

Summary
Recently, lipid metabolism reprogramming has been further evidenced in malignancies via the observation of large amounts of lipid droplets (LDs) in human tumors, including in glioblastoma (GBM), the most lethal primary brain tumor. However, the role played by LDs in tumor cells remains unknown. Here, we show that triglycerides (TG), the major components of LDs, serve as a critical energy reservoir to support GBM cell survival. TG/LDs rapidly diminished in GBM cells upon glucose reduction, whereas inhibiting fatty acid oxidation or autophagy resulted in the accumulation of TG/LDs and strongly potentiated GBM cell death. Immunofluorescence imaging and time-lapse videos showed that LDs are hydrolyzed by autophagy to release free fatty acids that mobilize into mitochondria for energy production. Our study demonstrates that autophagy-mediated hydrolysis of TG/LDs maintains energy homeostasis and GBM survival upon glucose reduction, suggesting that limiting TG/LDs utilization might be necessary upon treating GBM.
Subject Areas: Cellular Physiology, Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
TG/LDs function as energy reservoir for GBM tumors
-
•
TG/LDs are hydrolyzed by autophagy to maintain GBM survival when glucose levels decrease
-
•
TG/LD hydrolysis releases fatty acids that enter into mitochondria for energy production
-
•
Inhibiting autophagy causes TG/LD accumulation and GBM cell death
Cellular Physiology; Cell Biology; Cancer
Introduction
Metabolic alteration has been recognized as a hallmark of cancer (Hanahan and Weinberg, 2011; Intlekofer and Finley, 2019), but how lipid metabolism is reprogrammed in tumor cells remains poorly understood. A significant amount of effort has been put into understanding the underlying mechanisms elevating de novo fatty acid synthesis and its therapeutic potential in malignancies (Cheng et al., 2015, 2018a, 2018b; Guo, 2016; Guo et al., 2013, 2014; Menendez and Lupu, 2007; Ru et al., 2013; Talebi et al., 2018). However, whether other lipid metabolism pathways are altered in malignancies has not been extensively explored. Recently, our group and several others have found that human tumor tissues exhibit abundant lipid droplets (LDs), including in glioblastoma (GBM), the most lethal primary brain tumor (Cheng et al., 2020; Omuro and DeAngelis, 2013; Wen and Kesari, 2008), and in prostate, pancreas, breast, and clear cell renal cell carcinoma (Accioly et al., 2008; Du et al., 2017; Geng et al., 2016; Geng and Guo, 2017; Mitra et al., 2017; Pucer et al., 2013; Sevinsky et al., 2018; Sunami et al., 2017).
LDs are specific lipids-storage organelles, which are mainly located in adipose tissues (Murphy, 2001; Pond, 1999). A single LD contains a neutral lipid core that comprises triglycerides (TG) and cholesteryl esters, which are surrounded by a monolayer of phospholipid membrane that is associated with multiple proteins (Tauchi-Sato et al., 2002; Walther et al., 2017). A molecule of TG consists of a glycerol molecule with three hydroxyl groups that are each esterified by fatty acids (Olzmann and Carvalho, 2019; Paar et al., 2012). The presence of abundant LDs in cancer cells suggest that storing lipids in tumor tissues may be a common feature of malignancies. However, the role played by LDs in tumor cells is unknown.
In the tumor microenvironment, nutrient supplies are fluctuant due to variation in vascular development (Lyssiotis and Kimmelman, 2017). The mechanism used by tumor cells to solve this challenge and maintain growth has yet to be elucidated. We hypothesized that tumor cells utilize the fatty acids stored in LDs to support their survival and growth upon reduction in nutrient levels. To test this, we investigated the response of GBM cells to glucose deprivation and unveiled that TG/LDs serve as critical energy reservoirs to maintain cell survival upon energetic stress. Furthermore, we showed that GBM cells rapidly break down TG/LDs through an autophagic process that releases their stored fatty acids for energy production when glucose level decreases. Our study suggests that TG/LDs play an important role in the regulation of GBM growth.
Results
GBM Tissues Contain Large Amounts of TG and LDs
We examined TG levels in paired tumor and normal tissues of autopsy samples from GBM patients using lipid extraction and thin-layer chromatography (TLC). The data show that GBM tumor tissues contained large amounts of TG, which were rarely detected in the contralateral normal brain tissues (Figure 1A), consistent with the prevalence of LDs in GBM tumor tissues (Geng et al., 2016; Geng and Guo, 2017), as observed by confocal microscopy after BODIPY 493/503 staining and transmission electron microscopy (TEM) (Figure 1B). In agreement with these results, GBM patient primary GBM57 cells- or U87 cells-derived orthotopic xenograft models showed that TG/LDs are prominently formed in GBM tumor tissues but rarely formed in normal mouse brain tissues (Figures 1C–1F). In addition, by staining a variety of cancer cell types with BODIPY 493/503, we found that LDs are prevalent in various types of cancer cells, including breast, liver, pancreatic, cervical, skin, and sarcoma cancers (Figure S1A).
Figure 1.

GBM Tumor Tissues Contain Large Amounts of TG and LDs
(A) Thin-layer chromatography (TLC) analysis of total lipid extracts from the same weight (4 mg) of normal (N) versus tumor (T) tissues from GBM patient autopsy samples (middle panel). TG standard loaded on the left. The intensity of TLC TG bands for each sample was quantified by the ImageJ software and normalized to the average intensity of TG in six tumor tissues to determine the relative TG levels between normal brain and GBM tissue (mean ± SD, n = 6) (right panel). Significance was determined by unpaired Student's t test.
(B) Representative images of tumor tissues from GBM patient biopsy stained by H&E (left panel), BODIPY 493/503 (green), and DAPI (blue) (middle panel) or visualized by transmission electron microscopy (TEM) (right panel). Red arrows in TEM image indicate LDs. Scale bar: 50 μm for H&E, 10 μm for fluorescence imaging, 500 nm for TEM.
(C–F) Representative images of normal and tumor tissues from primary GBM57- (C) or U87-derived (E) orthotopic mouse models stained by H&E or BODIPY 493/503 (green)/DAPI (blue). Scale bar: 50 μm for H&E; 10 μm for fluorescence imaging. TLC analysis of total lipid extracts from normal and tumor tissues from GBM57 (D) or U87 (F) orthotopic mice. Relative TG levels were quantified by ImageJ software and normalized to the average TG levels in tumor tissues (mean ± SD, n = 4).
Significance was determined by an unpaired Student's t test. Please also see Figure S1A.
Glucose Deprivation Triggers GBM Cells Hydrolyzing TG/LDs to Support Cell Survival
We then investigated whether TG/LDs play a role in supporting GBM cell growth or survival by examining 3 GBM cell lines, i.e., U87, T98, and U251 that contain different amounts of TG (Figures 2A and 2B). Fluorescence imaging shows that U87 cells contain the highest number of LDs compared with the other two cell lines (Figure 2A), consistent with their highest TG levels (Figure 2B). We further compared LDs and TG levels between these 3 GBM cell lines and transformed astrocytes. The data show that GBM cells have dramatically more LDs and TG than the astrocytes, which are transformed cells that form noticeable amount of LDs/TG (Figure S1B). Whether LDs/TG are formed in normal astrocytes in vivo needs further investigation. We then removed glucose from the cell medium to examine the cell response and observed that LDs and TG quickly diminished in all GBM cells within a few hours (Figures 2A and 2B). Through careful examination, we observed that it took 8 h of glucose starvation for LDs and TG present in U87 cells to be greatly reduced, whereas it took only 4 h in T98 and U251 cells where LDs and TG levels are significantly lower (Figures 2A and 2B). Moreover, following the disappearance of TG/LDs, the viability of T98 and U251 cells quickly decreased to around 50% after 4-h glucose starvation and rapidly dropped to ∼5% after 8 h (Figures 2C and S1C). In contrast, the reduction of viability in U87 cells was much slower with ∼85% viability at 4 h, ∼78% at 8 h, and still ∼20% at 12 h (Figures 2C and S1C), suggesting that greater TG/LDs levels confer longer GBM cell survival upon glucose starvation.
Figure 2.

Glucose Deprivation Triggers GBM Cells Hydrolyzing TG/LDs to Support Cell Survival
(A–C) U87, T98, and U251 GBM cells were cultured in medium with 25 mM glucose or no glucose for 4, 8, and 12 h. Cells were stained with BODIPY 493/503 (green)/Hoechst 33342 (blue) and observed by confocal microscopy (A). Quantification of LDs/cell for over 30 cells was conducted using the ImageJ software (mean ± SD). Total lipids were extracted and analyzed by TLC (B). TG levels at different time points and in various cell lines were normalized to U87 cells cultured in 25 mM glucose medium (mean ± SD, n = 3). Live cell percentage at different time points after glucose withdrawal was determined after trypan blue staining (mean ± SD, n = 3) (C). Significance between different cell lines was determined by two-way ANOVA. ∗∗p < 0.001; ∗p < 0.01. Chol, cholesterol.
(D–F) U251 and T98 cells were cultured in medium with/without palmitic acid (PA, C16:0, 5 μM) and oleic acid (OA, C18:1, 5 μM) mixtures (1:1) for 24 h and then placed in fresh medium in the absence or presence of glucose (Gluc, 25 mM) for 4 h. Cells were stained with BODIPY 493/503 (green)/Hoechst 33342 (blue) and observed by confocal microscopy (D). Quantification of LDs/cell in over 30 cells was conducted using ImageJ (mean ± SEM). Total lipid extracts from these cells were analyzed by TLC (E). Relative TG levels were quantified by ImageJ and normalized to the control cells (mean ± SD, n = 3) (E). Cell death was determined by trypan blue exclusion (mean ± SD, n = 3) (F).
Significance was determined by one-way ANOVA. ∗∗p < 0.01, ∗p < 0.05. Please also see Figure S1B.
To validate that TG/LDs can support GBM cell survival, we increased LDs and TG amounts in U251 and T98 cells by supplementing cells with palmitic acid (PA, C16:0) and oleic acid (OA, C18:1) mixtures (1:1) for 24 h before glucose starvation (Figures 2D and 2E). Cells were then placed into fresh media with/without glucose (25 mM) for 4 h. The data show that increasing TG/LDs levels significantly elevated U251 and T98 survival upon glucose starvation as compared with control cells that did not receive PA/OA supplementation (Figures 2D–2F). These data strongly suggest that TG/LDs support GBM cell survival when glucose supply is limited.
TG/LDs Accumulate upon the Inhibition of Fatty Acid β-oxidation under Glucose-free Conditions, Leading to Increased GBM Cell Death
We next examined whether fatty acid β-oxidation plays a role in TG/LDs-supported GBM survival upon glucose starvation. GBM cells were treated with Etomoxir (ETO), an inhibitor of carnitine palmitoyltransferase 1 (CPT1) that transports fatty acids into mitochondria for β-oxidation and energy production (Raud et al., 2018). The data show that under glucose-rich conditions, ETO treatment did not affect LD and TG levels in GBM cells (Figures 3A and 3B) or their survival rate (Figure 3C). In contrast, inhibition of fatty acid oxidation by ETO in the absence of glucose significantly blocked the hydrolysis of LDs and TG (Figures 3A and 3B) and dramatically enhanced GBM cell death (Figure 3C).
Figure 3.

TG/LDs Accumulate upon the Inhibition of Fatty Acid β-Oxidation under Glucose-Free Conditions, Leading to Increased GBM Cell Death
(A and B) Representative fluorescence images of LDs stained with BODIPY 493/503 (green)/Hoechst 33342 (blue) (A). TLC analysis of total lipids (B) in U87 (8 h), U251, and T98 cells (4 h) after treatment with/without ETO (100 μM) in the absence or presence of glucose (25 mM). LDs/cell (mean ± SEM, n = 30) and TG levels (mean ± SD, n = 3) were quantified by ImageJ and normalized to the control cells.
(C) Cell death was determined in U87 (8 h), U251, and T98 cells (3 h) after treatment with/without ETO (100 μM) in the absence or presence of glucose (25 mM) by trypan blue staining (mean ± SD, n = 3).
(D–G) U87 cells were transfected with CPT1A shRNA-expressing lentivirus for 24 h and then cultured in glucose-free or 25 mM glucose medium for 8 h. Cells were analyzed by western blotting (D), observed by confocal microcopy after BODIPY 493/503 and Hoechst 33342 staining (E), or analyzed by TLC for lipid levels (F). LDs/cell (mean ± SEM, n = 30) or relative TG levels (mean ± SD, n = 3) were determined by ImageJ. Cell death was determined after trypan blue staining (mean ± SD, n = 3) (G). FA, fatty acids.
Significance was determined by one-way ANOVA. ∗∗p < 0.01, ∗p < 0.05. Please also see Figure S2.
We then used a genetic approach to silence CPT1A expression with lentivirus-expressing shRNA and block fatty acid β-oxidation. As with pharmacological inhibition (Figures 3A and 3B), CPT1A knockdown did not affect TG/LDs levels and GBM survival under glucose-rich conditions, but it blocked the hydrolysis of TG/LDs induced by glucose starvation (Figures 3D–3F, S2A and S2B) and significantly elevated GBM cell death (Figures 3G and S2C).
TG/LDs Are Hydrolyzed by Autophagy to Support GBM Cell Survival upon Glucose Starvation
We next examined how TG/LDs are hydrolyzed in GBM cells upon glucose starvation. Western blotting showed that autophagy is induced by glucose starvation in GBM cells, as demonstrated by an increase of the LC3 lipidation band (lower band, LC3-II), a marker of autophagosome formation (Kabeya et al., 2000) (Figure S3A). We then co-stained LDs and autophagosomes with BODIPY 493/503 (green) and anti-LC3 antibody (red). The data show that glucose starvation significantly increased the co-localization of LDs with LC3-stained autophagosomes, which was more prominent when lysosomal activity was suppressed by chloroquine (CQ) (Figure 4A), a lysotropic drug that reduces lysosomal acidification and thereby suppresses the activities of lysosomal hydrolases (Amaravadi et al., 2007; Mauthe et al., 2018). Moreover, using stably expressing red fluorescent protein (RFP)-LC3 U251 cells and time-lapse videos, we observed that glucose starvation, but not glucose-rich conditions, induces the direct association of LDs and RFP-LC3-labeled autophagosomes (Videos S1 and S2).
Figure 4.

TG/LDs are Hydrolyzed by Autophagy to Support GBM Cell Survival upon Glucose Deprivation
(A) Representative fluorescence images of U87 cells stained with BODIPY 493/503 (green) and anti-LC3 antibody (red) in the absence or presence of glucose (25 mM) and chloroquine (CQ, 5 μM) for 6 h. The co-localization was quantified by ImageJ (mean ± SEM) in over 30 cells.
(B and C) Representative fluorescence images of U87 cells co-stained with BODIPY 493/503 (green) and LysoTracker (red) (B) or anti-LAMP1 antibody (red) (C) in the presence or absence of glucose (25 mM) and CQ (5 μM) for 6 h. The co-localization was quantified by ImageJ (mean ± SEM, n = 30).
(D–F) Representative fluorescence images of U87 cells stained with BODIPY 493/503 (green) and Hoechst 33342 (blue) in the absence or presence of glucose (25 mM) and CQ (5 μM) for 8 h. LDs/cell were quantified in 30 cells (mean ± SEM, n = 30) (D). Their total lipids were analyzed by TLC (mean ± SD, n = 3) (E). Cell death was determined after trypan blue staining (mean ± SD, n = 3) (F).
(G–J) U87 cells were transfected with ATG5 shRNA-expressing lentivirus for 24 h and then cultured in medium without/with glucose (25 mM) for 8 h. Cell lysates were subjected to western blotting analysis using the indicated antibodies (G). Cells were stained with BODIPY 493/503 (green) and LysoTracker (red) and observed by confocal microscopy (H). Total cellular lipids were analyzed by TLC (I). Cell death was determined after trypan blue staining (J). Quantification and significance determination of LD number, relative TG levels, and cell death were same as in panels D–F.
Statistical significance was determined by one-way ANOVA for this entire figure; ∗∗p < 0.01, ∗p < 0.05. Please also see Figures S3 and S4, and Videos S1, S2, S3, and S4.
The videos show the association of LDs labeled by BODIPY-PA (green) and RFP-LC3-labeled autophagosomes (red) upon glucose deprivation. U251/RFP-LC3 cells were placed into glucose-rich (25 mM) (Video S1) or glucose-free (Video S2) fresh medium for 3 h. Cells were then imaged at 5 s intervals for 600 frames (1.5 h total) by confocal microscopy under 5% CO2, 95% air, and 37°C conditions. Arrows indicate the co-localization events of LDs and RFP-LC3-labeled puncta.
The videos show the association of LDs labeled by BODIPY-PA (green) and RFP-LC3-labeled autophagosomes (red) upon glucose deprivation. U251/RFP-LC3 cells were placed into glucose-rich (25 mM) (Video S1) or glucose-free (Video S2) fresh medium for 3 h. Cells were then imaged at 5 s intervals for 600 frames (1.5 h total) by confocal microscopy under 5% CO2, 95% air, and 37°C conditions. Arrows indicate the co-localization events of LDs and RFP-LC3-labeled puncta.
We then examined whether TG/LDs are hydrolyzed in lysosomes. We co-stained LDs and lysosomes with BODIPY 493/503 (green) and LysoTraker (red) in U87 cells after glucose starvation for 6 h. Live cell imaging and time-lapse video demonstrated that glucose starvation dramatically induced the mobilization of LDs into the lysosomes for hydrolysis (Figures 4B and S3B and Videos S3 and S4). The co-localization of LDs and lysosomes was more prominent when lysosomal activity was suppressed by CQ under glucose-poor conditions (Figure 4B). Co-localization was further confirmed by immunofluorescence staining of lysosomal-associated membrane protein 1 (LAMP1) together with LD staining by BODIPY 493/503 (Figure 4C). Accordingly, inhibition of lysosomal activity by CQ or the specific inhibitor of vacuolar H + ATPase that controls lysosomal acidification, bafilomycin (Mauvezin and Neufeld, 2015), blocked the hydrolysis of LDs and TG triggered by glucose starvation (Figures 4D, 4E, S4A, S4B, and S4D), resulting in a significant increase of GBM cell death (Figures 4F, S4C, and S4E), which was reduced by supplementing the media with OA/PA (Figure S4C). Moreover, we examined the effects of glucose starvation in neurospheres from GBM-patient-derived primary GBM83 cells. Similar as in U87 cells, confocal microscopy imaging showed that glucose starvation in GBM83 cells promoted the co-locolization of LDs with LC3-stained autophagosomes and LysoTracker-stained lysosomes (Figures S4F and S4G), and CQ treatment resulted in LD accumulation (Figure S4H).
The videos show the association of LDs (stained with BODIPY 493/503; green) with lysosomes (stained with LysoTracker; red). U87 cells were placed into glucose-rich (25 mM) (Video S3) or glucose-free fresh medium (Video S4) for 6 h. Cells were then imaged at 5 s intervals for 200 frames (30 min total) with confocal microscopy. Arrows indicate the co-localization event of BODIPY 493/503-stained LDs and LysoTracker-stained lysosomes.
The videos show the association of LDs (stained with BODIPY 493/503; green) with lysosomes (stained with LysoTracker; red). U87 cells were placed into glucose-rich (25 mM) (Video S3) or glucose-free fresh medium (Video S4) for 6 h. Cells were then imaged at 5 s intervals for 200 frames (30 min total) with confocal microscopy. Arrows indicate the co-localization event of BODIPY 493/503-stained LDs and LysoTracker-stained lysosomes.
We next genetically silenced the expression of ATG5 (Figure 4G), the key gene controlling autophagosome formation (Mizushima et al., 1998), to confirm the role of autophagy in TG/LDs hydrolysis. Western blotting showed that knockdown of ATG5 using lentivirus-mediated shRNA dramatically blocked the lipidation of LC3 (LC3-II) in GBM cells (Figure 4G), demonstrating the blockade of the autophagic process by ATG5 silencing. Moreover, ATG5 knockdown prominently suppressed glucose-deprivation-induced TG/LDs hydrolysis (Figures 4H and 4I), and significantly increased GBM cell death (Figure 4J), demonstrating that TG/LDs are hydrolyzed by autophagy to support GBM survival.
LD-Stored Fatty Acids Released by Autophagy Move into Mitochondria for Energy Production
We next examined the mobilization of fatty acids released from LDs. We first used BODIPY-labeled palmitic acid (BODIPY-PA, green) to culture U87 cells to form fluorescently labeled LDs (Figure 5A, left panel). After 12-h cultures, BODIPY-PA entered the cells and was stored into LDs (green dots), which were stained by Nile red, a fluorescent dye that stains neutral lipids and LDs (Figure 5A, middle and right panels). Moreover, BODIPY-PA-labeled LDs co-stained with TIP47, a specific LD membrane protein (Geng et al., 2016) (Figure 5B). These data demonstrate that BODIPY-labeled PA was stored in LDs in GBM cells. We then washed the cells with PBS to remove free BODIPY-PA and placed these cells into glucose-rich (25 mM) or glucose-free media for 8 h in the presence or absence of CQ or ETO. The cells were then stained with MitoTracker (red) to visualize the mitochondria. Under glucose-rich conditions, fluorescence imaging showed that BODIPY-labeled PA remained within LDs and did not move into the mitochondria and that CQ or ETO treatment had no effect (Figure 5C, left panels). In contrast, after glucose withdrawal for 8 h, green-labeled LDs were rapidly broken down and BODIPY-labeled PA diffused into the cytosol and trafficked into the mitochondria (Figure 5C, right panels), as demonstrated by the greatly increased co-localization of BODIPY-PA and MitoTracker-labeled mitochondria (Figure 5C, right and left panels). Blocking lysosomal activity by CQ or blocking fatty acid oxidation with ETO strongly suppressed glucose-starvation-induced breakdown of LDs and inhibited BODIPY-PA moving into the mitochondria (Figure 5C).
Figure 5.

LD-Stored Fatty Acids Released by Autophagy Move into Mitochondria for Energy Production
(A and B) Representative fluorescence images of U87 cells after culturing with BODIPY-labeled PA (C16:0; Cat# D3821, Life Technologies) (0.5 μM) for 12 h and then staining with Nile red and Hoechst 33342 (blue) (A) or with anti-TIP47 antibody (red) (B). Scale bar: 10 μm.
(C) U87 cells were cultured with BODIPY-PA (0.5 μM) for 12 h and then treated with/without CQ (5 μM) or ETO (100 μM) in the absence or presence of glucose (25 mM) for 8 h. Cells were then stained with MitoTracker (Red) and Hoechst 33342 (blue) and visualized by confocal microscopy. The co-localization of diffused BODIPY-PA and mitochondria was determined by the ratio of co-localized red and green signal (yellow) to total red signal using the ImageJ software (mean ± SEM, n = 30). Significance was determined by one-way ANOVA. ∗p < 0.01.
(D) U87 cells were transfected with shRNA-expressing lentivirus against ATG5 or CPT1A for 24 h and then cultured with BODIPY-labeled PA (0.5 μM) for 12 h. Cells were then placed into fresh medium without/with glucose (25 mM) for 8 h. Cell staining and quantification of the co-localization of BODIPY-PA and mitochondria were the same as in Panel C. Significance was determined by one-way ANOVA. ∗p < 0.01.
We then used a genetic approach to confirm that the trafficking of BODIPY-PA from LDs to mitochondria is mediated by autophagy. We first infected U87 cells with shRNA-expressing lentivirus for 24 h to knockdown ATG5 or CPT1A and then cultured these cells with BODIPY-labeled PA for an additional 12 h. The cells were then washed with PBS and placed in fresh medium in the presence (25 mM) or absence of glucose for 8 h. As shown in Figure 5D, BODIPY-labeled PA remained within the LDs in GBM cells infected with scramble shRNA and in ATG5 and CPT1A knockdown cells under glucose-rich conditions (Figure 5D, left panels). In contrast, glucose withdrawal induced the breakdown of LDs and promoted BODIPY-PA diffusing into the cytosol and trafficking into mitochondria in control-shRNA-infected cells (Figure 5D, right panels), whereas inhibition of autophagy by ATG5 knockdown or blocking fatty acid oxidation via CPT1A silencing dramatically suppressed LD hydrolysis and blocked BODIPY-PA trafficking from LDs into mitochondria (Figure 5D, right panels). Taken together, these results demonstrate that LD-stored fatty acids are released by autophagy and then move into mitochondria for β-oxidation upon glucose starvation in GBM cells.
Discussion
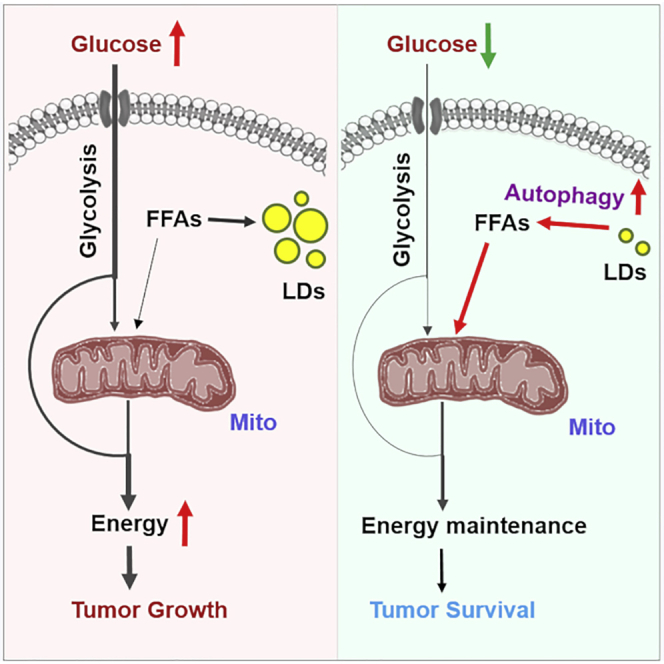
In the past decade, metabolic reprogramming in cancer cells has been extensively investigated; however, the identification of effective metabolic targets for cancer therapy has only incrementally progressed. In this study, we show that TG/LDs, functioning as critical energy reservoirs, support GBM cell survival when glucose supply decreases (Figure 6). We demonstrate that TG/LDs are hydrolyzed by autophagy to release their stored fatty acids for energy production and support GBM cell survival upon glucose starvation, suggesting that blocking TG/LDs utilization may be necessary upon targeting GBM (Figure 6).
Figure 6.

Schematic Model of TG/LDs Hydrolysis Mediated by Autophagy, Maintaining GBM Cell Energy Homeostasis and Survival
(A–C) GBM cells maintain energy homeostasis by utilizing glucose and TG/LDs. Under glucose-rich conditions, GBM cells mainly use glucose for energy production (A). Upon glucose starvation, autophagy is activated, which breaks down TG/LDs and releases the stored fatty acids that enter mitochondria for energy production (B). Blocking TG/LDs hydrolysis via inhibiting autophagy upon glucose reduction conditions will induce dramatic GBM cell death (C).
In the tumor microenvironment, nutrient supplies are frequently scarce due to the poor vascularization (Wellen and Thompson, 2010). Our results show that GBM cells promptly utilize TG/LDs to provide abundant fatty acids for energy production upon glucose starvation, demonstrating the high plasticity and resilience of malignant cells at times of energetic stress. It has been shown that GBM cells have developed efficient machineries to quickly import nutrients and synthesize macromolecules (Cosset et al., 2017; Flavahan et al., 2013; Geng et al., 2016; Guo et al., 2009, 2011; Vlashi et al., 2011). Under nutrient-rich conditions, GBM cells can absorb abundant glucose, which is converted to fatty acids that are then stored into LDs when in excess. Upon external nutrient paucity, we showed that GBM cells induced LDs breakdown and TG hydrolysis via autophagy (Figure 6). Autophagy is a critical cellular process that degrades protein aggregates and damaged organelles and recycles nutrients by hydrolyzing cytoplasmic components, thereby maintaining cellular homeostasis (Klionsky and Emr, 2000; Mizushima and Komatsu, 2011; Mizushima et al., 2008). Given the importance of autophagy in mediating the turnover of cytoplasmic components, autophagy has been shown to be one of the cellular defense mechanisms under nutrient stress (Komatsu et al., 2005; Lee et al., 2014). The first study showing a link between LDs and autophagy was in hepatic cells, which suggested that autophagy played a critical role in LD hydrolysis (Singh et al., 2009). A later study showed that chaperone-mediated autophagy can degrade LD-associated proteins, perilipin 2 (PLIN2) and perilipin 3 (PLIN3), and facilitate LD breakdown in mice livers upon nutrient shortage (Kaushik and Cuervo, 2015). However, the interaction between LDs and autophagy has not been reported in tumor tissues, and its role in human cancer is unclear. Our data showed that GBM cells can utilize autophagy to quickly hydrolyze TG/LDs to release fatty acids, demonstrating the importance of autophagy in maintaining energy homeostasis by hydrolyzing internally stored lipids. Thus, identifying an approach to block the utilization of LDs-stored fatty acids could be an effective strategy to inhibit GBM growth.
GBM is the most lethal brain tumor and is a very difficult cancer to treat (Omuro and DeAngelis, 2013; Wen and Kesari, 2008), with a median survival of only about 15 months after initial diagnosis despite extensive therapies (Stupp et al., 2009; Wen and Kesari, 2008). The greatest challenge in treating GBM is the unavoidable resistance to treatments developed by tumor cells (Bao et al., 2006; Nguyen et al., 2018). The standard therapy for GBM is radiation together with chemotherapy after surgery (Weller et al., 2014). Resistance to these treatments requires tumor cells to quickly repair their damaged DNA, which consumes large amounts of energy (Aird et al., 2015; Brace et al., 2016). Our data show that GBM cells can rapidly break down TG/LDs by autophagy for energy production. Given that radiation and chemotherapies have been shown to induce autophagy in GBM cells (Chaurasia et al., 2016; Classen et al., 2019; Jiang et al., 2017, 2019), we speculate that TG/LDs via autophagy contribute great amounts of energy to support GBM resistance to these therapies. Thus, blocking TG/LDs breakdown might increase GBM sensitivity to treatments and overcome resistance. Further studies are needed that should explore whether combining radiation and chemotherapies with blockade of TG/LDs utilization could effectively reduce resistance and suppress GBM growth.
Recent studies have shown that LDs are being formed in various cancers, including breast, prostate, and clear renal cell carcinoma among others (Accioly et al., 2008; Du et al., 2017; Geng et al., 2016; Mitra et al., 2017; Pucer et al., 2013; Sevinsky et al., 2018; Sunami et al., 2017). Autophagic release of LDs-stored fatty acids is highly likely to occur in different cancer types, which may be a common mechanism to support tumor cell survival upon nutrient reduction. Our study identifying the role of TG/LDs in maintaining energy homeostasis via autophagy will have a broad impact on cancers other than GBM. Therefore, it will be important in the future to further explore the role of LDs in different cancers, as targeting TG/LDs hydrolysis may be an effective means to sensitize tumor cells to therapies in a variety of cancer types.
Limitations of the Study
Our study unveiled the critical bioenergetic role played by TG/LDs in GBM cells and demonstrated that autophagy mediates TG/LD hydrolysis, also named lipophagy, to release their stored fatty acids for energy production. Nevertheless, the study was mainly performed in vitro in GBM cell lines, although multiple cell lines were examined. Future research needs to include in vivo studies to validate the bioenergetic role of TG/LDs in GBM and to demonstrate autophagic hydrolysis of TG/LDs in tumor tissues. In addition, how the autophagy machinery can specifically recognize and target LDs to hydrolyze TG remains unknown. Certainly, further research is needed to fully uncover the function and regulation of TG/LDs in human cancers.
Resource Availability
Lead Contact
Further information and requests for resources and materials should be directed to and will be fulfilled by the Lead Contact, Deliang Guo (deliang.guo@osumc.edu).
Materials Availability
All reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate/analyze datasets or computer code/algorithm.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by NINDS and NCI grants NS104332, NS112935 and R01CA240726 to DG, CA227874 to DG/AC, and American Cancer Society Research Scholar Grant RSG-14-228-01–CSM to DG. TFC and WHY are supported in part by the UCLA SPORE in Brain Cancer (NIH/NCI 1P50CA211015-01A1) and the Art of the Brain Foundation. We also appreciate the support from OSUCCC-Pelotonia Idea grant and start-up funds to DG. The authors wish to thank Dr. Martine Torres for her editorial assistance.
Author Contributions
D.G. conceived the ideas. X.W., F.G., and D.G. designed the experiments. X.W., F.G., X.C., Q.G., and Y.Z. performed the experiments. X.W., F.G., X.C., A.C., and D.G. analyzed the data. W.Y. and T.C. procured and provided normal brain and GBM autopsy samples. X.W. and D.G. wrote the manuscript, and all authors reviewed and approved the manuscript for publication.
Declaration of Interests
The authors declare that there are no conflicts of interest for this manuscript.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101569.
Supplemental Information
References
- Accioly M.T., Pacheco P., Maya-Monteiro C.M., Carrossini N., Robbs B.K., Oliveira S.S., Kaufmann C., Morgado-Diaz J.A., Bozza P.T., Viola J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–1740. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- Aird K.M., Worth A.J., Snyder N.W., Lee J.V., Sivanand S., Liu Q., Blair I.A., Wellen K.E., Zhang R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015;11:893–901. doi: 10.1016/j.celrep.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi R.K., Yu D., Lum J.J., Bui T., Christophorou M.A., Evan G.I., Thomas-Tikhonenko A., Thompson C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Brace L.E., Vose S.C., Stanya K., Gathungu R.M., Marur V.R., Longchamp A., Trevino-Villarreal H., Mejia P., Vargas D., Inouye K. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016;2:16022. doi: 10.1038/npjamd.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia M., Bhatt A.N., Das A., Dwarakanath B.S., Sharma K. Radiation-induced autophagy: mechanisms and consequences. Free Radic. Res. 2016;50:273–290. doi: 10.3109/10715762.2015.1129534. [DOI] [PubMed] [Google Scholar]
- Cheng C., Geng F., Cheng X., Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond). 2018;38:27. doi: 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Li J., Guo D. SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr. Top. Med. Chem. 2018;18:484–493. doi: 10.2174/1568026618666180523104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Ru P., Geng F., Liu J., Yoo J.Y., Wu X., Cheng X., Euthine V., Hu P., Guo J.Y. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. 2015;28:569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Geng F., Pan M., Wu X., Zhong Y., Wang C., Tian Z., Cheng C., Zhang R., Puduvalli V. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020;32:229–242 e228. doi: 10.1016/j.cmet.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen F., Kranz P., Riffkin H., Pompsch M., Wolf A., Gopelt K., Baumann M., Baumann J., Brockmeier U., Metzen E. Autophagy induced by ionizing radiation promotes cell death over survival in human colorectal cancer cells. Exp. Cell Res. 2019;374:29–37. doi: 10.1016/j.yexcr.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Cosset E., Ilmjarv S., Dutoit V., Elliott K., von Schalscha T., Camargo M.F., Reiss A., Moroishi T., Seguin L., Gomez G. Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell. 2017;32:856–868.e855. doi: 10.1016/j.ccell.2017.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W., Zhang L., Brett-Morris A., Aguila B., Kerner J., Hoppel C.L., Puchowicz M., Serra D., Herrero L., Rini B.I. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017;8:1769. doi: 10.1038/s41467-017-01965-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan W.A., Wu Q., Hitomi M., Rahim N., Kim Y., Sloan A.E., Weil R.J., Nakano I., Sarkaria J.N., Stringer B.W. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013;16:1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F., Cheng X., Wu X., Yoo J.Y., Cheng C., Guo J.Y., Mo X., Ru P., Hurwitz B., Kim S.H. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 2016;22:5337–5348. doi: 10.1158/1078-0432.CCR-15-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F., Guo D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. (Wash D C) 2017;3 doi: 10.18103/imr.v3i5.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D. SCAP links glucose to lipid metabolism in cancer cells. Mol. Cell Oncol. 2016;3:e1132120. doi: 10.1080/23723556.2015.1132120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D., Bell E.H., Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013;2:289–299. doi: 10.2217/cns.13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D., Bell E.H., Mischel P., Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014;20:2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D., Prins R.M., Dang J., Kuga D., Iwanami A., Soto H., Lin K.Y., Huang T.T., Akhavan D., Hock M.B. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D., Reinitz F., Youssef M., Hong C., Nathanson D., Akhavan D., Kuga D., Amzajerdi A.N., Soto H., Zhu S. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Intlekofer A.M., Finley L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019;1:177–188. doi: 10.1038/s42255-019-0032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C., Jiang L., Li Q., Liu X., Zhang T., Yang G., Zhang C., Wang N., Sun X., Jiang L. Pyrroloquinoline quinine ameliorates doxorubicin-induced autophagy-dependent apoptosis via lysosomal-mitochondrial axis in vascular endothelial cells. Toxicology. 2019;425:152238. doi: 10.1016/j.tox.2019.152238. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Ji F., Liu Y., He M., Zhang Z., Yang J., Wang N., Zhong C., Jin Q., Ye X., Chen T. Cisplatin-induced autophagy protects breast cancer cells from apoptosis by regulating yes-associated protein. Oncol. Rep. 2017;38:3668–3676. doi: 10.3892/or.2017.6035. [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015;17:759–770. doi: 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Emr S.D. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.M., Wagner M., Xiao R., Kim K.H., Feng D., Lazar M.A., Moore D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis C.A., Kimmelman A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017;27:863–875. doi: 10.1016/j.tcb.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.J., Coppes R.P., Engedal N., Mari M., Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C., Neufeld T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J.A., Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Mitra R., Le T.T., Gorjala P., Goodman O.B., Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer. 2017;17:631. doi: 10.1186/s12885-017-3589-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Levine B., Cuervo A.M., Klionsky D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Murphy D.J. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 2001;40:325–438. doi: 10.1016/s0163-7827(01)00013-3. [DOI] [PubMed] [Google Scholar]
- Nguyen H.S., Shabani S., Awad A.J., Kaushal M., Doan N. Molecular markers of therapy-resistant glioblastoma and potential strategy to combat resistance. Int. J. Mol. Sci. 2018;19:1765. doi: 10.3390/ijms19061765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J.A., Carvalho P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019;20:137–155. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omuro A., DeAngelis L.M. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- Paar M., Jungst C., Steiner N.A., Magnes C., Sinner F., Kolb D., Lass A., Zimmermann R., Zumbusch A., Kohlwein S.D., Wolinski H. Remodeling of lipid droplets during lipolysis and growth in adipocytes. J. Biol. Chem. 2012;287:11164–11173. doi: 10.1074/jbc.M111.316794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond C.M. Physiological specialisation of adipose tissue. Prog. lipid Res. 1999;38:225–248. doi: 10.1016/s0163-7827(99)00003-x. [DOI] [PubMed] [Google Scholar]
- Pucer A., Brglez V., Payre C., Pungercar J., Lambeau G., Petan T. Group X secreted phospholipase A(2) induces lipid droplet formation and prolongs breast cancer cell survival. Mol. Cancer. 2013;12:111. doi: 10.1186/1476-4598-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B., Roy D.G., Divakaruni A.S., Tarasenko T.N., Franke R., Ma E.H., Samborska B., Hsieh W.Y., Wong A.H., Stuve P. Etomoxir actions on regulatory and memory T cells are independent of cpt1a-mediated fatty acid oxidation. Cell Metab. 2018;28:504–515.e507. doi: 10.1016/j.cmet.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru P., Williams T.M., Chakravarti A., Guo D. Tumor metabolism of malignant gliomas. Cancers (Basel) 2013;5:1469–1484. doi: 10.3390/cancers5041469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevinsky C.J., Khan F., Kokabee L., Darehshouri A., Maddipati K.R., Conklin D.S. NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res. 2018;20:55. doi: 10.1186/s13058-018-0980-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R., Hegi M.E., Mason W.P., van den Bent M.J., Taphoorn M.J., Janzer R.C., Ludwin S.K., Allgeier A., Fisher B., Belanger K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Sunami Y., Rebelo A., Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers. 2017;10:3. doi: 10.3390/cancers10010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebi A., Dehairs J., Rambow F., Rogiers A., Nittner D., Derua R., Vanderhoydonc F., Duarte J.A.G., Bosisio F., Van den Eynde K. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018;9:2500. doi: 10.1038/s41467-018-04664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi-Sato K., Ozeki S., Houjou T., Taguchi R., Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J. Biol. Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- Vlashi E., Lagadec C., Vergnes L., Matsutani T., Masui K., Poulou M., Popescu R., Della Donna L., Evers P., Dekmezian C. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. U S A. 2011;108:16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther T.C., Chung J., Farese R.V., Jr. Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen K.E., Thompson C.B. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M., van den Bent M., Hopkins K., Tonn J.C., Stupp R., Falini A., Cohen-Jonathan-Moyal E., Frappaz D., Henriksson R., Balana C. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15 doi: 10.1016/S1470-2045(14)70011-7. e395–403. [DOI] [PubMed] [Google Scholar]
- Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The videos show the association of LDs labeled by BODIPY-PA (green) and RFP-LC3-labeled autophagosomes (red) upon glucose deprivation. U251/RFP-LC3 cells were placed into glucose-rich (25 mM) (Video S1) or glucose-free (Video S2) fresh medium for 3 h. Cells were then imaged at 5 s intervals for 600 frames (1.5 h total) by confocal microscopy under 5% CO2, 95% air, and 37°C conditions. Arrows indicate the co-localization events of LDs and RFP-LC3-labeled puncta.
The videos show the association of LDs labeled by BODIPY-PA (green) and RFP-LC3-labeled autophagosomes (red) upon glucose deprivation. U251/RFP-LC3 cells were placed into glucose-rich (25 mM) (Video S1) or glucose-free (Video S2) fresh medium for 3 h. Cells were then imaged at 5 s intervals for 600 frames (1.5 h total) by confocal microscopy under 5% CO2, 95% air, and 37°C conditions. Arrows indicate the co-localization events of LDs and RFP-LC3-labeled puncta.
The videos show the association of LDs (stained with BODIPY 493/503; green) with lysosomes (stained with LysoTracker; red). U87 cells were placed into glucose-rich (25 mM) (Video S3) or glucose-free fresh medium (Video S4) for 6 h. Cells were then imaged at 5 s intervals for 200 frames (30 min total) with confocal microscopy. Arrows indicate the co-localization event of BODIPY 493/503-stained LDs and LysoTracker-stained lysosomes.
The videos show the association of LDs (stained with BODIPY 493/503; green) with lysosomes (stained with LysoTracker; red). U87 cells were placed into glucose-rich (25 mM) (Video S3) or glucose-free fresh medium (Video S4) for 6 h. Cells were then imaged at 5 s intervals for 200 frames (30 min total) with confocal microscopy. Arrows indicate the co-localization event of BODIPY 493/503-stained LDs and LysoTracker-stained lysosomes.
Data Availability Statement
This study did not generate/analyze datasets or computer code/algorithm.