroglobulinemia (WM).13−17 However, the indication development of RA was limited due to possible off-target side effects.18 Subsequently, several other irreversible BTK inhibitors,19−23 such as HM71224,19 ONO-4029,20 CC-292,21 and evobrutinib,23 have been also advanced into clinical trials for the treatment of autoimmune diseases (Figure 1).
Figure 1.

Representative chemical structures of irreversible BTK inhibitors.
The BTK inhibitors in Figure 1 contain different pyrimidine scaffolds with an acrylamide group as Michael acceptor in the structures, which can form a covalent bond with Cys481. Inspired by these pioneering works, our medicinal strategy was to keep the acrylamide moiety as a Michael acceptor unchanged initially and search for a novel structural motif as the central core, hoping to generate irreversible BTK inhibitor with potent activity, improved PK property, and safety. Herein we describe the discovery of a novel 4-amino-1H-pyrazolo[3,4-d]pyridazin-7(6H)-one scaffold and our early lead optimization efforts in this series, resulting in compound 8 as a highly potent BTK inhibitor for RA (Figure 2).24
Figure 2.
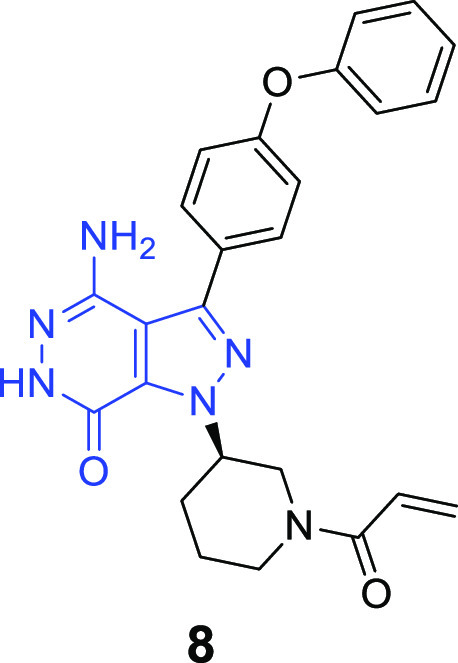
Structure of compound 8.
Our primary optimization is focused on probing different scaffolds that replace a pyrazolo[3,4-d]pyrimidine in the ibrutinib structure (Figure 3). Compound 1 was initially synthesized by cleaving the pyrazolo ring of ibrutinib and evaluated in vitro activity against the BTK enzyme. Unfortunately, compound 1 displayed weak activity against BTK with IC50 values of 921 nM. Cyclization of compound 1 led to different products 2–4, which also exhibited poor inhibition of the enzyme (>10 μM, 1675 nM, and >10 μM, respectively). In addition, the introduction of a phenyl group into the pyrazolo[3,4-d]pyrimidine showed similarly poor potency to inhibit the BTK kinase, possessing an IC50 value of 1244 nM (5). Changing the pyrimidinamine of compound 5 into isoxazol-3-amine resulted in a 3.7-fold decrease in activity (6, IC50 = 4664 nM). The replacement of pyrazolo[3,4-d]pyrimidine of ibrutinib with pyrazolo[3,4-d]oxazinone also afforded weaker potency against BTK (7). To our delight, further replacing the “O” of the pyrazolo[3,4-d]oxazinone ring with a “NH” led to pyrazolo[3,4-d]pyridazinone derivative 8, showing a significantly increased enzymatic activity with IC50 of 2.1 nM.
Figure 3.

Study of novel core and BTK IC50. BTK IC50 values are shown as single determinations. BTK IC50 of ibrutinib is 2.0 nM.
Docking study of compound 8 with BTK was carried out via covalent docking protocol of Glide.25 As illustrated in Figure 4, compound 8 could covalently bind to Cys481 and formed an HB network with hinge key residues Met477, Glu475, and gatekeeper Thr474 (Figure 4a). The binding mode of compound 8 could overlay well with that of ibrutinib (cocrystallized with BTK, PDB code 5P9J) (Figure 4b). The piperidine and diphenyl ether groups of compound 8 formed the same orientation with that of ibrutinib, and the terminal phenyl group inserted into a hydrophobic pocket to form π–π stacking with Phe540. Although compound 8 can potentially bind well to the BTK model, the calculated strain energy of its binding mode is high. Therefore, originally it was not recommended with high priority for the synthesis. However, the high potency of compound 8 reflected that it was sometimes tricky to evaluate compounds by using ligand strain energy.
Figure 4.
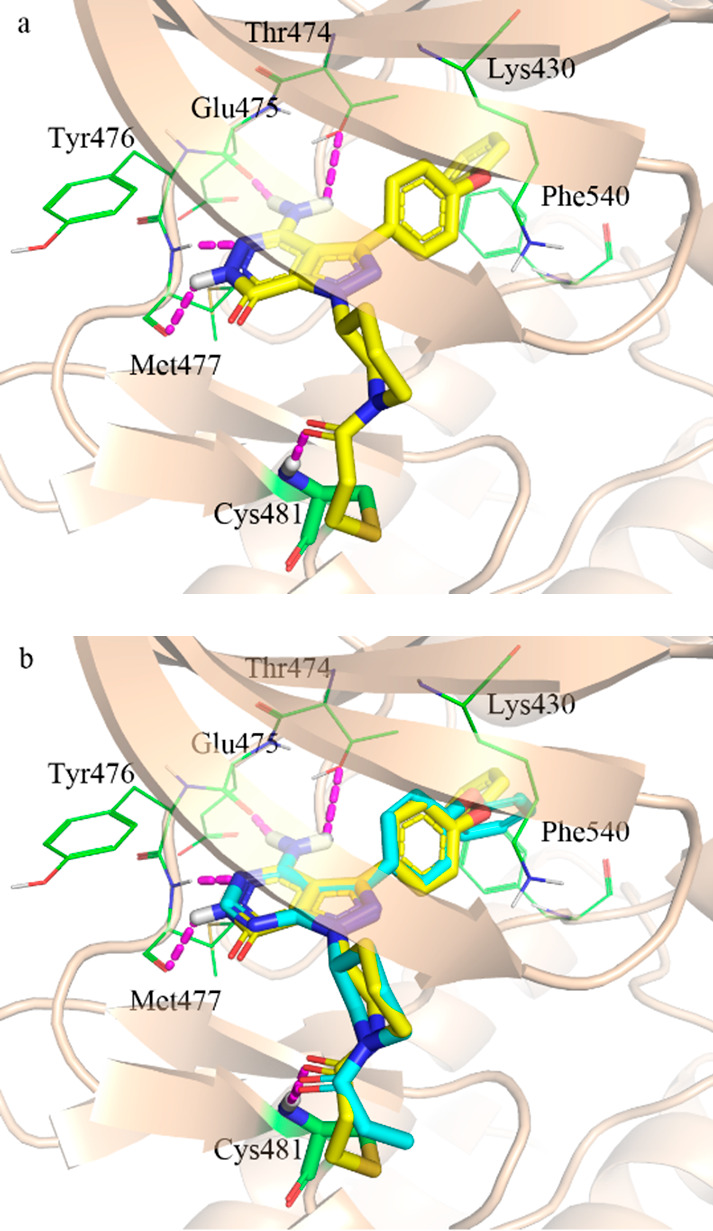
Predicted binding mode of compound 8 (a) and overlaid compound 8 with ibrutinib (b) on BTK (PDB code 5P9J).
Interestingly, the originally predicted binding modes indicated that compounds 5 and 6 could covalently bind well on 3PIY,26 while both compounds showed poor potency. The biological data are not consistent with the computational modeling results. The reason was not clear until the cocrystal structure of ibrutinib (5P9J) was released in 2017.27 Compounds 5 and 6 could not bind well on 5P9J, which has a typical DFG-in conformation, while 3PIY has an atypical DFG conformation. In 5P9J, the Asp539 formed a salt bridge with Lys430, which narrowed down the binding pocket. Therefore, the tricycles core of both compounds could not be accommodated in the binding pocket. The docking template with atypical DFG conformation seemed not appropriate for the prediction in this case. Different from compounds 5 and 6, the core structure of compound 8 is bicyclic and comparably small. Compound 8 could bind well on both typical DFG-in 5P9J and atypical DFG-in 3PIY with high docking score (−11.66 and −10.40 kcal/mol). In atypical DFG-in 3PIY, Phe540 is out of typical DFG position. The terminal phenyl group of compound 8 inserted into the hydrophobic pocket but could not form π–π stacking with Phe540, which led to the decreased docking score (−10.40 vs −11.66 kcal/mol).
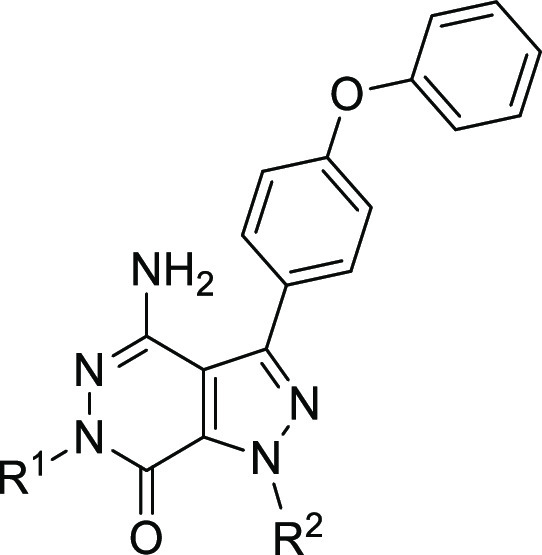
Encouraged by this result, we commenced with investigation on N-substituent groups in pyridazinone ring (R1) and acrylamide moiety containing different alkyl rings (R2) to obtain more potent irreversible BTK inhibitors. The biological and pharmacokinetics data for compounds 8–19 are summarized in Table 1. Replacing the proton of the pyridazinone ring with a methyl led to a decrease of potency for compound 9 (2.1 vs 160 nM). Introduction of larger substituent groups, such as −Et (10) and −CH2CF3 (11), led to complete loss of activity. This indicated that an extra hydrogen bond interaction of NH in the pyridazinone ring with the carbonyl group of Met477 played a critical role for maintaining BTK inhibitory activity. We next turned to optimize the Michael acceptor moiety of the molecule. First, the ring size of the cyclic amino group was investigated. When piperidine in compound 8 was replaced by pyrrolidine, the resulting compound 12 was also very active (IC50 = 1.4 nM). However, this change reduced oral Cmax (252 vs 137 nM) and exposure (128 vs 68 ng·h/mL). Changing the size of ring into four-membered (13) or seven-membered (14) ring resulted in 3- to 5-fold decrease in activity (IC50 = 9.8 nM and 6.7 nM). With regard to compound 15, containing an aza-spiroheptane framework as acrylamide moiety, it exhibited a decreased BTK enzymatic potency, with an IC50 value of 46 nM. In addition, the enzymatic activity of compound 16 bearing acrylamide moiety at the para-position in cyclohexane group was similar to that of compound 8 (1.6 vs 2.1 nM). Compound 16 had better Cmax and AUC, suggesting better PK profile than compound 8; however, a poor solubility affected its druggability, and compound 16 demonstrated a higher human plasma protein binding rate (see Supporting Information p S41). By movement of the para-position acrylamide group of compound 16 to the meta-position, the resulting compound 17 showed 6.3-fold reduced BTK activity. The combination of a short carbon linker and N-methylacrylamide (18) appeared to be beneficial in inhibiting the BTK (IC50 = 1.2 nM) enzyme, albeit with a lower Cmax (123 vs 252 nM) and oral AUC exposure (49.5 vs 128 ng·h/mL). An attempt to introduce dimethyl aminomethyl group at the alkene terminus of compound 18 led to decreased potency, with an IC50 value of 35 nM (19).
Table 1. Structure–Activity Relationship with R1 and R2 Groups.

IC50 values are shown as single determinations.
Oral Cmax (maximum concentration) after 2 mg/kg oral dosing in male Balb/C mice.
Oral AUC (area under the curve) after 2 mg/kg oral dosing in male Balb/C mice.
On the basis of the structure of compound 8, we conducted further SAR studies on the Michael acceptor moiety of this compound. As shown in Table 2, we examined the effect of α,β-unsaturated amide groups on potency. Replacing the acrylamide with vinyl sulfonamide resulted in compound 20, which displayed a 2-fold decrease in activity against BTK (IC50 = 4.2 nM). When a dimethyl aminomethyl group was introduced at the terminal alkene of acrylamide of compound 8, the potency of the resulting compound 21 was similar to that of compound 8 (IC50 = 2.6 nM). However, a larger morpholine group attached to the same position (22) showed a decreased activity relative to compound 21 (13 vs 2.6 nM). In addition, other analogues containing cyano cyclopropylacrylamide (23) or 2-butynamide (24) showed IC50 values of 22 nM and 12 nM, respectively.
Table 2. Structure–Activity Relationship with R3 Group.
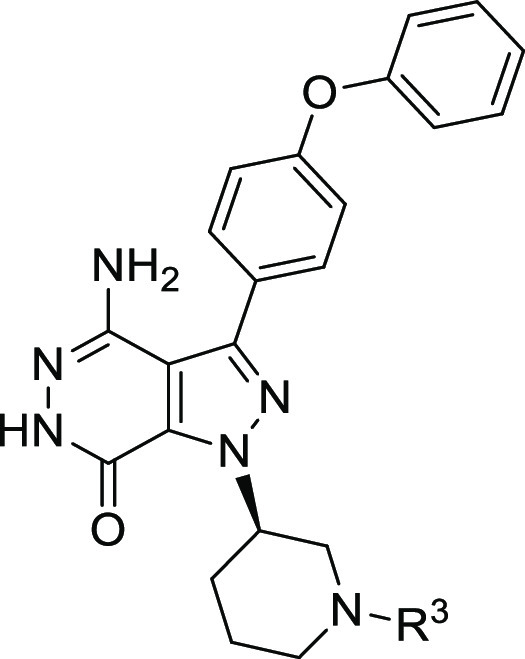

IC50 values are shown as single determinations.
After this preliminary optimization of enzymatic activities, we next examined in vitro and in vivo pharmacokinetic (PK) profiles of compound 8. For comparison, the approved drug ibrutinib was also evaluated as reference. As presented in Table 3, compound 8 and ibrutinib exhibited equivalent potency against BTK kinase (2.1 nM vs 2.0 nM). They also showed >95% plasma protein binding across three species of human, rat, and mouse. No major drug–drug interaction liability was detected since inhibition against all 5 CYP enzyme was IC50 of >10 μM except for ibrutinib’s IC50 of 9.7 μM against 2C9. Similar hERG liability was observed in compound 8 and ibrutinib (5.6 vs 7.7 μM).
Table 3. In Vitro Profiling Data for Compound 8 and Ibrutinib.
| parameter | 8 | ibrutinib |
|---|---|---|
| enzyme IC50 (nM) | 2.1 | 2.0 |
| PPB % bound (h, r, m) | 97.5, 99.7, 95.3 | 96.9, 97.8, 96.2 |
| CYP inhibition (μM) | ||
| 1A2 | >100 | >100 |
| 2C9 | 39.6 | 9.69 |
| 2C19 | 43.9 | 33.5 |
| 2D6 | >100 | >100 |
| 3A4 | 37.3 | 40.8 |
| hERG IC50 (μM) | 5.6 | 7.7 |
Pharmacokinetic parameters of compound 8 and ibrutinib in the rat and mice are summarized in Table 4. After an intravenous injection, similar pharmacokinetic properties with half-life (rat, 0.32 vs 0.32 h; mice, 0.42 vs 0.34 h), clearance (rat, 54.6 vs 53.2 (mL/min0/kg; mice, 31.3 vs 27.4 (mL/min)/kg), volume of distribution (rat, 1.55 vs 1.44 L/kg; mice, 0.82 vs 0.55 L/kg), and AUC exposure (rat, 604 vs 643 ng.h/mL; mice, 576 vs 628 ng.h/mL) were observed in two species. After oral administration, compound 8 exhibited higher Cmax (rat, 466 vs 150 ng/mL; mice, 252 vs 55.3 ng/mL) and plasma exposure (rat, 642 vs 318 ng.h/mL; mice, 128 vs 21.4 ng.h/mL) with a favorable oral bioavailability (rat, 23.7% vs 10.3%; mice, 11.2% vs 1.8%) compared to ibrutinib.
Table 4. Pharmacokinetic Parameters of Compound 8 and Ibrutinib.
|
8 |
ibrutinib |
|||
|---|---|---|---|---|
| for rat | iv | po | iv | po |
| dose (mg/kg) | 2 | 10 | 2 | 10 |
| T1/2 (h) | 0.32 | 0.32 | ||
| Cl ((mL/min)/kg) | 54.6 | 53.2 | ||
| Vdss (L/kg) | 1.55 | 1.44 | ||
| Cmax (ng/mL) | 466 | 150 | ||
| AUC0–last (ng·h/mL) | 604 | 642 | 643 | 318 |
| F (%) | 23.7 | 10.3 | ||
|
8 |
ibrutinib |
|||
|---|---|---|---|---|
| for mice | iv | po | iv | po |
| dose (mg/kg) | 1 | 2 | 1 | 2 |
| T1/2 (h) | 0.42 | 0.34 | ||
| Cl ((mL/min)/kg) | 31.3 | 27.4 | ||
| Vdss (L/kg) | 0.82 | 0.55 | ||
| Cmax (ng/mL) | 252 | 55.3 | ||
| AUC0–last (ng·h/mL) | 576 | 128 | 628 | 21.4 |
| F (%) | 11.2 | 1.8 | ||
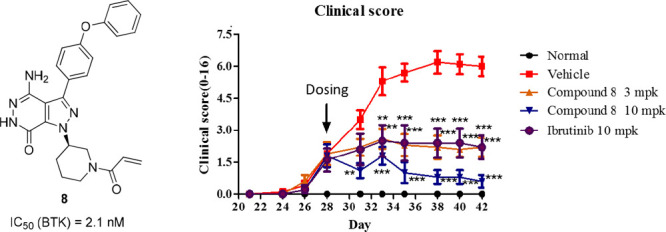
On the basis of its enzymatic potency and pharmacokinetic profile, compound 8 was taken forward for in vivo efficacy study in a mouse collagen-induced arthritis (CIA) model.28 In this study, compound 8 (3 mg/kg or 10 mg/kg) or ibrutinib (10 mg/kg) was dosed by oral gavage, q.d., from day 28 (10 mice per group). As shown in Figure 5, the treatment with compound 8 or ibrutinib for 14 days inhibited the significant progression of the disease compared to the vehicle control, and compound 8 exhibited a clear dose-dependent reduction per paw clinical scores. Notably, the 3 mg/kg regimen of compound 8 provided equivalent efficacy to ibrutinib at 10 mg/kg, and the best in vivo efficacy was observed when dosed 10 mg/kg of compound 8 (Figure 5). Furthermore, compound 8 was also well tolerated, and no significant body weight loss was observed for all different dosages.
Figure 5.

In vivo efficacy of compound 8 and ibrutinib in a mouse collagen-induced arthritis (CIA) model.
Compound 8, which is exemplified in the present article, was prepared starting from the commercially available materials, as outlined in Scheme 1. Compound 25 as the starting material was treated by a series of reactions, including substitution, elimination, and cyclization, to provide the key intermediate 28. The treatment of compound 28 with NIS in the presence of CAN gave the corresponding product 29. Compound 29 was reacted with CuCN in the presence of Pd2(dba)3 and Pd(dppf)2Cl2 in DMF at 100 °C to afford the desired product 30. The treatment of compound 30 with (S)-N-Boc-piperidin-3-ol under Mitsunobu reaction conditions generated the product 31, followed by cyclization in refluxing hydrazine hydrate leading to compound 32. Boc deprotection of compound 32 with hydrochloric acid in EtOAc afforded compound 33. Finally, the product 8 was obtained by the condensation reaction of compound 33 with acrylic acid. This synthetic sequence can be applied to the synthesis of compounds 9–24 by varying the corresponding materials under similar reaction conditions (for details for the synthesis of compounds 1–7 and 9–24, see Supporting Information).
Scheme 1. Synthetic Route of Compound 8.

(i) PhOH, t-BuOK; (ii) diethyl oxalate, NaH, toluene; (iii) hydrazine hydrate, AcOH, EtOH; (iv) NIS, CAN, CH3CN; (v) CuCN, Pd(dppf)Cl2, Pd2(dba)3, DMF; (vi) PPh3, DIAD, THF; (vii) hydrazine hydrate; (viii) HCl/EtOAc; (x) HATU, DIPEA, DCM.
In summary, we designed and synthesized a new series of pyrazolo[3,4-d]pyridazinone derivatives as potent and orally active irreversible BTK inhibitors. Through the exploration of different N-substituent groups in pyridazinone ring and Michael acceptor moieties in pyrazolo[3,4-d]pyridazinone, we successfully developed the SAR profile around this series, resulting in the discovery of compound 8, which has shown high potency against BTK enzyme and acceptable oral PK. Furthermore, compound 8 demonstrated prominent inhibition of arthritis in a mouse collagen-induced arthritis (CIA) model.
Glossary
Abbreviations
- PK
pharmacokinetics
- SAR
structure–activity relationship
- PPB
plasma protein binding
- CYP
cytochrome P450
- hERG
human ether-a-go-go-related gene
- iv
intravenous
- po
per oral
- AUC
area under curve
- mpk
mg/kg
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00395.
Experimental and characterization data for all compounds and all biological data (PDF)
Author Contributions
The manuscript was written by Y.L. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Vetrie D.; Vorechovsky I.; Sideras P.; Holland J.; Davies A.; Flinter F.; Hammarstrom L.; Kinnon C.; Levinsky R.; Bobrow M.; Smith C. I.; Bentley D. R. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- Tsukada S.; Saffran D. C.; Rawlings D. J.; Parolini O.; Allen R. C.; Klisak I.; Sparkes R. S.; Kubagawa H.; Mohandas T.; Quan S.; Belmont J. W.; Cooper M. D.; Conley M. E.; Witte O. N. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. 10.1016/0092-8674(93)90667-F. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A. B.; Witte O. N. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol. Rev. 2000, 175, 120–127. [PubMed] [Google Scholar]
- Buggy J. J.; Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int. Rev. Immunol. 2012, 31, 119–132. 10.3109/08830185.2012.664797. [DOI] [PubMed] [Google Scholar]
- Lou Y.; Owens T. D.; Kuglstatter A.; Kondru R. K.; Goldstein D. M. Bruton’s tyrosine kinase inhibitors: approaches to potent and selective inhibition, preclinical and clinical evaluation for inflammatory diseases and B cell malignancies. J. Med. Chem. 2012, 55, 4539–4550. 10.1021/jm300035p. [DOI] [PubMed] [Google Scholar]
- Hendriks R. W.; Yuvaraj S.; Kil L. P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- Akinleye A.; Chen Y.; Mukhi N.; Song Y.; Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J. Hematol. Oncol. 2013, 6, 59–67. 10.1186/1756-8722-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Sun X. Y.; Jin S. Y.; Yang L.; Xu H. J.; Rao Y. Discovery of 4-Aminoquinoline-3-carboxamide Derivatives as Potent Reversible Bruton’s Tyrosine Kinase Inhibitors for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2019, 62, 6561–6574. 10.1021/acs.jmedchem.9b00329. [DOI] [PubMed] [Google Scholar]
- Crawford J. J.; Johnson A. R.; Misner D. L.; Belmont L. D.; Castanedo G.; Choy R.; Coraggio M.; Dong L. M.; Eigenbrot C.; Erickson R.; Ghilardi N.; Hau J.; Katewa A.; Kohli P. B.; Lee W.; Lubach J. W.; McKenzie B. S.; Ortwine D. F.; Schutt L.; Tay S.; Wei B. Q.; Reif K.; Liu L. C.; Wong H.; Young W. B. Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development. J. Med. Chem. 2018, 61, 2227–2245. 10.1021/acs.jmedchem.7b01712. [DOI] [PubMed] [Google Scholar]
- Liu J.; Guiadeen D.; Krikorian A.; Gao X. L.; Wang J.; Boga S. B.; Alhassan A. B.; Yu Y. N.; Vaccaro H.; Liu S. L.; Yang C. D.; Wu H.; Cooper A.; de Man J.; Kaptein A.; Maloney K.; Hornak V.; Gao Y. D.; Fischmann T. O.; Raaijmakers H.; Vu-Pham D.; Presland J.; Mansueto M.; Xu Z. W.; Leccese E.; Zhang-Hoover J.; Knemeyer I.; Garlisi C. G.; Bays N.; Stivers P.; Brandish P. E.; Hicks A.; Kim R.; Kozlowski J. A. Discovery of 8-amino-imidazo[1,5-a]pyrazines as reversible BTK inhibitors for the treatment of rheumatoid arthritis. ACS Med. Chem. Lett. 2016, 7, 198–203. 10.1021/acsmedchemlett.5b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y.; Han X. C.; Kuglstatter A.; Kondru R. K.; Sweeney Z. K.; Soth M.; McIntosh J.; Litman R.; Suh J.; Kocer B.; Davis D.; Park J.; Frauchiger S.; Dewdney N.; Zecic H.; Taygerly J. P.; Sarma K.; Hong J.; Hill R. J.; Gabriel T.; Goldstein D. M.; Owens T. D. Structure-based drug design of RN486, a potent and selective Bruton’s tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. J. Med. Chem. 2015, 58, 512–516. 10.1021/jm500305p. [DOI] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S. J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd J. C.; Furman R. R.; Coutre S. E.; Flinn I. W.; Burger J. A.; Blum K. A.; Grant B.; Sharman J. P.; Coleman M.; Wierda W. G.; Jones J. A.; Zhao W.; Heerema N. A.; Johnson A. J.; Sukbuntherng J.; Chang B. Y.; Clow F.; Hedrick E.; Buggy J. J.; James D. F.; O’Brien S. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. L.; Simon R.; Peter M.; Andre G.; Rebecca A.; Kahl B. S.; Wojciech J.; Advani R. H.; Romaguera J. E.; Williams M. E.; et al. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treon S. P.; Tripsas C. K.; Meid K.; Warren D.; Varma G.; Green R.; Argyropoulos K. V.; Yang G.; Cao Y.; Xu L.; Patterson C. J.; Rodig S.; Zehnder J. L.; Aster J. C.; Harris N. L.; Kanan S.; Ghobrial I.; Castillo J. J.; Laubach J. P.; Hunter Z. R.; Salman Z.; Li J.; Cheng M.; Clow F.; Graef T.; Palomba M. L.; Advani R. H. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- Caldwell R. D.; Qiu H.; Askew B. C.; Bender A. T.; Brugger T.; Camps M.; Dhanabal M.; Dutt V.; Eichhorn T.; Gardberg A. S.; Goutopoulos A.; Grenningloh R.; Head J.; Healey B.; Hodous B. L.; Huck B. R.; Johnson T. L.; Jones C.; Jones R. C.; Mochalkin I.; Morandi F.; Nguyen N.; Meyring M.; Potnick J. R.; Santos D. C.; Schmidt R.; Sherer B.; Shutes A.; Urbahns K.; Follis A. V.; Wegener A. A.; Zimmerli S. C.; Liu-Bujalski L. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. 10.1021/acs.jmedchem.9b00794. [DOI] [PubMed] [Google Scholar]
- Park J. K.; Park J. A.; Lee Y. J.; Song J.; Oh J. I.; Lee Y.-M.; Suh K. H.; Son J.; Lee E. B. THU0499 HM71224, a novel oral BTK inhibitor, inhibits human immune cell activation: new drug candidate to treat B-cell associated autoimmune diseases. Ann. Rheum. Dis. 2014, 73, 355–356. 10.1136/annrheumdis-2014-eular.2783. [DOI] [Google Scholar]
- Yasuhiro T.; Yoshizawa T.; Birkett J. T. P.; Kawabata K. ONO-4059, A novel Bruton’s tyrosine kinase (Btk) inhibitor: synergistic activity in combination with chemotherapy in a ABC-DLBCL Cell line. Blood 2013, 122, 5151–5151. 10.1182/blood.V122.21.5151.5151. [DOI] [Google Scholar]
- Evans E. K.; Tester R.; Aslanian S.; Karp R.; Sheets M.; Labenski M. T.; Witowski S. R.; Lounsbury H.; Chaturvedi P.; Mazdiyasni H.; Zhu Z.; Nacht M.; Freed M. L.; Petter R. C.; Dubrovskiy A.; Singh J.; Westlin W. F. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- Byrd J. C.; Harrington B.; O’Brien S.; Jones J. A.; Schuh A.; Devereux S.; Chaves J.; Wierda W. G.; Awan F. T.; Brown J. R.; Hillmen P.; Stephens D. M.; Ghia P.; Barrientos J. C.; Pagel J. M.; Woyach J.; Johnson D.; Huang J.; Wang X.; Kaptein A.; Lannutti B. J.; Covey T.; Fardis M.; McGreivy J.; Hamdy A.; Rothbaum W.; Izumi R.; Diacovo T. G.; Johnson A. J.; Furman R. R. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselmayer P.; Camps M.; Liu-Bujalski L.; Nguyen N.; Morandi F.; Head J.; O’Mahony A.; Zimmerli S. C.; Bruns L.; Bender A. T.; Schroeder P.; Grenningloh R. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J. Immunol. 2019, 202, 2888–2906. 10.4049/jimmunol.1800583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. H.; Wu C. D.; Xu Y.; Shen C. L.; Li L. E.; Hu G. P.; Yue Y.; Li J.; Guo D.; Shi N. Y.; Huang L.; Chen S. H.; Tu R. H.; Yang Z. W.; Zhang X. W.; Xiao Q.; Tian H.; Yu Y. P.; Chen H. L.; Sun W. J.; He Z. Y.; Shen J.; Yang J.; Tang J.; Zhou W.; Yu J.; Zhang Y.; Liu Q.. BTK Inhibitor. WO 2016112637 A1, July 21, 2016.
- Schrödinger Release 2016-3: Glide; Schrödinger, LLC: New York, NY, 2016.
- Kuglstatter A.; Wong A.; Tsing S.; Lee S. W.; Lou Y.; Villaseñor A. G.; Bradshaw J. M.; Shaw D.; Barnett J. W.; Browner M. F. Insights into the conformational flexibility of Bruton’s tyrosine kinase from multiple ligand complex structures. Protein Sci. 2011, 20, 428–436. 10.1002/pro.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T.; Gardberg A.; Pereira A.; Johnson T.; Wu Y.; Grenningloh R.; Head J.; Morandi F.; Haselmayer P.; Liu-Bujalski L. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 2017, 91, 208–219. 10.1124/mol.116.107037. [DOI] [PubMed] [Google Scholar]
- Clinical arthritis scores from daily oral treatment from day 28 to day 42 with different doses of compound 8 or ibrutinib are presented as the mean ± SEM (n = 10). Disease control mice received vehicle only. Scores from ibrutinib (10 mg/kg) or 8 treatments at 3 and 10 mg/kg were significantly different compared with control (*p < 0.05, **p < 0.01, ***p < 0.001 vs control).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.