

Abstract
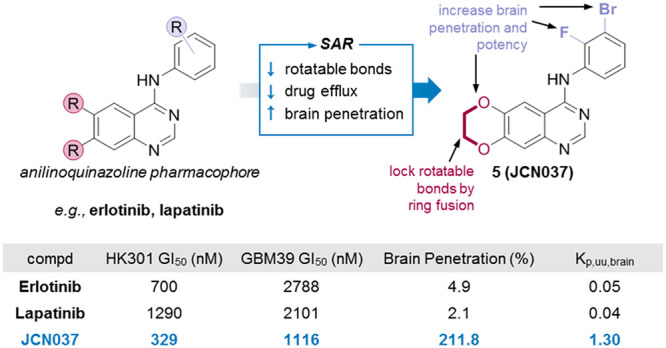
The epidermal growth factor receptor (EGFR) is genetically altered in nearly 60% of glioblastoma tumors; however, tyrosine kinase inhibitors (TKIs) against EGFR have failed to show efficacy for patients with these lethal brain tumors. This failure is attributed to the inability of clinically tested EGFR TKIs to cross the blood–brain barrier (BBB) and achieve adequate pharmacological levels to inhibit various oncogenic forms of EGFR that drive glioblastoma. Through SAR analysis, we developed compound 5 (JCN037) from an anilinoquinazoline scaffold by ring fusion of the 6,7-dialkoxy groups to reduce the number of rotatable bonds and polar surface area and by introduction of an ortho-fluorine and meta-bromine on the aniline ring for improved potency and BBB penetration. Relative to the conventional EGFR TKIs erlotinib and lapatinib, JCN037 displayed potent activity against EGFR amplified/mutant patient-derived cell cultures, significant BBB penetration (2:1 brain-to-plasma ratio), and superior efficacy in an EGFR-driven orthotopic glioblastoma xenograft model.
Keywords: EGFR, EGFRvIII, kinase inhibitor, glioblastoma, brain-penetration
Malignant gliomas, including the universally lethal glioblastoma (GBM), are the most common and the deadliest primary brain tumors. The epidermal growth factor receptor (EGFR) is mutated and/or amplified in ∼60% of GBM tumors.1 Of these tumors with genetically altered EGFR, approximately 50% consist of amplified wild-type EGFR (wtEGFR) with no mutations, while the remaining tumors in this cohort have an amplification with an activating extracellular domain mutation. The most prominent activating mutation is a deletion of exons 2–7 in EGFR [EGFRvIII].1 Both amplified wtEGFR and EGFRvIII play important roles in tumor growth, proliferation, and survival.2 Moreover, in EGFRvIII expressing tumors, wtEGFR is diffusely expressed and cooperates with EGFRvIII to promote tumorigenesis.2,3 Given the importance of both mutant and wtEGFR as drivers of malignant glioma, numerous clinical trials using first generation EGFR tyrosine kinase inhibitors (TKIs) (i.e., erlotinib, lapatinib, and gefitinib) have been evaluated in GBM patients. However, all studies using these EGFR TKIs failed to improve the outcomes of patients with GBM.4,5
Significant evidence suggests that all the first generation EGFR TKIs do not cross the blood–brain barrier (BBB) in concentrations sufficient to achieve therapeutic consequences in GBM tumors.4−6 Although next-generation EGFR inhibitors, such as afatinib, dacomitinib, and neratinib, are still under clinical investigation for GBM, early data suggest minimal clinical activity for those EGFR TKIs in which patient outcomes are available.7,8 The limited efficacy observed in GBM patients with these next-generation EGFR inhibitors may also be due to their inadequate brain exposure.9,10 While the EGFR TKI osimertinib—developed for EGFR-mutated lung cancer—has reported high brain penetration,9 it has yet to be thoroughly evaluated either preclinically or clinically for GBM. Moreover, osimertinib does not effectively inhibit wtEGFR,11 which is presumably required to effectively target EGFR-driven GBMs.2 Thus, obtaining pharmacological levels of EGFR TKIs within GBM tumors, while also having potent activity against both wtEGFR and EGFRvIII, remains a major obstacle for their effective treatment.
A potential contributor to the low brain exposures of currently used clinical EGFR TKIs (and for the FDA-approved kinase inhibitors lacking brain-penetration) is that they do not conform to the physicochemical properties that are associated with BBB penetration (Table 1).12 Specifically, for clinically available EGFR TKIs, the molecular weight (MW), the number of hydrogen bond donors (HBD) and acceptors (HBA), the polar surface area (TPSA), and the number of rotatable bonds (NRB) fall outside the desired ranges recommended by Ghose et al.13 and Wager et al.14 (Table 1). Finally, these physicochemical properties have also been shown to influence the ability of the P-glycoprotein (P-gp)—a prominent drug efflux transporter found in brain capillary endothelial cells—to recognize drugs that include currently clinically used EGFR TKIs and thus limit drug exposure in the brain.15
Table 1. Comparison of Physicochemical Properties for CNS Drugs, FDA-Approved Kinase Inhibitors, and EGFR Kinase Inhibitors.
| Physicochemical property | CNS drugs (preferred range, n = 317)a | CNS drugs (median, n = 119)b | FDA-approved protein kinase inhibitors (median, n = 49)c,d | Clinical EGFR kinase inhibitors (median, n = 25)c,d,e |
|---|---|---|---|---|
| MW | 250–355 | 305 | 486 | 491 |
| clogP | 2.1–4.4 | 2.8 | 4.2 | 4.5 |
| clogD7.4 | 1.2–3.1 | 1.7 | 3.3 | 3.9 |
| HBD | 0–1 | 1 | 2 | 2 |
| HBA | 2–3 | N/A | 7 | 7 |
| TPSA (Å2) | 25–60 | 45 | 94 | 89 |
| NRB | 1–4 | N/A | 6 | 8 |
| Most basic center (pKa) | 7.9–10.7 | 8.4 | 7.1 | 7.7 |
Preferred ranges for physicochemical properties from ref (13).
Median values from ref (14).
See Supporting Information for more details on how these values were compiled.
For at least 7 out of 49 FDA-approved kinase inhibitors, brain-penetration data has been reported.12
EGFR kinase inhibitors approved by any agency or in clinical development (noncomprehensive). N/A, no data provided.
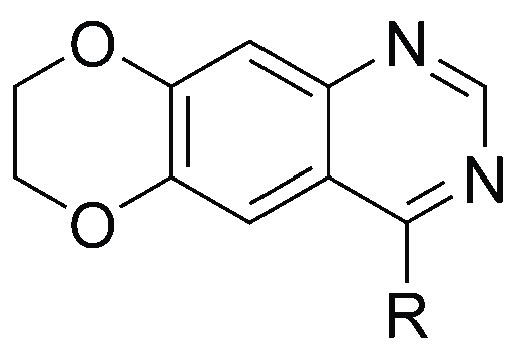
To address this problem of low brain penetration of EGFR TKIs, we modified the 4-anilinoquinazoline scaffold of first generation EGFR TKIs with the goal of obtaining an EGFR TKI with the desired physicochemical properties for BBB penetration, while having activity against both wtEGFR and EGFRvIII. We report the synthesis and characterization of 5 (JCN037), a noncovalent EGFR TKI that demonstrated both nanomolar potency against both mutant EGFRvIII and wtEGFR in cellular assays and greater than 2:1 brain to plasma levels. Moreover, 5 was effective at inhibiting the growth of EGFR-driven primary GBM cells, both in cell culture and in orthotopic xenografts. Importantly, the outcomes of in vivo treatment of xenografted malignant glioma with 5 were superior to that of both erlotinib and lapatinib.
Like other type I EGFR TKIs (e.g., gefitinib), erlotinib can potently target both the active confirmation of wtEGFR and has the capacity to bind, although with less affinity, to mutant EGFRvIII.6 Conversely, type II EGFR TKIs (e.g., lapatinib, neratinib)—which favor the inactive form of EGFR—can have high affinity for EGFRvIII yet are extremely ineffective at targeting activated wtEGFR.6 Our goal was to have a compound that could potently inhibit both wtEGFR and EGFRvIII; for this reason we selected erlotinib as our starting point to initiate our structure actvity relationship (SAR) studies.
Erlotinib penetrates the brain at a very low level of 7%.16 Physicochemical features of erlotinib that could make it a poor brain-penetrating drug include a large NRB (10), several HBA (7), and a high TPSA (75 Å), and many of these liabilities derive from the flexible alkyl ether tails. We hypothesized that improving these physicochemical properties linked to brain penetration might be achievable by modifying positions that may not be essential for binding to the EGFR kinase domain.
From the extensive SAR work performed on the 4-anilinoquinazoline pharmacophore, as well as from the wealth of available structural information on the EGFR kinase domain, the essential binding interactions of this TKI scaffold are well-known.11,17,18 An overview of the type I binding mode of erlotinib and, for comparison, the nonhydrolyzable ATP-analogue AMP-PNP is depicted in Figure S1.
Based on these considerations, we hypothesized that closing the flexible alkoxy chains at C6 and C7 to form a 1,4-dioxane ring fused onto the quinazoline scaffold—a modification that has been investigated previously as a means to increase the solubility of these compounds19—may increase BBB penetrance without affecting binding of the molecule to EGFR. This modification yielded 1, which contains a reduced NRB (10 to 2), HBA (7 to 5), and TPSA (75 Å to 56 Å) relative to erlotinib (Table 2). Importantly, the more optimal physicochemical properties of 1 were associated with, perhaps unpredictably, a nearly ten times increase in BBB penetration relative to erlotinib. Following a single oral dose of 1 (10 mg/kg) in healthy CD-1 mice, the brain/plasma ratio was 0.71; in contrast, and in line with previous reports,16 the brain/plasma ratio of erlotinib was 0.085 (Table 2).
Table 2. Compared to Erlotinib, a Fused Dioxane Ring Improves Brain Penetration, but Reduces Potency, while the Addition of an ortho-Fluorine on the Aniline Ring Improves Potency while Retaining BBB Penetrationa.

| Compound | MW | clogP | HBA | TPSA (Å) | NRB | EGFR IC50 (nM) | p-wtEGFR IC50 (nM) | p-EGFRvIII IC50 (nM) | HK301 GI50 (nM) | GBM39 GI50 (nM) |
|---|---|---|---|---|---|---|---|---|---|---|
| Erlotinib | 393 | 2.39 | 7 | 75 | 10 | 2.17 ± 0.58 | 3.90 ± 0.24 | 12.5 ± 1.1 | 700.2 ± 76.8 | 2788 ± 179 |
| 1 | 303 | 2.98 | 5 | 56 | 2 | 48.7 ± 7.8 | 59.8 ± 1.6 | 188.4 ± 26.9 | 8824 ± 1109 | 20536 ± 1212 |
| 2 | 321 | 3.71 | 5 | 56 | 2 | 19.7 ± 3.7 | 20.5 ± 0.1 | 32.2 ± 1.2 | 791.1 ± 108.1 | 2946 ± 353 |
All EGFR inhibition data are represented as mean ± SEM from n = 2 or more independent replicates. All growth inhibition data are represented as mean ± SEM from n = 3 or more independent replicates.
To determine how the fusion of the dioxane ring impacts activity against wild-type and EGFRvIII, 1 was tested in enzymatic and cellular biochemical assays (Table 2). Despite our prediction that this modification would not affect activity, 1 was significantly less potent than erlotinib against both wild-type and mutant EGFRvIII. The reduced potency of 1 relative to erlotinib was also reflected in a lower activity against two EGFRvIII mutant patient derived GBM cells, HK301 and GBM39; the half maximal growth inhibitory concentration (GI50) for 1 was 10-fold worse than erlotinib against these GBM cell lines. Thus, the surprisingly remarkable brain penetration achieved by fusing the alkyl ether tails of erlotinib came at an unexpected loss of inhibitor potency.
To improve upon potency, we considered modifications of the aniline ring through the introduction of a second substituent. As the binding pocket of the aniline ring—the apolar hole—only permits small, lipophilic substituents, we considered the strategic placement of a halide next to the alkyne. For several known EGFR TKIs, halogenated aniline rings are common with, in particular, fluorine or chlorine substituents. Moreover, a 2′-fluorine with a 3′-substituent on the aniline ring has been shown to increase activity against EGFR.20,21 As such, we hypothesized that the addition of a 2′-fluorine substituent to 1 would improve its affinity for EGFR. Indeed, cell-free enzymatic activity assays and cellular wtEGFR and EGFRvIII phosphorylation studies revealed an increased potency from 1 against EGFR kinase activity for 2 (Table 2). Additional in vitro profiling of cellular growth and proliferation showed a marked improvement in the GI50 of 2 on HK301 and GBM39 patient-derived GBM lines, suggesting the ortho-fluorine improved the protein–ligand interaction with EGFR.
Fluorine substituents are known to affect biological activity and absorption, distribution, metabolism, and excretion (ADME) properties of a drug by modulating lipophilicity while preserving hydrogen bonds and total polar surface area.22 Specifically, an ortho-fluorine on an aniline ring has been observed in various reports to mitigate the strength of an adjacent HBD and can potentially improve brain penetration and membrane permeability by reducing the strength of hydrogen bond interactions.21,22 To determine the influence of an ortho-fluorine on the aniline ring on brain penetration, we profiled 2in vivo in healthy CD-1 mice (Table 2). Although the exposure and maximum concentration of 2 was considerably improved relative to 1, the increase in BBB permeability was modest, with a change in brain/plasma AUC0–7h from 0.65 (1) to 0.85 (2).
Closure of the alkoxy chains and adding a fluorine to the 2′-position on the aniline ring led to a compound that was more brain penetrant and potent than 1 yet still less active than clinically available EGFR inhibitors (such as erlotinib). Constrained by the environment of the apolar hole as explained before, we focused our efforts on the SAR analysis of halogen as well as similar bioisosteric substituents on the aniline ring. In particular, we carried out a fluorine scan (S1–S8 in Table S1) on the aniline ring to identify the optimal substitution pattern for EGFR inhibition (while retaining the 2′-fluorine).23 We observed that the 2′,3′-difluoro substitution pattern (S3) was the most potent in cellular wtEGFR and EGFRvIII phosphorylation studies of these compounds and, consistent with these biochemical assays, was the most potent of the multifluoro substituted compounds at inhibiting both HK301 and GBM39 patient-derived GBM lines. Collectively, these results suggest a 2′,3′-disubstitution pattern on the aniline ring is the most active of fluoro-substituted derivatives against both wtEGFR and EGFRvIII biochemically and in cellular proliferation assays. One potential rationale is the favorable dipolar and lipophilic character created on the aniline by this substitution pattern, which fits well with the possible lipophilic and electrostatic environment of the apolar hole (Figure S2).
Next, we proceeded to test additional isostere substituents to improve potency against EGFR (Table 3). We focused on substituents that were not expected to interfere with the properties that we had previously optimized to obtain significant brain penetrance, including NRB, TPSA, and HBA. To mimic the 3′-ethynyl substituent of 2, a related isostere consisting of a 3′-cyano group was also synthesized (3). However, the efficacy of the TKI activity was reduced by the introduction of the 3′-cyano group, suggesting that the anisotropic electron-density distribution of the apolar ethynyl group with a partially positive charged region (hydrogen atom) is more favorable at this position than the partially negatively charged region of the more polar cyano group (the lone electron pair on the nitrogen). Therefore, we decided to test all additional halide substituents at this 3′-position on the aniline ring, as they should provide a better isosteric replacement for the ethynyl group.24
Table 3. Comparison of the 2′- and 3′-Position of 4-Anilinoquinazolinesa.
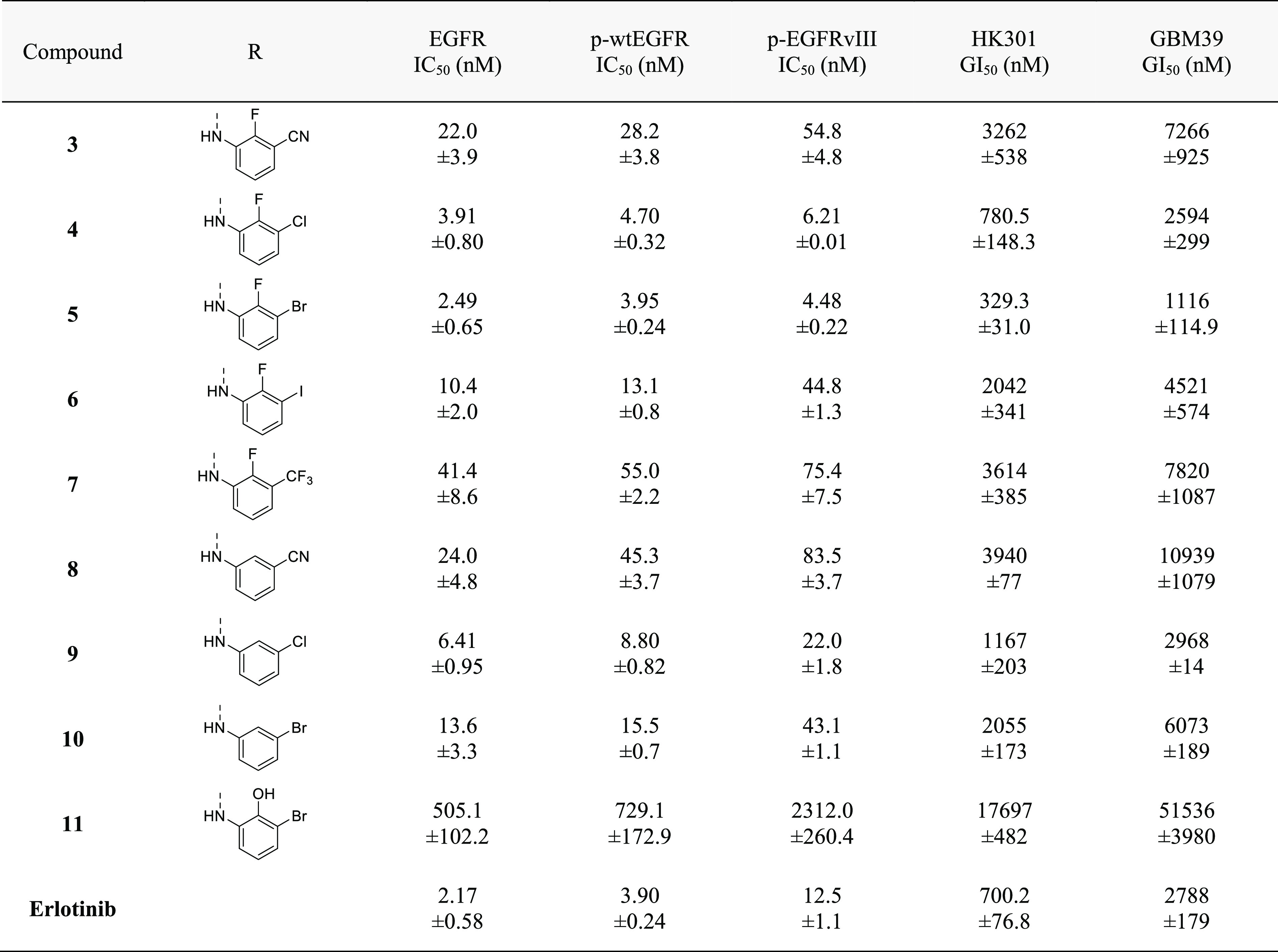
All EGFR inhibition data are represented as mean ± SEM from n = 2 or more independent replicates. All growth inhibition data are represented as mean ± SEM from n = 3 or more independent replicates.
Surprisingly, the affinity toward EGFR increased with the size of the 3′-halogen substituent on the aniline ring up to a maximum with a 3′-bromine (S3, 4, 5, 6) (cell-free IC50 of 18.9 nM, 3.91 nM, 2.49 nM, and 10.4 nM, respectively) (Table S2). This result may imply unspecific lipophilic interactions and a possible size limitation of the 3′ position on the aniline ring.22 In biochemical cellular phosphorylation and cell proliferation assays, the same trend in potency was also observed with the most potent among them being 5. Together, exploration of 3′-substituents on the aniline ring revealed a bias toward a 2′-fluorine and 3′-halide as the most potent inhibitors of EGFR, with the Cl or Br substitution in the 3′-position having the most activity.
To further test the importance of the 2′-fluorine, derivatives of 3–5 lacking the 2′-fluorine were evaluated (Table 3). Although 8 was similar in potency as 3 against EGFR, it was inferior to the 3′-halide substituted compounds (S3, 4, 5, 6). Moreover, compounds 9 and 10(25) had reduced efficacy against EGFR compared to their 2′-fluorinated counterparts. The significance of the 2′-fluorine in protein–ligand binding was further accentuated by the substitution with a polar 2′-hydroxy group for the fluorine (11), which greatly reduced EGFR affinity. These results suggest a limitation of size and polarity of the 2′ substituent which is in line with previous studies.17
We next investigated trisubstituted anilines as they can be potent inhibitors of EGFR.26 Retaining the 2′-fluorine and 3′-bromine on the aniline ring, we examined the effect of an additional halide in either the 4′- or 5′-position (12–17). In particular, the trisubstituted aniline ring of 15 resulted in a potent EGFR inhibitor in both biochemical and cell-based proliferation assays (Table 4). Since an ortho-fluorine was identified as important to improve potency, we also asked if a 6′- instead of a 2′-fluorine, or two ortho-fluorines would influence potency against EGFR (18 and 19). Although antiproliferative effects against patient-derived GBM lines were on par with those of 5, the ability to inhibit EGFR in cell-free and cell-based assays was reduced, suggesting potential off-target effects of 18 and 19.
Table 4. Modifications of the 4′, 5′, and 6′-Positions of 3′-Bromo-2′-fluoro-Substituted 4-Anilinoquinazolinesa.
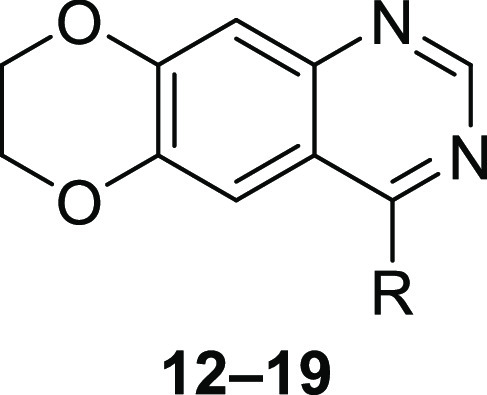

All EGFR inhibition data are represented as mean ± SEM from n = 2 or more independent replicates. All growth inhibition data are represented as mean ± SEM from n = 3 or more independent replicates.
To further differentiate our lead EGFR inhibitors, we next examined their selectivity as well as BBB penetrance. First, to examine potential off-target activity, compounds were screened against endogenous cells of the brain, normal human astrocytes (NHA), which, in contrast to EGFR-altered GBM cells, lack a dependency on EGFR for growth (Figure S3). As predicted, compounds 15 and 18, as well as 19, had a low NHA/GBM GI50 ratio supporting their potential for off-target effects (Table S3). Conversely, 1, 2, S3, and 4–6 displayed high potency against primary GBM cells relative to NHAs (Table S3). Next, we sought to ascertain the brain penetrance of those compounds with a high NHA/GBM GI50 ratio. Pharmacokinetic analysis of brain/plasma ratios in healthy CD-1 mice revealed an improved brain penetrance with a 3′-halide over a 3′-alkyne substituent, with the most penetrant unexpectedly containing a bromine substitution (Table 5). Brain penetration of the most potent compounds, 4 and 5, achieved brain/plasma ratios of 1.064 and 2.118 and Kp,uu of 1.04 and 1.30, respectively (Figure S4).
Table 5. Brain Penetration and in Vivo Parameters of Select Compoundsa.
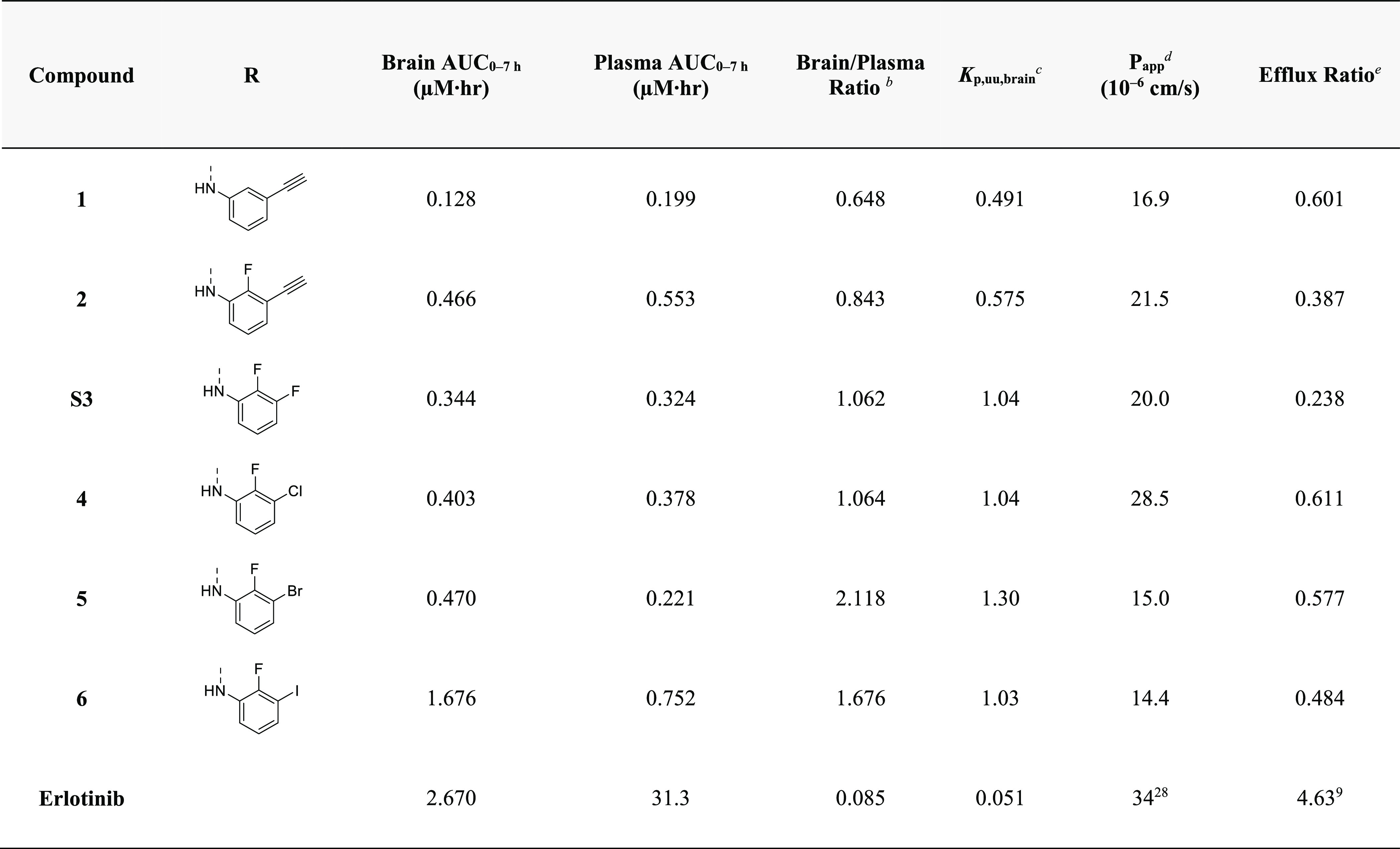
All brain and plasma AUC0–7h determined after oral administration of 10 mg/kg in male CD-1 mice.
Brain/plasma ratios determined over 0–7 h.
Ratio of the unbound concentration in the brain to that of plasma.
Permeability determined using MDCK-MDR1 cells.
Ratio of Papp B-A/Papp A-B.
Given the relatively high BBB penetration of our dioxane-containing EGFR TKIs, we explored the potential molecular rationale for this observation. Reducing the MW, HBD, HBA, TPSA, and NRB can increase brain penetration through circumventing recognition by the P-gp or breast cancer resistance protein (BCRP) drug efflux pumps on brain capillary endothelial cells.15,27,28 We hypothesized that the fused dioxane ring of our EGFR TKIs may limit substrate identification by P-gp or BCRP. Evaluation of compounds 1, 2, S3, and 4–6 by transwell culture with MDCK-MDR1 cells revealed that our compounds are highly permeable (>10 × 10–6 cm/s), with a low efflux ratio, indicating that these new EGFR TKIs are not strong substrates of P-gp or BCRP (Table 5, Table S4). Together, these data suggest that replacement of the alkoxy tails by the fused dioxane ring reduces P-gp and BCRP substrate identification—potentially by the disruption of a recognized pattern of HBA. This reduced substrate affinity may contribute to the enhanced BBB penetration observed with our EGFR TKIs. Based on the high potency against both wtEGFR and EGFRvIII in cell-based biochemical assays, the strong antiproliferative effects against multiple EGFR-driven patient-derived lines, the low activity against the NHA cell line, and the high brain penetrance, 5 was chosen as the lead candidate for additional in vitro and in vivo evaluations.
To determine the specificity of 5, kinome profiling was performed at 1 μM across 485 wild-type and mutant kinases (Thermofisher). 5 strongly (>90%) inhibited EGFR and most EGFR kinase domain mutants with few off-target kinases (Figure S5). Only 14 kinases were inhibited by greater than 50%, of which eight were EGFR and EGFR mutant kinases. Moreover, IC50 values of all wild-type kinases with greater than 50% inhibition at 1 μM of 5 revealed nearly 400× selectivity for EGFR (0.6 nM) relative to the next closest kinase (RIPK3: 226 nM) (Figure S5).
To further evaluate 5, we biochemically profiled it against both erlotinib and lapatinib in two EGFR-altered patient-derived gliomaspheres: GBM39 (EGFRvIII mutant) and GS025 (amplified EGFR). In GBM39, all three TKIs potently inhibited EGFRvIII activation as well as the RAS-MAPK (via p-ERK) and PI3K-AKT-mTOR (via pS6) signaling pathways downstream of EGFR in a dose-dependent manner (Figure 1A). Modulation of these pathways with the various EGFR TKIs occurred to a similar degree, albeit lapatinib and 5 demonstrated slightly more signaling inhibition in the 100–333 nM concentrations relative to erlotinib. Consistent with these signaling results, we observed all three TKIs inhibited growth of EGFRvIII mutant GBM39 cells, with lapatinib and 5 showing more robust growth inhibition than erlotinib at 100–300 nM (Figure 1C). For GS025, we observed that erlotinib and 5 had nearly identical effects on wtEGFR signaling and, consequently, growth inhibition (Figure 1B and D). Conversely, we observed a notable loss in biochemical and functional efficacy for lapatinib in GS025 compared to the other two TKIs (Figure 1B and D). These data are consistent with lapatinib having lower activity against active wtEGFR.6 Together, these results indicate that 5 can potently inhibit the signaling and growth of EGFRvIII mutant and EGFR amplified primary GBM cells at levels on par with or better than that of both erlotinib and lapatinib.
Figure 1.

Biochemical and functional activity of 5, lapatinib, and erlotinib on EGFRvIII mutant and EGFR amplified patient-derived GBM cells. Immunoblot of EGFR signaling components in (A) EGFRvIII mutant patient-derived GBM39 cells and (B) amplified EGFR patient-derived GS025 cells. Growth inhibition of (C) GBM39 and (D) GS025 cells relative to vehicle. All growth inhibition data are represented as mean ± SEM from n = 3 independent replicates. *p < 0.05, **p < 0.01, ***p < 0.001.
We next carried out a similar evaluation in an orthotopic GBM xenograft model. To do this comparison in the most clinically relevant manner, we first established the clinically relevant dose of erlotinib and lapatinib in which the plasma exposures in mice matches that of human plasma levels at the standard clinical dose.29,30 Erlotinib and lapatinib administered at 10 mg/kg and 80 mg/kg in nontumor bearing mice reached plasma exposures of 51,689 nM·h and 44,807 nM·h over 24 h, respectively, which mirrors the 24-h human clinical plasma exposures for both drugs.31,32 However, due to the low bioavailability of 5 of approximately 4.7% (Figure S4), we dosed at 300 mg/kg BID to achieve plasma exposures of 5 similar to that of the clinically relevant doses of both erlotinib and lapatinib in nontumor bearing mice (Figure 2A).
Figure 2.

In vivo pharmacodynamics and efficacy of erlotinib, lapatinib, and 5 against EGFRvIII mutant patient-derived orthotopic GBM39 xenografts. (A) Plasma and brain exposures of erlotinib (10 mg/kg), lapatinib (80 mg/kg), and 5 (300 mg/kg) in mice. Below are the published human 24-h plasma exposures of erlotinib and lapatinib at clinical doses. (B) Immunoblot of EGFR signaling components of orthotopic GBM39 xenografts following 3 days of oral administration of the indicated drugs or vehicle. (C) Quantification of immunoblot in (B). All quantified immunoblot data are represented as mean ± SEM of n = 3 independent replicates. *p < 0.05, ** p < 0.01, ***p < 0.001. Intracranial GBM39 growth of daily (D) erlotinib, (E) lapatinib, or (F) twice daily treatment of 5. (G) Survival of mice from (D)–(F).
With the relevant doses established, we next implanted EGFRvIII mutant GBM39 into the brains of NOD-SCID Gamma mice. Once tumors reached exponential growth, as determined by secreted gaussia luciferase,33 tumors were analyzed by immunoblotting for activation of EGFRvIII and its downstream signaling effectors. In comparison to vehicle treated tumors, erlotinib and lapatinib treatment showed no significant difference in EGFRvIII activation (Figure 2B). Similarly, erlotinib and lapatinib did not significantly inhibit signaling pathways downstream of EGFR, including RAS-MAPK (via p-ERK) or PI3K-AKT-mTOR (via pAKT and pS6) signaling (Figure 2B and C). These observations are in agreement with clinical data suggesting that erlotinib and lapatinib do not reach sufficient levels in glioblastoma tumors to consistently inhibit EGFR signaling.4,6,34 Conversely, tumors from 5-treated mice showed a significant decrease in EGFRvIII activity that was associated with reduced RAS-MAPK and PI3K-AKT-mTOR signaling (Figure 2B and C). These data support the hypothesis that the heightened BBB penetration of 5 would result in a greater capacity to inhibit EGFR signaling in an orthotopic GBM xenograft model.
Next, to compare the antitumor efficacy of 5 against erlotinib and lapatinib, a second cohort of orthotopic GBM39 tumor-bearing mice was established with the same doses and schedules as the above pharmacodynamic studies with the various EGFR TKIs. We observed no significant differences in tumor growth nor survival with erlotinib or lapatinib treatment (Figure 2D and E). In contrast, a notable reduction in tumor proliferation was identified in 5-treated mice (Figure 2F), with no significant loss in body weight (Figure S6). Moreover, 5 treatment provided a significant survival benefit, whereby median survival increased by 47% from 37.5 days to 55 days with 5 treatment (Figure 2G). Taken together, these data show that, in contrast to clinically relevant doses of erlotinib and lapatinib, 5 robustly inhibits EGFR signaling and tumor growth and prolongs the survival of mice bearing EGFR mutant, orthotopic GBM xenografts.
To gain greater insight into the low bioavailability and potential metabolic liabilities of 5, we investigated its in vitro clearance using liver microsomes. We observed a rapid hydroxylation of the fused 1,4-dioxane ring, suggesting first pass metabolism contributed to low oral bioavailability (Figure S7). Our hypothesis was confirmed by the coadministration of the cytochrome p450 inhibitor, 1-aminobenzotriazole, which resulted in a 3-fold increase in exposure of 5 (Figure S8). To explore this issue, we made modifications at the metabolic labile site of the fused 1,4-dioxane ring moiety by perdeuteration, as well as adding vicinal methyl groups on the 1,4-dioxane ring (Figure S9). Perdeuteration was unable to alter the bioavailability in mice compared to 5. Conversely, the addition of vicinal methyl groups on the 1,4-dioxane ring significantly improved plasma exposures and bioavailability in mice suggesting a potential location for future modifications on this scaffold.
Recent evidence suggests that Type I EGFR TKI inhibitors—which favor the active confirmation of EGFR—have less affinity for mutant EGFRvIII relative to Type II EGFR TKIs, which prefer the inactive form of the receptor.6 Given that compound 5 can potently inhibit both wtEGFR and EGFRvIII, we performed a molecular docking study to elucidate on a molecular level how this dual specificity of 5 is achieved. The docking results of 5, displayed in Figure 3 (and Figure S10), suggest the typical type I TKI binding mode occurs as is also observed for erlotinib (cf. Figure S1), through hydrogen bond interactions with hinge residues and gatekeeper residues mediated through crystallographic water molecule(s). According to our docking results, no clear difference in the binding to the active and inactive EGFR conformations can be discerned, except for a slightly closer fit of the dioxane and aniline part of 5 into the binding pocket of the inactive conformation. The gain in efficacy and selectivity upon introduction of the 2′-fluorine might be attributed to several orthogonal multipolar interactions of this fluorine to nearby apolar residues including a hypothesized C–F···C=O contact with Ala719/743 (active/inactive).23 Collectively, although the conformation selectivity for EGFR TKIs is an intriguing effect that is not yet fully understood,35 we observed 5 may have the ability to bind both the active and inactive conformations of EGFR, which may contribute to its potency for both wtEGFR and EGFRvIII, respectively.
Figure 3.

Predicted binding mode of 5 to active and inactive wtEGFR kinase domain. 5 was docked with AutoDock Vina to active EGFR (PDB 1M17)18 as shown in (A), (B), and (C), and to inactive EGFR (PDB 1XKK),36 as shown in (D), (E), and (F). Color code: Cenzyme gray, O red, N blue, Cligand green, F light blue, Br dark red.
The synthesis of analogues 1–19 is summarized in Scheme 1. The quinazoline core was made according to the Niementowski quinazoline synthesis from methyl 3,4-dimethoxyanthranilate (20). The dimethoxy groups of quinazolinone 21 were replaced with pivaloyl groups to obtain 22. Chlorination with POCl3, followed by deprotection gave intermediate 23, which was alkylated with 1-bromo-2-chloroethane to obtain the 1,4-dioxane-fused 4-chloroquinazoline 24.
Scheme 1. Synthesis of 7,8-Dihydro[1,4]dioxino[2,3-g]quinazolin-4-amines 1–19.

Preparation of the final analogues was accomplished by SNAr of 24 with the respective anilines (see also Scheme S1) or by transition-metal catalyzed transformations of 4 or 5. To prepare sufficient material of our lead compound 5 for all in vivo testing, we devised the shorter route of Scheme S2, which comprises five steps and is based on a Dimroth cyclization.
In summary, herein we have described the synthesis of a novel, brain-penetrant EGFR TKI with high activity against EGFR altered primary GBM cells both in culture and in orthotopic xenografts. Compound 5 was developed by first modifying erlotinib via ring fusion of the 6,7-alkoxy groups. Similar dioxane-containing anilinoquinazoline compounds have been described before for the purpose of improved solubility,25,37 yet here we determined that this modification also leads to unforeseen BBB penetration, potentially as a result of the more optimal physicochemical properties and impaired P-gp and BCRP substrate identification. Moreover, 5 contains both a 2′-fluorine and 3′-bromine on the aniline ring; these substitutions further improved brain penetration, while providing nearly equipotent activity against both oncogenic activated wtEGFR and mutant EGFRvIII. While the EGFR TKIs developed specifically for EGFR-mutated lung cancer, osimertinib and AZD3759, both have reported high brain penetration (Table S5),9,19 osimertinib lacks the ability to effectively inhibit wtEGFR that is prevalent across GBM3 (Figure S11), and AZD3759 has reduced activity against EGFRvIII relative to 5 (Figure S12). These differences may explain the improved potency of 5 against EGFR altered primary GBM cells compared to either osimertinib or AZD3759 (Figures S11 and S12).
In contrast to the observation that some EGFR TKIs may promote paradoxical induction of cell growth,38 compound 5—and any other EGFR TKI tested—did not (Figure S13). Rather, despite the low bioavailability of 5 (4.7%), it significantly suppressed in vivo tumor growth via oral administration. As we have shown (Figure S7), the main liability of compound 5 is fast clearance through first-pass metabolism. Therefore, future work will be aimed at identifying drug candidates with improved bioavailability and other ADME properties to obtain an optimal clinical EGFR TKI for GBM tumors with EGFR alterations.
Acknowledgments
We thank Professor Harvey R. Herschman for proof reading of this manuscript. We also thank all members of the Nathanson Lab for their assistance and constructive comments throughout the course of this project.
Glossary
Abbreviations
- AUC
area under the curve
- BBB
blood–brain-barrier
- EGFR
epidermal growth factor receptor
- wtEGFR
wild-type EGFR
- GBM
glioblastoma
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- NRB
number of rotatable bonds
- TPSA
total polar surface area
- NSG
NOD-SCID Gamma
- P-gp
P-glycoprotein
- BCRP
breast cancer resistance protein
- TKI
tyrosine kinase inhibitor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00599.
Additional figures, tables, and schemes; experimental procedures and analytical characterization; NMR spectra of all final analogues; X-ray crystal structure data of compounds 4 and 5; tables with physicochemical properties of protein kinase inhibitors (PDF)
Author Contributions
# J.E.T. and L.M.U. contributed equally to this work. J.E.T., T.F.C., M.E.J., and D.A.N. conceived the study. J.E.T., L.M.U., and D.A.N. wrote the manuscript. J.E.T., L.M.U., P.M.C., M.E.J., and D.A.N. designed the compounds. J.E.T., K.C., L.B., and K.F. performed biological assays. L.M.U. and G.K. performed synthesis and characterization of compounds. All authors read and edited the manuscript.
This work was funded by the Uncle Kory Foundation, the B Hasso Family Foundation, the Spiegelman Family Foundation in Memory of Barry Spiegelman, the Art of the Brain, the Ziering Family Foundation, Sharpe/NBTS Grant, Chip Rosenbloom and Harry Zimmerman, Margaret M Bloomfield/Hunter Family Foundation. This work was also partially supported by grants from the National Institutes of Health (P50 CA211015; R01 CA213133).
The authors declare the following competing financial interest(s): J.E.T., L.M.U., G.K., T.F.C., P.M.C., M.E.J., and D.A.N. are inventors on a patent application covering chemical matter in this publication. T.F.C., M.E.J., and D.A.N. are founders and shareholders of Katmai Pharmaceuticals which has licensed chemical matter in this publication.
Supplementary Material
References
- Brennan C. W.; Verhaak R. G.; McKenna A.; Campos B.; Noushmehr H.; Salama S. R.; Zheng S.; Chakravarty D.; Sanborn J. Z.; Berman S. H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q. W.; Cheng C. K.; Gustafson W. C.; Charron E.; Zipper P.; Wong R. A.; Chen J.; Lau J.; Knobbe-Thomsen C.; Weller M.; et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013, 24, 438–449. 10.1016/j.ccr.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor R. B.; Zhu H. J.; Walker F.; Vitali A. A.; Perera R. M.; Burgess A. W.; Scott A. M.; Johns T. G. The tumor-specific de2–7 epidermal growth factor receptor (EGFR) promotes cells survival and heterodimerizes with the wild-type EGFR. Oncogene 2004, 23, 6095–6104. 10.1038/sj.onc.1207870. [DOI] [PubMed] [Google Scholar]
- Lassman A. B.; Rossi M. R.; Razier J. R.; Abrey L. E.; Lieberman F. S.; Grefe C. N.; Lamborn K.; Pao W.; Shih A. H.; Kuhn J. G.; et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01–03 and 00–01. Clin. Cancer Res. 2005, 11, 7841–7850. 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- Thiessen B.; Stewart C.; Tsao M.; Kamel-Reid S.; Schaiquevich P.; Mason W.; Easaw J.; Belanger K.; Forsyth P.; McIntosh L.; Eisenhauer E. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- Vivanco I.; Robins H. I.; Rohle D.; Campos C.; Grommes C.; Nghiemphu P. L.; Kubek S.; Oldrini B.; Chheda M. G.; Yannuzzi N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discovery 2012, 2, 458–471. 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon D. A.; Nabors L. B.; Mason W. P.; Perry J. R.; Shapiro W.; Kavan P.; Mathieu D.; Phuphanich S.; Cseh A.; Fu Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted Temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2014, 17, 430–439. 10.1093/neuonc/nou160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A.; Hernández-Laín A.; Balañá C.; Vicente E.; Reynés G.; Benavides M.; Martínez-García M.; Quindós M.; Vaz M. Á.; Gil-Gil M.; et al. Phase II trial of dacomitinib, a pan–human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology 2017, 19, 1522–1531. 10.1093/neuonc/nox105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard P.; Yates J. W. T.; Yang Z.; Kim D.-W.; Yang J. C.-H.; Cantarini M.; Pickup K.; Jordan A.; Hickey M.; Grist M.; et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin. Cancer Res. 2016, 22, 5130–5140. 10.1158/1078-0432.CCR-16-0399. [DOI] [PubMed] [Google Scholar]
- Freedman R. A.; Gelman R. S.; Agar N. Y.; Santagata S.; Randall E. C.; Lopez B. G. C.; Connolly R. M.; Dunn I. F.; Van Poznak C. H.; Anders C. K.; et al. Pre-and Postoperative Neratinib for HER2-Positive Breast Cancer Brain Metastases: Translational Breast Cancer Research Consortium 022. Clin. Breast Cancer 2020, 20, 145. 10.1016/j.clbc.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay M. R. V.; Ward R. A. Small molecule inhibitors of the epidermal growth factor receptor. Top. Med. Chem. 2017, 28, 39–74. 10.1007/7355_2016_26. [DOI] [Google Scholar]
- Heffron T. P. Small molecule kinase inhibitors for the treatment of brain cancer. J. Med. Chem. 2016, 59, 10030–10066. 10.1021/acs.jmedchem.6b00618. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Hudkins R. L.; Dorsey B. D.; Mallamo J. P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Chandrasekaran R. Y.; Hou X.; Troutman M. D.; Verhoest P. R.; Villalobos A.; Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci. 2010, 1, 420–434. 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raub T. J. P-glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharmaceutics 2006, 3, 3–25. 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- Broniscer A.; Panetta J. C.; O’Shaughnessy M.; Fraga C.; Bai F.; Krasin M. J.; Gajjar A.; Stewart C. F. Plasma and cerebrospinal fluid pharmacokinetics of erlotinib and its active metabolite OSI-420. Clin. Cancer Res. 2007, 13, 1511–1515. 10.1158/1078-0432.CCR-06-2372. [DOI] [PubMed] [Google Scholar]
- Denny W. A. The 4-anilinoquinazoline class of inhibitors of the erbB family of receptor tyrosine kinases. Farmaco 2001, 56, 51–56. 10.1016/S0014-827X(01)01026-6. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Barker A. J.; Zeneca Pharmaceuticals. Quinazoline derivatives. Eur. Pat. Appl. EP0566226, 1993.
- Ballard P.; Bradbury R. H.; Harris C. S.; Hennequin L. F.; Hickinson M.; Kettle J. G.; Kendrew J.; Klinowska T.; Ogilvie D. J.; Pearson S. E.; et al. Inhibitors of epidermal growth factor receptor tyrosine kinase: optimization of potency and in vivo pharmacokinetics. Bioorg. Med. Chem. Lett. 2006, 16, 4908–4912. 10.1016/j.bmcl.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Zeng Q.; Wang J.; Cheng Z.; Chen K.; Johnström P.; Varnäs K.; Li D. Y.; Yang Z. F.; Zhang X. Discovery and evaluation of clinical candidate AZD3759, a potent, oral active, central nervous system-penetrant, epidermal growth factor receptor tyrosine kinase inhibitor. J. Med. Chem. 2015, 58, 8200–8215. 10.1021/acs.jmedchem.5b01073. [DOI] [PubMed] [Google Scholar]
- Böhm H. J.; Banner D.; Bendels S.; Kansy M.; Kuhn B.; Müller K.; Obst-Sander U.; Stahl M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Bauer M. R.; Rutherford T. J.; Fersht A. R.; Joerger A. C.; Boeckler F. M. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem. Biol. 2015, 10, 2725–2732. 10.1021/acschembio.5b00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Park Y. K.; Seo S. H.; So I.-S.; Chung H.-K.; Yang B.-S.; Lee S.-J.; Park H.; Lee Y. S. 1,4-Dioxane-fused 4-anilinoquinazoline as inhibitors of epidermal growth factor receptor kinase. Arch. Pharm. 2001, 334, 357–360. . [DOI] [PubMed] [Google Scholar]
- Cha M. Y.; Lee K. O.; Kim M.; Song J. Y.; Lee K. H.; Park J.; Chae Y. J.; Kim Y. H.; Suh K. H.; Lee G. S.; et al. Antitumor activity of HM781–36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int. J. Cancer 2012, 130, 2445–2454. 10.1002/ijc.26276. [DOI] [PubMed] [Google Scholar]
- Szafraniec M. J.; Szczygieł M.; Urbanska K.; Fiedor L. Determinants of the activity and substrate recognition of breast cancer resistance protein (ABCG2). Drug Metab. Rev. 2014, 46, 459–474. 10.3109/03602532.2014.942037. [DOI] [PubMed] [Google Scholar]
- O’Brien Z.; Moghaddam M. F. A systematic analysis of physicochemical and ADME properties of all small molecule kinase inhibitors approved by US FDA from January 2001 to October 2015. Curr. Med. Chem. 2017, 24, 3159–3184. 10.2174/0929867324666170523124441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Korn J. M.; Ferretti S.; Monahan J. E.; Wang Y.; Singh M.; Zhang C.; Schnell C.; Yang G.; Zhang Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Spilker M. E.; Chen X.; Visswanathan R.; Vage C.; Yamazaki S.; Li G.; Lucas J.; Bradshaw-Pierce E. L.; Vicini P. Found in translation: maximizing the clinical relevance of nonclinical oncology studies. Clin. Cancer Res. 2017, 23, 1080–1090. 10.1158/1078-0432.CCR-16-1164. [DOI] [PubMed] [Google Scholar]
- Ling J.; Johnson K. A.; Miao Z.; Rakhit A.; Pantze M. P.; Hamilton M.; Lum B. L.; Prakash C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 2006, 34, 420–426. 10.1124/dmd.105.007765. [DOI] [PubMed] [Google Scholar]
- Burris H. A. III; Taylor C. W.; Jones S. F.; Koch K. M.; Versola M. J.; Arya N.; Fleming R. A.; Smith D. A.; Pandite L.; Spector N.; Wilding G. A phase I and pharmacokinetic study of oral lapatinib administered once or twice daily in patients with solid malignancies. Clin. Cancer Res. 2009, 15, 6702–6708. 10.1158/1078-0432.CCR-09-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai W. X.; Gosa L.; Daniels V. W.; Ta L.; Tsang J. E.; Higgins B.; Gilmore W. B.; Bayley N. A.; Harati M. D.; Lee J. T.; et al. Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat. Med. 2017, 23, 1342–1351. 10.1038/nm.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall E. C.; Emdal K. B.; Laramy J. K.; Kim M.; Roos A.; Calligaris D.; Regan M. S.; Gupta S. K.; Mladek A. C.; Carlson B. L.; et al. Integrated mapping of pharmacokinetics and pharmacodynamics in a patient-derived xenograft model of glioblastoma. Nat. Commun. 2018, 9, 4904. 10.1038/s41467-018-07334-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H.; Liu Y.; Lemmon M. A.; Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood E. R.; Truesdale A. T.; McDonald O. B.; Yuan D.; Hassell A.; Dickerson S. H.; Ellis B.; Pennisi C.; Horne E.; Lackey K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Chilin A.; Conconi M. T.; Marzaro G.; Guiotto A.; Urbani L.; Tonus F.; Parnigotto P. Exploring epidermal growth factor receptor (EGFR) inhibitor features: the role of fused dioxygenated rings on the quinazoline scaffold. J. Med. Chem. 2010, 53, 1862–1866. 10.1021/jm901338g. [DOI] [PubMed] [Google Scholar]
- O’Connor M.; Zhang J.; Markovic S.; Romashko D.; Salomatov A.; Ishiyama N.; Iacone R.; Flohr A.; Nicolaides T.; Mayweg A.; et al. EGFR Oncogenes Expressed In Glioblastoma Are Activated As Covalent Dimers And Paradoxically Stimulated By Erlotinib. bioRxiv 2019, 810721.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.