

Abstract

Focal adhesion kinase (FAK), a cytoplasmic protein tyrosine kinase, exerts kinase-dependent enzymatic functions and kinase-independent scaffolding functions, both of which are crucial in cancer development, early embryonic development, and reproduction. However, previous efforts for FAK blocking mainly focus on kinase inhibitors. Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules that allow direct post-translational knockdown of proteins via ubiquitination of a target protein by E3 ubiquitin ligase and subsequent proteasomal degradation. Here, we designed and synthesized a FAK PROTAC library with FAK inhibitor (PF562271 or VS6063) and CRBN E3 ligand. A novel FAK-targeting PROTAC, FC-11, showed a rapid and reversible FAK degradation with a picomolar of DC50 in various cell lines in vitro, which imply that FAK-PROTACs could be useful as expand tools for studying functions of FAK in biological system and as potential therapeutic agents.
Keywords: FAK, PROTAC, chemical knockdown, protein degradation
Focal adhesion kinase (FAK, ∼125 kDa), also called protein tyrosine kinase 2 (PTK2), was first reported in 1992 as a member of the nonreceptor protein tyrosine kinases (PTKs) subfamily.1 FAK is widely expressed in a variety of species, including human, rodent, chicken, frog, drosophila, and Xenopus, which has a greater than 90% homology in amino acid sequence.2 FAK contains four linearly arranged functional domains from its N terminus (Figure 1a): the FERM (band 4.1, Ezrin, Radixin, Moesin) domain, the catalytic kinase domain, three proline-rich regions (PRI, PRII, PRIII), and the focal adhesion targeting (FAT) domain.3,4 FAK FERM domain contains a nuclear localization sequence (NLS), which has an important role in cellular regulation by binding to membrane proteins (growth factor receptors and chemokine receptors) and nuclear proteins.5−7 FAK kinase domain contains the activation loop and tyrosine sites, which ultimately regulate FAK kinase activity.8 The PRI-III regions and FAT domain mainly participate in various protein–protein interactions, similar to FAK FERM domain.9 Thus, FAK exerts kinase-dependent enzymatic functions and kinase-independent scaffolding functions, both types of function are crucial in cancer development, early embryonic development, reproduction, and so on.10−13
Figure 1.
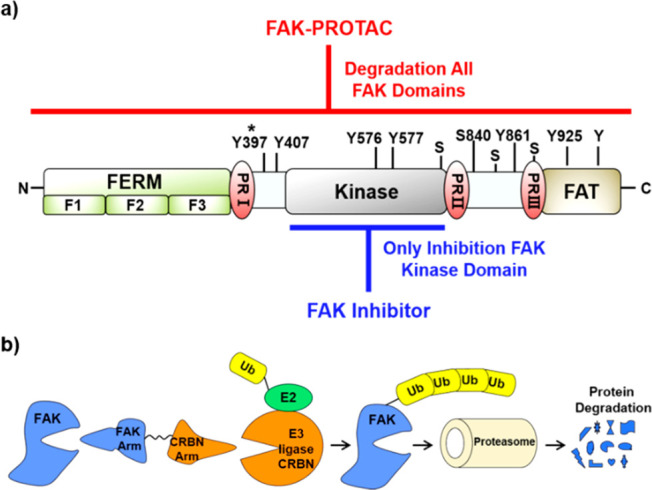
Introduction to FAK protein and FAK-PROTACs. (a) FAK protein schematic. FAK-PROTACs can act on both enzymatic and nonenzymatic functions of FAK, while FAK inhibitor only acts on the enzymatic function of FAK. Y, phosphotyrosine; S, phosphoserine; *, autophosphorylation site (Tyr397); PR, proline-rich regions. (b) Schematic depiction of the PROTAC strategy.
Although a few FAK small molecule inhibitors were developed by major pharmaceutical companies, and some of them have reached clinical trials for varieties of malignant cancers,14−16 essential nonenzymatic functions of FAK cannot be investigated or blocked with reported FAK kinase inhibitors. As conventional kinase inhibitors can only act on the protein kinase domain and may lead to drug resistance. Thus, developing a practical strategy against both kinase-dependent enzymatic functions and kinase-independent scaffolding functions of FAK is an urgent and meaningful need for FAK-related diseases.
Proteolysis targeting chimera (PROTAC) is a novel chemical knockdown technology for the post-translational proteins of interest. PROTACs are heterobifunctional small molecules containing two recognition moieties: one specifically binds an E3 ubiquitin ligase, and the other specifically binds the target protein. PROTAC molecules can drive E3 ubiquitin ligase to the target protein, which results in ubiquitination of the target protein and consequent proteasome-mediated degradation (Figure 1b).17,18 Unlike classic inhibitors, PROTAC eliminates rather than inhibits both enzymatic and nonenzymatic protein functions (Figure 1a). Although two FAK-targeting PROTACs have been reported in previous studies,19,20 the combination of different E3 ligase ligands with different inhibitors of FAK could be valuable for improving the activities and drug-like properties and exploring structure–activity relationships (SARs). Herein, we designed and synthesized a series of different FAK-targeting PROTACs with FAK inhibitor (PF562271 or VS6063) and ligand of CRBN E3 ligase, which could contribute to development of potent tools or potential therapeutic agents for specifically degrading FAK.
Based on the previous studies of our laboratory, linker length, mode of binding to the target protein, and relative spatial orientations of the target protein and E3 ubiquitin ligase are three major critical factors for achieving efficient degradation of the target protein. Optimizing the combination of these three factors is the key to the design of potent and specific PROTACs. To identify the suitable linker length in our FAK-PROTAC library, we designed linkers of different lengths by changing the number units of ethylene glycol (e.g., using diethylene or triethylene glycol). Both the position of the linker connecting to binders and the rigidity of the linker were also investigated to modulate the interaction of the target protein and E3 ligase, and thus directly determine degradation efficacy.
In this study, we chose two FAK inhibitors, VS6063 and PF562271,21 as FAK binders, and one PF562271 analogue as a negative control for FAK binding (Figure 2a). According to the cocrystal structure of FAK and PF562271 (PDB: 5TOB, Figure 2b), the lactam ring of PF562271 was exposed to the solvent region and does not play a significant role in protein binding; therefore, it presented a suitable site to link with ligands of E3 ligase. Based on the above design principles, we constructed a series of FAK targeting PROTACs with a combination of different FAK ligands, diverse linkers, and the ligand of CRBN based E3 ligase22 (Figure 2c) following a general chemical synthetic route (Scheme 1). In brief, the F atom was substituted by N-methylmethanesulfonamide to generate intermediates 1 or 2. Then the cyano group was reduced to a primary amine, which was used to substitute a chloride atom in 2,4-dichloro-5-(trifluoromethyl) pyrimidine. The selectivity of this step was not very good, such that two analogues were obtained. The remained chloride atom was substituted by intermediates 9 or 10 subsequently, and the final PROTACs were produced via click chemistry.
Figure 2.

Design of FAK-PROTACs. (a) Chemical structures of FAK inhibitors. (b) Binding model of FAK inhibitor PF562271 with FAK protein. (c) General structure of the designed FAK-PROTACs.
Scheme 1. Synthetic Route of FAK-PROTACs.
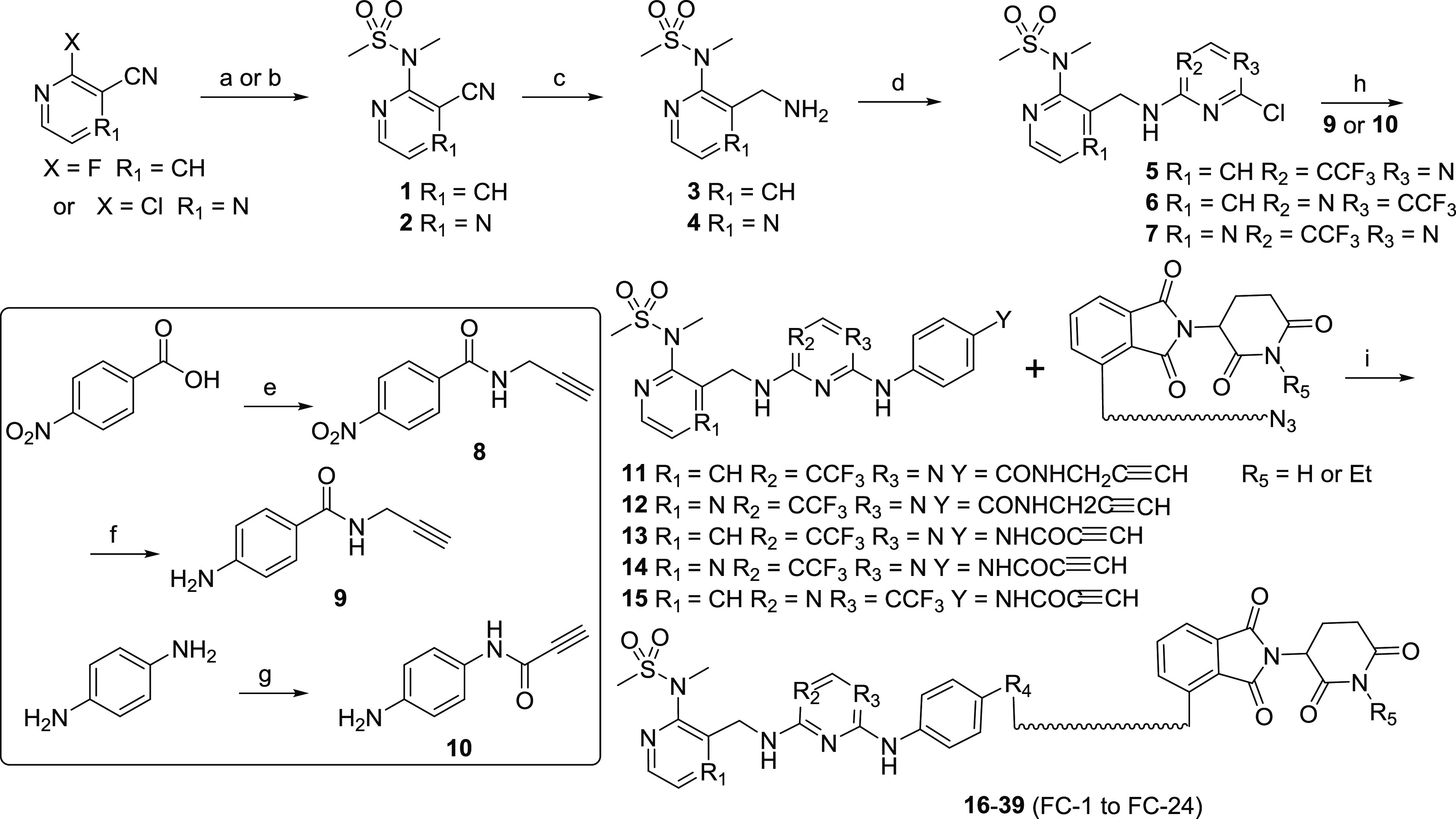
Reaction conditions: (a) t-BuOK, DMF, reflux, 2 h for compound 1; (b) Cs2CO3, MeCN, 70 °C, 20 h for compound 2; (c) Pd/C, H2, EtOH/DMF, rt, 16 h; (d) 2,4-dichloro-5-(trifluoromethyl) pyrimidine, TEA, MeOH, rt, overnight; (e) i: SOCl2, reflux, ii: propargulamine, K2CO3, THF, rt, 16 h; (f) Fe, NH4Cl, EtOH/H2O, reflux, 4 h; (g) propiolic acid, DCC, DMAP, DEE/DMF/CHCl3, rt, 1 h; (h) AcOH, t-amyl alcohol, reflux, 4 h; (i) CuSO4, sodium ascorbate, t-BuOH/H2O, 70 °C, 8 h.
The degradation efficiency of FAK-PROTACs was first evaluated in the human ovarian cancer cell line PA1 at 1 and 10 nM with 8 h treatment. We found that FAK-PROTACs derived from VS6063 or PF562271 with shorter diethylene or triethylene glycol linkers exhibited higher degradation activity: FC-1, DR (protein degradation relative to DMSO)10 nM = 71%; FC-4, DR10 nM = 90%; FC-8, DR10 nM = 96%; FC-11, DR10 nM = 99% (Table 1 and Figure S1). These results indicate that a shorter FAK-PROTAC linker is more conducive to promoting a close proximity between CRBN and FAK.
Table 1. Structures and Protein Degradation Activities of FAK-PROTACsa.

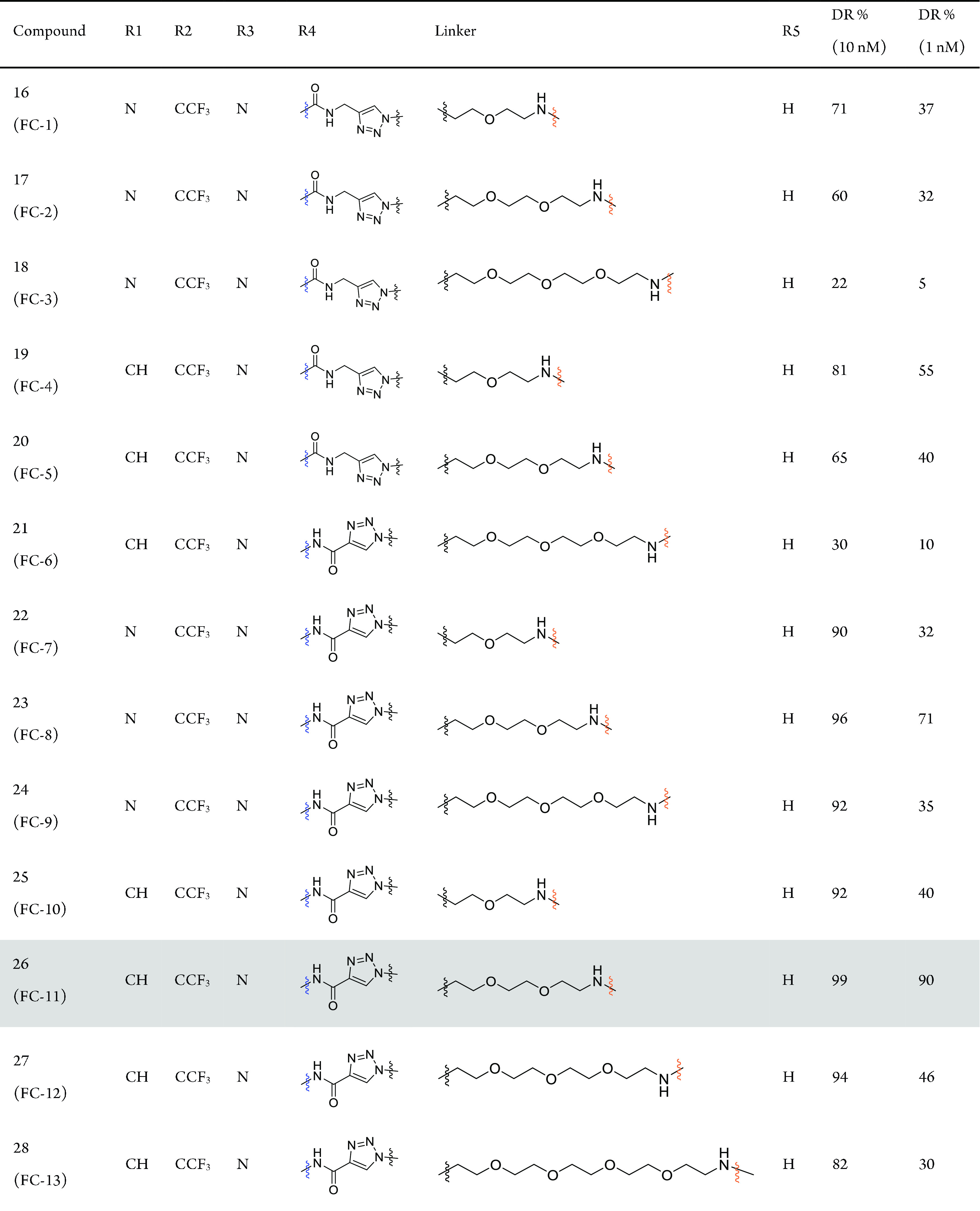
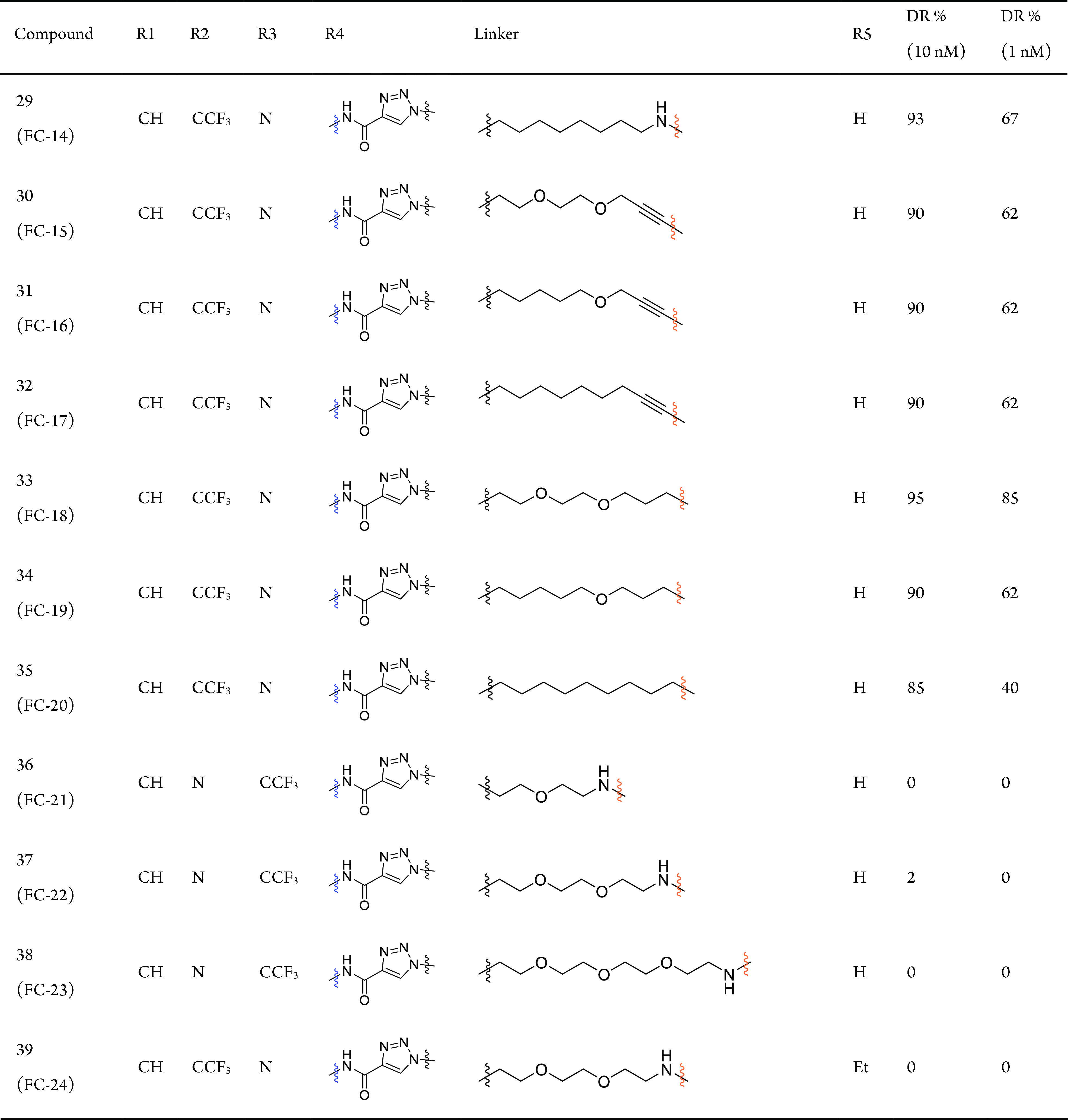
DR, target protein degradation relative to DMSO. The data were repeated in three independent experiments by Western blot method.
In our FAK-PROTAC library, the FAK ligands could present two different orientations of the amide bond (−NHCO– and −CONH−) for extension (Table 1). Our results suggest that PROTACs with a carbonyl group closer to triazole exhibited higher degradation activity (FC-7–FC-9 better than FC-1–FC-3; FC-10–FC-12 better than FC-4–FC-6, Table 1). Therefore, the −NHCO– extending group provided a better angle and spatial orientation for the interaction of FAK and CRBN (Figure S2). At the same time, we observed that the linker length with the highest degradation efficacy was different for these two binding modes: diethylene glycol and triethylene glycol linkers yielded higher degradation activity for −CONH– and −NHCO–, respectively, which seems that different proper linkers formed a special conformation with the highest degradation in these two binding modes.
When the amino group was replaced with a more rigid alkynyl group in the ligand for binding E3 ligase, degradation activity was significantly decreased (FC-15–FC-17 vs FC-11; DR1 nM, 62%, 62%, 62% vs 90%, Table 1 and Figure S1), whereas substitution with a methylene group bearing similar flexibility to the amide group maintained degradation activity (FC-18 vs FC-11; DR1 nM, 85% vs 90%,). These results suggest that the introduction of rigid linking groups on the side of the CRBN ligand will restrict the spatial orientation of CRBN, preventing it from getting close to FAK. Flexible linking groups, however, allow CRBN to swing toward FAK at a favorable angle to form a special conformation and further result in increased degradation potency.
Finally, the composition of linkers also influences degradation activity. A comparison of FC-11 (DR1 nM = 90%), FC-14 (DR1 nM = 67%), FC-19 (DR1 nM = 62%), and FC-20 (DR1 nM = 40%) illustrated that a linker with oxygen atoms could lead to more efficient degradation, which may be related to the special conformation forms, cell permeability, and other possible factors of PROTACs. Taken together, our results revealed that the balance of linkers and binding modes between FAK and CRBN is critical in the design of efficient PROTACs.
Due to the potent degradation efficiency, we chose FC-11 (Figure 3a–c) as the probe to establish chemical FAK-knockdown models in our remaining investigations. We confirmed that it induced FAK degradation via CRBN-mediated and proteasome-dependent mechanisms (Figure 3d,e). PA1 cells were treated with an excess of the CRBN ligand pomalidomide or the FAK ligand PF562271 to compete with FC-11 for binding to E3 ligase and FAK, respectively (Figure 3d). As expected, the excess pomalidomide or PF562271 successfully reduced FAK degradation induced by FC-11, which confirmed that the degradation was mediated by CRBN E3 ubiquitin ligase and required the binding of FC-11 to FAK and CRBN. Pomalidomide or PF562271 treatment alone (at the same concentration) can not result in degradation. Furthermore, a combination of proteasome inhibitor (MG132 or carfilzomib) and FC-11 treatment completely blocked FC-11 induced FAK degradation (Figure 3e). Overall, these results demonstrate that the fast and efficient FAK degradation induced by FC-11 is based on a CRBN-mediated and proteasome-dependent mechanism.
Figure 3.

FC-11 induced degradation of FAK via ubiquitin-proteasome system. (a) Chemical structure of FC-11. (b) FAK degradation at the indicated dose of FC-11. (c) Quantitative analysis of FAK levels after FC-11 treatment. The cells were treated by FC-11 at the indicated doses for 8 h in PA1 cells, and the data was collected from three independent experiments. (d) Confirmation of CRBN-based mechanism in driving degradation of FAK upon FC-11 treatment. PA1 cells were treated for 8 h with FC-11 (+, 1 nM; ++, 10 nM; +++ 100 nM) alone, or pomalidomide (10 μM) or PF562271 (10 μM) alone, or combination of FC-11 with pomalidomide or with PF562271. (e) Confirmation of proteasome-based mechanism in driving degradation of FAK upon FC-11 treatment. PA1 cells were treated for 8 h with FC-11 (+, 1 nM; ++, 10 nM; +++ 100 nM) alone, MG132 (5 μM) or carfilzomib (5 μM) alone, or a combination of FC-11 with MG132 or with carfizomib.
To further assess the efficiency and broad applicability of FC-11, five different cell lines from different species were used (Figure 4a and Figure S3). Surprisingly, after 8 h incubation, the half-maximal degradation concentration (DC50) of all the tested cell lines (both normal and cancer cell lines) were all at the scale of picomolar. The DC50 showed 310 pM in TM3, 80 pM in PA1, 330 pM in MDA-MB-436, 370 pM in LNCaP, and 40 pM in Ramos cells. As with previously reported PROTACs,23 excess FC-11 exhibited the hook effect in some cell lines (Figure S3). Eight hours of FC-11 treatment in PA1 cells resulted in profound FAK degradation, which recovered to normal levels at least one-week post-washout (Figure 4b). More importantly, FC-11 significantly outperformed the FAK inhibitor PF562271 in the reduction of autophosphorylation of FAK (pFAKtyr397) under the same concentration and treatment conditions (Figure 4c). PF562271 showed an inhibitory effect on pFAKtyr397 only at a high dose (3 μM), while FC-11 exhibited a significant effect below 1 nM. Furthermore, in a time-course experiment, FC-11 rapidly decreased FAK and pFAKtyr397 levels, leading to more than 50% protein loss within 1 h at 100 nM in TM3 cells (Figure 4d). The result demonstrated that the potent FAK PROTAC (FC-11) not only has a broad application but also with high degradation activity which dramatically outperforms the FAK inhibitor PF562271 in the reduction of autophosphorylation of FAK (pFAKtyr397).
Figure 4.

Highly potent and broad applicability of FC-11. (a) Half-maximal degradation concentrations (DC50) of FC-11 for FAK in different sources of cell lines. (b) Cellular FAK recovery levels after washout of FC-11 from PA1 cell culture medium. The cells were treated for 8 h before washout. (c) Efficiently decreased pFAKtyr397 levels at the indicated dose of FC-11 for 8 h in TM3 cells. FC-11 significantly exceeds PF562271 on decreasing the levels of pFAKtyr397. (d) Time course reduction of pFAKtyr397 in TM3 cells by FC-11.
Furthermore, we also detected the effect of FC-11 induced FAK degradation on cell proliferation of the tested cell lines (Figure S4). Like the reported FAK PROTACs,19,20 the efficient knockdown of FAK by FC-11 did not more severely affect proliferation of the tested cell lines than the FAK inhibitor PF562271 in 3 days of drug incubation (Figure S4), which provides the question whether the kinase-independent scaffolding function of FAK is required for cell proliferation in vitro in the tested cell lines beyond the effect of inhibition by its kinase-dependent enzymatic activity.
In summary, we demonstrated that one potent FAK degrader, FC-11, exhibits rapid and highly efficient degradation of FAK in various cell lines with a DC50 at picomolar potencies for 8 h treatment. Furthermore, the degraded FAK proteins can be fully recovered after washout of PROTAC molecules, which need only about 1 week in in vitro cell lines. However, like the reported FAK PROTACs, the cell proliferation activity by FAK PROTAC does not significantly exceed beyond FAK inhibitor in the tested cell lines in vitro. Therefore, more in vivo work is required to understand and clarify the biological functions of FAK PROTACs. In addition, the chemical modifications in PROTACs in comparison with the parental inhibitors could reduce the binding affinity with targets and improve the selectivity under most conditions. In particular, PROTACs employ the ubiquitin-proteasome system with a few components and steps, which naturally introduces the essential matching conditions for target degradation and thus improves the selectivity.17,19,20,24,25 Therefore, FC-11 may also be used as a highly specific and potent tool to study FAK-related biology, which may not only help us to understand the FAK biology but could also lead to the development of new therapeutic agents for the therapy of FAK-related diseases.
Acknowledgments
We thank Dr. Wei Wu, Dr. Xiuyun Sun, and Zimo Yang from Tsinghua University for their kind help. This work was supported by the National Natural Science Foundation of China (#81573277, 81622042, 81773567), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (#SQ2017ZX095003, 2018ZX09711001), the CAMS Innovation Fund for Medical Sciences (CIFMS; No. 2019-I2M-1-003), the China Postdoctoral Science Foundation (No. 2015M571027), and the Tsinghua-Peking University Life Science Center postdoctoral fellowship.
Glossary
ABBREVIATIONS
- FAK
focal adhesion kinase
- PTK2
protein tyrosine kinase 2
- PTKs
protein tyrosine kinases
- PR
proline-rich regions
- FAT
focal adhesion targeting
- NLS
nuclear localization sequence
- PROTACs
proteolysis targeting chimeras
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00372.
Figure S1. The screening of all FAK-PROTACs. Figure S2. The model of FAK-PROTACs with FAK protein and cereblon protein. Figure S3. FAK degradation at the indicated dose of FC-11 in different cell lines for 8 h incubation. Figure S4. Cell proliferation activities on the tested cell lines. Details of cell culture, antibodies, chemical materials, synthesis of all compounds, and 1H NMR spectra of FC-11 (PDF)
Author Contributions
⊥ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Zachary I.; Sinnett-Smith J.; Rozengurt E. Bombesin, vasopressin, and endothelin stimulation of tyrosine phosphorylation in Swiss 3T3 cells. Identification of a novel tyrosine kinase as a major substrate. J. Biol. Chem. 1992, 267, 19031–4. [PubMed] [Google Scholar]
- van Nimwegen M. J.; van de Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem. Pharmacol. 2007, 73, 597–609. 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Li S. Y.; Mruk D. D.; Cheng C. Y. Focal adhesion kinase is a regulator of F-actin dynamics: New insights from studies in the testis. Spermatogenesis 2013, 3, e25385 10.4161/spmg.25385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. E.; Fu W.; Schaller M. D. Focal adhesion kinase: exploring Fak structure to gain insight into function. Int. Rev. Cell Mol. Biol. 2011, 288, 185–225. 10.1016/B978-0-12-386041-5.00005-4. [DOI] [PubMed] [Google Scholar]
- Jung Y.; McCarty J. H. Band 4.1 proteins regulate integrin-dependent cell spreading. Biochem. Biophys. Res. Commun. 2012, 426, 578–84. 10.1016/j.bbrc.2012.08.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. T.; Miller N. L.; Chen X. L.; Tancioni I.; Walsh C. T.; Lawson C.; Uryu S.; Weis S. M.; Cheresh D. A.; Schlaepfer D. D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012, 197, 907–19. 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M. C.; Patel H.; Serrels B.; Lietha D.; Eck M. J. The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–14. 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- Schaller M. D. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–13. 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- Brami-Cherrier K.; Gervasi N.; Arsenieva D.; Walkiewicz K.; Boutterin M. C.; Ortega A.; Leonard P. G.; Seantier B.; Gasmi L.; Bouceba T.; Kadare G.; Girault J. A.; Arold S. T. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014, 33, 356–70. 10.1002/embj.201386399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler B. E.; Sharma V.; Zhou Q.; Jing X.; Pike L. A.; Kerege A. A.; Sams S. B.; Schweppe R. E. FAK Expression, Not Kinase Activity, Is a Key Mediator of Thyroid Tumorigenesis and Protumorigenic Processes. Mol. Cancer Res. 2016, 14, 869–82. 10.1158/1541-7786.MCR-16-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud C.; Dormoy V.; Danilin S.; Lindner V.; Bethry A.; Hochane M.; Coquard C.; Barthelmebs M.; Jacqmin D.; Lang H.; Massfelder T. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth. Int. J. Cancer 2015, 137, 1549–59. 10.1002/ijc.29522. [DOI] [PubMed] [Google Scholar]
- Gogate P. N.; Kurenova E. V.; Ethirajan M.; Liao J.; Yemma M.; Sen A.; Pandey R. K.; Cance W. G. Targeting the C-terminal focal adhesion kinase scaffold in pancreatic cancer. Cancer Lett. 2014, 353, 281–9. 10.1016/j.canlet.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungor-Ordueri N. E.; Mruk D. D.; Wan H. T.; Wong E. W.; Celik-Ozenci C.; Lie P. P.; Cheng C. Y. New insights into FAK function and regulation during spermatogenesis. Histol Histopathol 2014, 29, 977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts W. G.; Ung E.; Whalen P.; Cooper B.; Hulford C.; Autry C.; Richter D.; Emerson E.; Lin J.; Kath J.; Coleman K.; Yao L.; Martinez-Alsina L.; Lorenzen M.; Berliner M.; Luzzio M.; Patel N.; Schmitt E.; LaGreca S.; Jani J.; Wessel M.; Marr E.; Griffor M.; Vajdos F. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–44. 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- Infante J. R.; Camidge D. R.; Mileshkin L. R.; Chen E. X.; Hicks R. J.; Rischin D.; Fingert H.; Pierce K. J.; Xu H.; Roberts W. G.; Shreeve S. M.; Burris H. A.; Siu L. L. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–33. 10.1200/JCO.2011.38.9346. [DOI] [PubMed] [Google Scholar]
- Lee B. Y.; Timpson P.; Horvath L. G.; Daly R. J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–49. 10.1016/j.pharmthera.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Zhao X.; Ding N.; Gao H.; Wu Y.; Yang Y.; Zhao M.; Hwang J.; Song Y.; Liu W.; Rao Y. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Crews C. M. Chemical inducers of targeted protein degradation. J. Biol. Chem. 2010, 285, 11057–60. 10.1074/jbc.R109.078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromm P. M.; Samarasinghe K. T. G.; Hines J.; Crews C. M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- Popow J.; Arnhof H.; Bader G.; Berger H.; Ciulli A.; Covini D.; Dank C.; Gmaschitz T.; Greb P.; Karolyi-Ozguer J.; Koegl M.; McConnell D. B.; Pearson M.; Rieger M.; Rinnenthal J.; Roessler V.; Schrenk A.; Spina M.; Steurer S.; Trainor N.; Traxler E.; Wieshofer C.; Zoephel A.; Ettmayer P. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 2019, 62, 2508–2520. 10.1021/acs.jmedchem.8b01826. [DOI] [PubMed] [Google Scholar]
- Schultze A.; Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Invest. Drugs 2010, 19, 777–88. 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- Fischer E. S.; Bohm K.; Lydeard J. R.; Yang H.; Stadler M. B.; Cavadini S.; Nagel J.; Serluca F.; Acker V.; Lingaraju G. M.; Tichkule R. B.; Schebesta M.; Forrester W. C.; Schirle M.; Hassiepen U.; Ottl J.; Hild M.; Beckwith R. E.; Harper J. W.; Jenkins J. L.; Thoma N. H. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–63. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z.; Lv W.; Su S.; Wu W.; Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.