

Abstract

Organophosphorus nerve agents (OPNAs) inhibit acetylcholinesterase (AChE) and, despite the Chemical Weapons Convention arms control treaty, continue to represent a threat to both military personnel and civilians. 2-Pralidoxime (2-PAM) is currently the only therapeutic countermeasure approved by the United States Food and Drug Administration for treating OPNA poisoning. However, 2-PAM is not centrally active due to its hydrophilicity and resulting poor blood–brain barrier permeability; hence, these deficiencies warrant the development of more hydrophobic analogs. Specifically, gaps exist in previously published structure activity relationship (SAR) studies for 2-PAM, thereby making it difficult to rationally design novel analogs that are concomitantly more permeable and more efficacious. In this study, we methodically performed a methyl scan on the core pyridinium of 2-PAM to identify ring positions that could tolerate both additional steric bulk and hydrophobicity. Subsequently, SAR-guided molecular docking was used to rationalize hydropathically feasible binding modes for 2-PAM and the reported derivatives. Overall, the data presented herein provide new insights that may facilitate the rational design of more efficacious 2-PAM analogs.
Keywords: Drug design, medicinal chemistry, structure−activity relationships, molecular modeling, antidotes, nerve agents
Acetylcholinesterase (AChE) catalyzes the hydrolysis of acetylcholine, a neurotransmitter responsible for sending activation signals resulting in muscle contraction.1 Structurally, the enzyme’s active site, a catalytic triad consisting of Ser-203, Glu-334, and His-447 (human AChE numbering), is buried in a 20 Å deep binding gorge lined primarily by aromatic residues.2 The nucleophilic Ser-203 residue, in conjunction with its Glu and His partners, is responsible for the homeostatic hydrolysis of acetylcholine.
Organophosphorus nerve agents (OPNAs) (Figure 1a) are some of the most toxic of synthetic substances; in small laboratory animals (e.g., rabbits, guinea pigs, and mice), 24 h subcutaneous LD50’s range from 10 to 165 μg/kg.3 Mechanistically, these agents phosphylate the catalytic Ser-203 residue, which consequently inhibits AChE’s function.4,5 Subsequently, toxic levels of neurotransmitter accumulate in the synaptic cleft and overstimulate cholinergic receptors. Resulting pathophysiology includes seizures, respiratory arrest, and often death.4
Figure 1.

AChE inhibitors and clinically used antidotes: (a) Structures of organophosphorus nerve agents. (b) Structures of clinically used antidotes. (c) Structure of AChE inhibitor, paraoxon ethyl, used in this study.
Current antidotes for OPNA poisoning (Figure 1b) provide only 24–34% reactivation of AChE in the peripheral nervous system; none offer global protection against OPNAs.4,5 While there are treaties and diplomatic efforts attempting to prevent the use of such agents, OPNAs are still being used during both acts of terrorism and, more recently, military conflicts. Consequently, there remains the need for antidotes that will more effectively pass the blood–brain barrier (BBB) and dephosphylate inhibited AChE with a greater efficacy than the currently used antidotes shown in Figure 1b.
With respect to 2-pralidoxime (2-PAM) efficacy, the majority of previously conducted synthetic studies have focused either on developing bis-pyridinium oximes (via alteration of the N-alkyl group) or on modifying the electrostatic character of the core pyridinium.5−7 However, these modifications appear to have been driven by synthetic accessibility rather than establishing systematic and rational structure–activity relationships (SAR)8 and, therefore, fail to clearly delineate optimal substitution patterns for AChE active site complementarity. To our knowledge, no studies to date have systematically explored SAR for alkyl group substitution on the pyridinium ring of 2-PAM, i.e., with the position of the alkyl group being the only variable. Therefore, in the current work, we conducted a methyl scan on the pyridinium ring of 2-PAM to (1) systematically probe the steric and electrostatic constraints of the AChE binding site to provide a foundation for the rational, structure-based design of new analogs and (2) determine whether the active site can tolerate an additional alkyl group on the pyridinium ring. The results of this study would allow for the rational design of a new, more hydrophobic, and therefore more BBB permeable, therapeutic candidate than 2-PAM.11−13 We chose 2-PAM because this is the only antidote approved by the United States Food and Drug Administration and because the simpler structure provides a more tractable template than related dimeric compounds, such as HI-6. For example, if we installed a methyl group in HI-6, the biological results would be uninterpretable because the biological results would be a sum of two modifications. However, the results presented herein will, in future studies, be extended to further optimize dimeric analogs, since we will understand the impact of a single methylation substitution.
Methyl scanning involves synthesizing a series of compounds in which a methyl group is systematically added to each modifiable position of a parent structure. This is a viable experimental method to delineate regions of a drug that are important or unimportant for biological activity, affording opportunities for structural modifications to improve potency and/or modify bioavailablilty.9 Specifically, for 2-PAM, we hypothesized that synthesizing the analogs shown in Figure 2 and subsequently testing their reactivation potencies against AChE from human (hAChE) and Electrophorus electricus (eel AChE) pretreated with an organophosphorus-based inhibitor would provide a better understanding of how such reactivators interact with surrounding AChE binding site residues, and hence provide a rational basis for future synthetic efforts. Although paraoxon ethyl (Figure 1c) may not fully represent nerve agents,10−12 it has been used in many studies as a simulant of OPNAs6,13−27 due to stringent government controls and, therefore, was used in the current study. Moreover, as shown in Table S2, by simply adding a methyl group, computational calculations predict slight increases in both ALogP and BBB permeability, and a slight decrease in solubility (i.e., an indicator of increased lipophilicity).
Figure 2.

Structures of target C-methylated 2-PAM analogs for this study.
The compounds shown in Figure 2 were synthesized via the route shown in Scheme 1. The first synthetic step involved the N-oxidation of commercially available dimethylpyridines following a method reported by Limnios and Kokotos (54–69% yields).28 Subsequently, a Boekelheide rearrangement29 afforded the corresponding hydroxymethylpyridines in 40–96% yields. The oxidation of the alcohols with MnO2 generated the corresponding aldehydes in 42–77% yields, and these aldehydes were then condensed with hydroxylamine to form the oximes in 52–86% yields. Finally, N-methylation with MeI provided the final products in 21–77% yields. N-Methylation of the 3-, 4-, and 5-methyl precursors was facile and went to completion. The N-methylation of the 6-methyl precursor required longer reaction time and required purification due to the formation of a side product.
Scheme 1. Synthesis of C-Methylated 2-PAM Analogs.
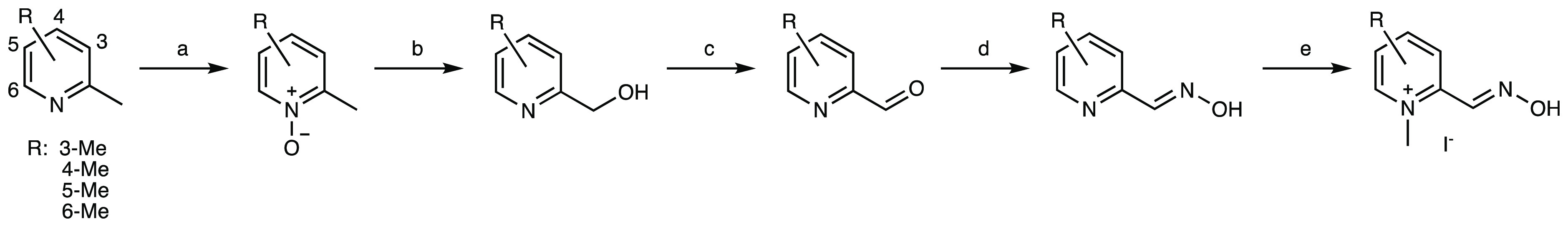
Reagents and conditions: (a) 2,2,2-trifluoroacetophenone (10 mol %), MeCN (1.5 equiv), 30% aq H2O2 (5 equiv), tBuOH, aq K2CO3, 23 °C, 54–69%; (b) TFAA (3 equiv), DCM, 23 °C, 40–96%; (c) MnO2 (5 equiv), DCE, reflux, 42–77%; (d) K2CO3 (1.2 equiv), NH2OH·HCl (1 equiv), MeOH, H2O, reflux, 52–86%; (e) MeI (10 equiv), acetone, 21–55 °C, 21–77%. tBu = tertiary butyl, TFAA = trifluoroacetic anhydride, Me = methyl, DCM = dichloromethane, DCE = 1,2-dichloroethane.
With these four 2-PAM analogs in hand, Ellman’s assay was employed to determine their reactivation potencies.30 The enzyme, either hAChE or eel AChE, was incubated with paraoxon ethyl for 10 min before being added to a clear 96-well plate containing the oximes, Ellman’s reagent, and acetylthiocholine. First, dose-dependent reactivation was determined by measuring the absorbance at 405 nm with hAChE (Figure 3a). At 18.8 μm, 3-Me-2-PAM is approximately 13% as efficient as 2-PAM as a reactivator. The relative reactivation efficiencies for 4-Me-2-PAM, 5-Me-2-PAM, and 6-Me-2-PAM were 67%, 34%, and 43%, respectively. When we compared the relative efficiency of reactivation at 75 μm, 3-, 4-, 5-, and 6-Me-2-PAM were 20%, 85%, 37%, and 62%, respectively.
Figure 3.

Reactivation of AChE by methyl scan analogs. (a) Dose response curve at 60 min with hAChE. (b) 18.8 μm oxime with hAChE. (c) Dose response curve at 20 min with eel AChE. (d) 18.8 μm oxime with eel AChE.
The kinetic data with 18.8 μm oximes are shown in Figure 3b. Our linear regression analysis suggests that the relative rates of 3-, 4-, 5-, and 6-Me-2-PAM (with 2-PAM being 100%) are 11%, 68%, 32%, and 45%, respectively. This is consistent with the above analysis with these oximes at 18.8 μm.
The dose response curve of the methyl scan analogs for eel AChE (Figure 3c) showed that 4-Me-2-PAM and 6-Me-2-PAM were nearly equivalent to 2-PAM until approximately 150 μm concentrations. At concentrations greater than 150 μm, the reactivation decreased for both analogs. 5-Me-2-PAM showed moderate reactivation efficiency, while 3-Me-2-PAM only slightly reactivated the inhibited enzyme at high concentrations. At 18.8 μm, both 4-Me-2-PAM and 6-Me-2-PAM reactivated paraoxon-ethyl-inhibited AChE (Figure 3d) and were found to be nearly equivalent to 2-PAM. Moreover, 5-Me-2-PAM showed reduced reactivation efficiency, while 3-Me-2-PAM showed negligible reactivation (Figure 3b). At 18.8 μm, the relative efficiencies of reactivation for 3-, 4-, 5-, and 6-Me-2-PAM in comparison to 2-PAM were 13%, 90%, 44%, and 87%, respectively.
The four analogs were also incubated with free hAChE and eel AChE to determine whether any of them might directly inhibit the enzyme. As Supporting Information (SI) Figure S1 shows, none significantly did so, thereby excluding the possibility that weaker reactivation potencies observed in Figure 3 were caused by the inhibition of AChE by the oxime. Interestingly, we reproducibly observed the bell-shape reactivation trends with 4-Me-2-PAM shown in Figure 3c and questioned if this might stem from compound aggregation. However, UV–vis spectroscopic analysis of 4-Me-2-PAM based on Beer’s law indicated that this was not the case (Supporting Information Table S1, Figure S2, and Figure S3). Therefore, we are not able to account for the bell-shape trends at this time at this point. However, with regard to therapeutic viability, the downward trend may not be relevant, as the compound concentration in vivo, especially in the central nervous system, might not exceed 100 μm.
Molecular modeling studies were performed to aid in rationalizing the AChE reactivation differences observed for the methyl scan analogs, which could not be explained based solely on analyses of changes in electrostatic potential (Supporting Information Figure S4). For AChE coordinates, PDB entry 5HFA was used.31 This is the human form of the enzyme inhibited by paraoxon and is cocrystallized with 2-PAM in an unproductive binding mode. Specifically, in the unmodified 5HFA coordinates, the pyridinium nitrogen of 2-PAM is in the same location as observed for the acetylcholine quaternary nitrogen in the active site cation box. This allows 2-PAM to be engaged in both face-to-face π-stacking and a cation−π interaction with Trp-86. However, while beneficial as a starting point for modeling studies, the oxime moiety in the 5HFA cocrystal is pointed away from phosphylated Ser-203, with the antidote’s oxygen atom located 8.42 Å away from the Ser-203-bound phosphorus atom (Supporting Information Figure S5a). Such a 2-PAM position neither rationalizes its reactivation efficacy nor the reactivation efficacies of the methyl scan analogs, hence the term “unproductive binding” (Supporting Information Figure S5b).
With respect to the species choice for the modeling studies, it is important to note that the human 5HFA X-ray structure, versus an Electrophorus electricus (eel) structure, was employed, despite using eel AChE as a secondary in vitro experiment to demonstrate consistency across species. The rationale for this choice was 2-fold: First, the highest available resolution apo-state X-ray structures for the two species are virtually identical (with a root-mean-square deviation (including α-helices, sheets, and loops) of only 0.642 Å (Supporting Information Figure S6a)), as are all species from phylum Chordata for which an AChE X-ray structure has been solved (Supporting Information Figure S6b). Second, residues surrounding the binding site are identical in human and eel (Supporting Information Figure S6c) AChE, as well as in other species (Supporting Information Figure 6d).
With AChE reactivation in mind, we considered a more therapeutically relevant, productive binding mode for 2-PAM, with the oxime oxygen atom placed within a reasonable distance from the phosphorus atom of the covalently bound paraoxon for the transesterification process. To aid in repositioning the 2-PAM orientation (i.e., versus the orientation observed in cocrystal 5HFA), a second X-ray structure, PDB entry code 2HA0, was used as a guide. This structure contains ketoamyltrimethylammonium (KTA), an acetylcholine mimetic, which forms a hemiketal bond with Ser-203.32 Hence, it provided a good approximation and distance required for an SN2 attack by 2-PAM on the paraoxon phosphorus. Therefore, the 5HFA and 2HA0 X-ray structures were superimposed (Supporting Information Figure S7). Next, we manually repositioned 2-PAM, via translational and rotational adjustments, over the three-dimensional coordinates of KTA, taking special care to position the oxime oxygen atom of 2-PAM directly over the KTA methyl group covalently bound to the hydroxy group of Ser-203 and the nitrogen atom of the 2-PAM pyridinium ring over the KTA ammonium nitrogen (Figure 4a). Following this alignment, the entire 2HA0 X-ray structure (including KTA) was completely removed, and iterative rounds of energy refinement, atom–atom contact scoring, and manual adjustments (to remove van der Waals violations) were performed to optimize the 2-PAM binding position in the PDB entry 5HFA structure (see the SI for method specifics). The energy refined binding mode for 2-PAM is shown in Figure 4b. With respect to the more applicable, pre-AChE rescue 2-PAM binding mode, three features are notable: First, the pyridinium nitrogen is engaged in a cation−π interaction with the Trp-86 side chain indole in a location that is comparable to that of the ammonium ion of bound KTA (i.e., the positive charge of 2-PAM is well positioned in the aromatic box motif of the binding site (Figures 4a and 4b)). Second, the pyridinium ring is engaged in favorable edge-to-face stacking with Trp-86. Third, the oxime oxygen is properly oriented and within a reasonable distance (2.3 Å) for SN2 attack on the paraoxon phosphorus that is covalently bound to Ser-203.
Figure 4.

Binding mode of 2-PAM and its methyl scan derivatives. PDB entry 5HFA(31) was used for all modeling and is shown in pale cyan cartoon. Select residues surrounding the active site are shown with green carbons, the carbons of both paraoxon-bound Ser-203 and paraoxon are shown in yellow carbons, and the paraoxon phosphorus is colored in orange in panels b–f. Yellow dashes indicate the interaction between the compound’s oxime oxygen and the paraoxon phosphorus prior to transesterification. (a) Proposed 2-PAM (magenta carbons) binding mode that places the oxime group in an orientation that is plausible for SN2 attack on the phosphorus atom of paraoxon when it is covalently bound to Ser-203. The proposed 2-PAM binding mode is superimposed onto that of covalently bound acetylcholine derivative KTA (orange carbons (both ligand and Trp-86), PDB entry 2HA0(32)) to show similarities in key, atomic-level positioning. (b) The same binding mode of 2-PAM as shown in (a), but without KTA, and with 2-PAM hydrogens shown. (c) The 3-methyl (cyan carbon) of 3-Me-2-PAM forces the oxime moiety to adopt an out-of-plane relative to the pyridinium ring. This conformation is predicted to significantly perturb the derivative’s ability to avoid engaging in unfavorable contacts with surrounding residues to achieve a binding orientation that allows for AChE reactivation. For example, in one potential binding pose, the 3-methyl would be engaged in a highly unfavorable hydrophobic-polar clash with Tyr 337 (red dashes, distance 2.4 Å). (d) The 4-methyl substituent (cyan carbon) of 4-Me-2-PAM points out the AChE active site and into the sterically unhindered AChE binding gorge. (e) The 5-methyl (cyan carbon) of 5-Me-2-PAM is located in a position within 3.4 Å of the side chain phenol oxygen of Tyr-337, resulting in a moderate hydrophobic-polar clash (pink dashes) that would perturb optimal binding. (f) The 6-methyl (cyan carbon) of 6-Me-2-PAM points into a hydrophobic location in the AChE binding site and is engaged in a favorable hydrophobic contact with the benzene ring of the Trp 86 side chain.
Our revised 2-PAM binding mode was iteratively methylated on the 3-, 4-, 5-, and 6-positions of the drug’s pyridinium. Each derivative was energetically refined in the binding site (see the SI for method specifics and SI Table S3 for quantitative, final HINT scores for active analogs 4-Me-2-PAM, 6-Me-2-PAM, and 2-PAM as a control for comparison). Subsequently, the optimized 2-PAM binding mode and those of the methyl scan analogs were compared to rationalize the reactivation data shown in Figure 3. Figure 4c shows a proposed binding mode of the inactive 3-Me-2 PAM derivative. Conformational analysis indicates that the 3-position methyl forces the oxime moiety of this analog out of the pyridinium plane. This twisted conformation significantly perturbs the derivative’s ability to achieve an orientation and distance that properly positions the oxime oxygen near the phosphorus atom of Ser203-bound paraoxon. When the oxime is oriented as observed for the optimized 2-PAM model (Figure 4b), the 3-methyl substituent experiences a highly unfavorable hydrophobic-polar clash with the hydroxy group of Tyr-124. Subsequently, if the pyridinium ring of 3-Me-2-PAM is rotated to moderate this highly unfavorable residue interaction, the 3-methyl is engaged in an unfavorable hydrophobic-polar and steric clash with the side chain of Tyr-337. Finally, when attempting to ameliorate both of these steric/electrostatic incompatibilities, the position of the 3-methyl-2-PAM oxime moiety cannot orient properly to perform the SN2 attack required to liberate Ser 203 from covalently bound paraoxon. Hence, due to its conformation, the 3-Me-2-PAM derivative is trapped in a negative feedback cycle that obviates its ability to act as an AChE rescue agent.
In contrast, the binding mode of 4-Me-2-PAM (Figure 4d) is promising for the incorporation of more structurally diverse substituents to improve 2-PAM binding with AChE and/or to increase BBB permeability. Specifically, the model indicates that the 4-methyl substituent points into sterically unhindered coordinate space that is mainly surrounded by aromatic residues of the enzyme’s binding gore. Hence, our computational analysis shows that this position on the 2-PAM pyridinium ring is optimal for further derivatizations.
The proposed binding mode of less potent 5-Me-2-PAM indicates that the 5-methyl group would experience a moderately unfavorable hydrophobic-polar clash with the side chain of Tyr-337 (Figure 4e). In contrast, the binding mode of 6-Me-2-PAM (Figure 4f) suggests that the 6-methyl group points into a desolvated subsite and is engaged in favorable hydrophobic contacts with the benzene component of the Trp-86 side chain. However, this subsite is sterically limited; therefore, when this derivative binds, Trp-86, which is located in a loop composed of residues Met-85 to Arg-90, is forced to slightly reorient to accommodate the bulk of the methyl group. Consequently, due to this structural adjustment, the binding affinity of 6-Me-2-PAM is less favorable than that of 2-PAM. Finally, as observed in Figure 4f, the pocket for this methyl position is in the interior of the acetylcholine binding site, and substituents larger than a methyl group will result in unfavorable steric clashes with surrounding residues.
In conclusion, a systematic methyl scanning study on 2-PAM was conducted for the first time, because although the 4-position appeared to be the most common position for the derivatization of 2-PAM, there was no convincing study to validate such an approach. This study revealed that the 4- and 6-positions can tolerate methylation, while the 3-position cannot. 5-Me-2-PAM showed reduced reactivation, indicating that this is not an ideal position for parent compound derivatization to develop more potent antidotes. The reactivation efficiency of 5- and 6-Me-2-PAM slightly differed between eel and hAChE. Varying reactivation profiles have previously been observed across species.15 A pictorial summary of the data is shown in Figure 5. Finally, computational analyses rationalized the assay results and indicated that the 4-position of 2-PAM provides the most promising location for future efforts to synthesize new derivatives to improve either binding affinity, AChE reactivation, and/or BBB permeability.
Figure 5.

Summary of methyl scan study.
Glossary
Abbreviations
- OPNAs
organophosphorus nerve agents
- AChE
acetylcholinesterase
- 2-PAM
2-pralidoxime
- SAR
structure activity relationship
- BBB
blood–brain barrier
- KTA
ketoamyltrimethylammonium
- PX
paraoxon
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00586.
Synthetic and biological experimental procedures, computational methods, supplementary figures, and NMR spectra (PDF)
This work was financially supported by the US Defense Threat Reduction Agency (HDTRA1-16-1-0041).
The authors declare no competing financial interest.
Supplementary Material
References
- Quinn D. M. Acetylcholinesterase: Enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. 10.1021/cr00081a005. [DOI] [Google Scholar]
- Sussman J. L.; Harel M.; Frolow F.; Oefner C.; Goldman A.; Toker L.; Silman I. Atomic structure of acetylcholinesterase from Torpedo californica:- A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- Guidelines for Chemical Warfare Agents in Military Field Drinking Water; The National Academies Press: Washington, DC, 1995; p 80. [PubMed] [Google Scholar]
- Acharya J.; Dubey D. K.; Srivastava A. K.; Raza S. K. In vitro reactivation of sarin-inhibited human acetylcholinesterase (AChE) by bis-pyridinium oximes connected by xylene linkers. Toxicol. In Vitro 2011, 25, 251–256. 10.1016/j.tiv.2010.07.024. [DOI] [PubMed] [Google Scholar]
- Mercey G.; Verdelet T.; Renou J.; Kliachyna M.; Baati R.; Nachon F.; Jean L.; Renard P.-Y. Reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. Acc. Chem. Res. 2012, 45, 756–766. 10.1021/ar2002864. [DOI] [PubMed] [Google Scholar]
- Gorecki L.; Korabecny J.; Musilek K.; Malinak D.; Nepovimova E.; Dolezal R.; Jun D.; Soukup O.; Kuca K. SAR study to find optimal cholinesterase reactivator against organophosphorous nerve agents and pesticides. Arch. Toxicol. 2016, 90, 2831–2859. 10.1007/s00204-016-1827-3. [DOI] [PubMed] [Google Scholar]
- Ohta H.; Ohmori T.; Suzuki S.; Ikegaya H.; Sakurada K.; Takatori T. New safe method for preparation of sarin-exposed human erythrocytes acetylcholinesterase using non-toxic and stable sarin analogue isopropyl p-nitrophenyl methylphosphonate and its application to evaluation of nerve agent antidotes. Pharm. Res. 2006, 23, 2827–2833. 10.1007/s11095-006-9123-1. [DOI] [PubMed] [Google Scholar]
- Pooja; Aggarwal S.; Tiwari A. K.; Kumar V.; Pratap R.; Singh G.; Mishra A. K. Novel pyridinium oximes: synthesis, molecular docking and in vitro reactivation studies. RSC Adv. 2015, 5, 23471–23480. 10.1039/C4RA14696D. [DOI] [Google Scholar]
- Pirrung M. C.; Liu Y.; Deng L.; Halstead D. K.; Li Z.; May J. F.; Wedel M.; Austin D. A.; Webster N. J. G. Methyl scanning: total synthesis of demethylasterriquinone B1 and derivatives for identification of sites of interaction with and isolation of its receptor(s). J. Am. Chem. Soc. 2005, 127, 4609–4624. 10.1021/ja044325h. [DOI] [PubMed] [Google Scholar]
- Radić Z.; Sit R. K.; Kovarik Z.; Berend S.; Garcia E.; Zhang L. M.; Amitai G.; Green C.; Radic B.; Fokin V. V.; Sharpless K. B.; Taylor P. Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases. J. Biol. Chem. 2012, 287, 11798–11809. 10.1074/jbc.M111.333732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worek F.; Thiermann H.; Szinicz L.; Eyer P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol. 2004, 68, 2237–2248. 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Sit R. K.; Kovarik Z.; Hrvat N. M.; Žunec S.; Green C.; Fokin V. V.; Sharpless K. B.; Radić Z.; Taylor P. Pharmacology, pharmacokinetics, and tissue disposition of zwitterionic hydroxyiminoacetamido alkylamines as reactivating antidotes for organophosphate exposure. J. Pharmacol. Exp. Ther. 2018, 367, 363–372. 10.1124/jpet.118.249383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musilek K.; Holas O.; Kuca K.; Jun D.; Dohnal V.; Opletalova V.; Dolezal M. Novel series of bispyridinium compounds bearing a (Z)-but-2-ene linker – Synthesis and evaluation of their reactivation activity against tabun and paraoxon-inhibited acetylcholinesterase. Bioorg. Med. Chem. Lett. 2007, 17, 3172–3176. 10.1016/j.bmcl.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Jeong H. C.; Park N. J.; Chae C. H.; Musilek K.; Kassa J.; Kuca K.; Jung Y. S. Fluorinated pyridinium oximes as potential reactivators for acetylcholinesterases inhibited by paraoxon organophosphorus agent. Bioorg. Med. Chem. 2009, 17, 6213–6217. 10.1016/j.bmc.2009.07.043. [DOI] [PubMed] [Google Scholar]
- Bharate S. B.; Guo L. L.; Reeves T. E.; Cerasoli D. M.; Thompson C. M. Bisquaternary pyridinium oximes: Comparison of in vitro reactivation potency of compounds bearing aliphatic linkers and heteroaromatic linkers for paraoxon-inhibited electric eel and recombinant human acetylcholinesterase. Bioorg. Med. Chem. 2010, 18, 787–794. 10.1016/j.bmc.2009.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit R. K.; Radic Z.; Gerardi V.; Zhang L. M.; Garcia E.; Katalinic M.; Amitai G.; Kovarik Z.; Fokin V. V.; Sharpless K. B.; Taylor P. New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases. J. Biol. Chem. 2011, 286, 19422–19430. 10.1074/jbc.M111.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercey G.; Renou J.; Verdelet T.; Kiachyna M.; Baati R.; Gilor E.; Arboleas M.; Loiodice M.; Nachon F.; Jean L.; Renard P. Y. Phenyltetrahydroisoquinoline-pyridinaldoxime conjugates as efficient uncharged reactivators for the dephosphylation of inhibited human acetylcholinesterase. J. Med. Chem. 2012, 55, 10791–10795. 10.1021/jm3015519. [DOI] [PubMed] [Google Scholar]
- Gupta B.; Singh N.; Sharm R.; Foretic B.; Musilek K.; Kuca K.; Acharya J.; Satnami M. L.; Ghosh K. K. Assessment of antidotal efficacy of cholinesterase reactivators against paraoxon: In vitro reactivation kinetics and physicochemical properties. Bioorg. Med. Chem. Lett. 2014, 24, 4743–4748. 10.1016/j.bmcl.2014.07.095. [DOI] [PubMed] [Google Scholar]
- Kliachyna M.; Santoni G.; Nussbaum V.; Renou J.; Sanson B.; Colletier J. P.; Arboléas M.; Loiodice M.; Weik M.; Jean L.; Renard P. Y.; Nachon F.; Baati R. Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- aldoxime and -amidoxime hybrids as efficient uncharged reactivators of nerve agent-inhibited human acetylcholinesterase. Eur. J. Med. Chem. 2014, 78, 455–467. 10.1016/j.ejmech.2014.03.044. [DOI] [PubMed] [Google Scholar]
- Renou J.; Loiodice M.; Arboleas M.; Baati R.; Jean L.; Nachon F.; Renard P. Y. Tryptoline-3-hydroxypyridinaldoxime conjugates as efficient reactivators of phosphylated human acetyl and butyrylcholinesterases. Chem. Commun. 2014, 50, 3947–3950. 10.1039/C4CC00561A. [DOI] [PubMed] [Google Scholar]
- Pashirova T. N.; Zueva I. V.; Petrov K. A.; Babaev V. M.; Lukashenko S. S.; Rizvanov I. K.; Souto E. B.; Nikolsky E. E.; Zakharova L. Y.; Masson P.; Sinyashin O. G. Nanoparticle-delivered 2-PAM for rat brain protection against paraoxon central toxicity. ACS Appl. Mater. Interfaces 2017, 9, 16923–16933. 10.1021/acsami.7b04163. [DOI] [PubMed] [Google Scholar]
- Rosenberg Y. J.; Mao L. J.; Jiang X. M.; Lees J.; Zhang L. M.; Radic Z.; Taylor P. Post-exposure treatment with the oxime RS194B rapidly reverses early and advanced symptoms in macaques exposed to sarin vapor. Chem.-Biol. Interact. 2017, 274, 50–57. 10.1016/j.cbi.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad M.; Fatmi M. Q.; Musilek K.; Hussain A.; Kuca K.; Petroianu G.; Kalasz H.; Nurulain S. M. In silico and in vitro evaluation of two novel oximes (K378 and K727) in comparison to K-27 and pralidoxime against paraoxon-ethyl intoxication. Toxicol. Mech. Methods 2018, 28, 62–68. 10.1080/15376516.2017.1357777. [DOI] [PubMed] [Google Scholar]
- de Koning M. C.; Horn G.; Worek F.; van Grol M. Discovery of a potent non-oxime reactivator of nerve agent inhibited human acetylcholinesterase. Eur. J. Med. Chem. 2018, 157, 151–160. 10.1016/j.ejmech.2018.08.016. [DOI] [PubMed] [Google Scholar]
- de Paula R. L.; de Almeida J. S. F. D.; Cavalcante S. F. A.; Goncalves A. S.; Simas A. B. C.; Franca T. C. C.; Valis M.; Kuca K.; Nepovimova E.; Granjeiro J. M., Molecular modeling and in vitro studies of a neutral oxime as a potential reactivator for acetylcholinesterase inhibited by paraoxon. Molecules 2018, 23, ARTN 2954. 10.3390/molecules23112954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Malpani Y. R.; Lee J.; Shin J. S.; Han S. B.; Jung Y.-S. Novel tacrine-pyridinium hybrid reactivators of organophosphorus-inhibited acetylcholinesterase: Synthesis, molecular docking, and in vitro reactivation study. Bioorg. Med. Chem. Lett. 2018, 28, 3784–3786. 10.1016/j.bmcl.2018.10.006. [DOI] [PubMed] [Google Scholar]
- Rosenberg Y. J.; Wang J.; Ooms T.; Rajendran N.; Mao L. J.; Jiang X. M.; Lees J.; Urban L.; Momper J. D.; Sepulveda Y.; Shyong Y. J.; Taylor P. Post-exposure treatment with the oxime RS194B rapidly reactivates and reverses advanced symptoms of lethal inhaled paraoxon in macaques. Toxicol. Lett. 2018, 293, 229–234. 10.1016/j.toxlet.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limnios D.; Kokotos C. G. 2,2,2-Trifluoroacetophenone as an organocatalyst for the oxidation of tertiary amines and azines to N-oxides. Chem. - Eur. J. 2014, 20, 559–563. 10.1002/chem.201303360. [DOI] [PubMed] [Google Scholar]
- Boekelheide V.; Linn W. J. Rearrangements of N-oxides. A novel synthesis of pyridyl carbinols and aldehydes. J. Am. Chem. Soc. 1954, 76, 1286–1291. 10.1021/ja01634a026. [DOI] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V. Jr.; Feather-Stone R. M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Franklin M. C.; Rudolph M. J.; Ginter C.; Cassidy M. S.; Cheung J. Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins: Struct., Funct., Genet. 2016, 84, 1246–1256. 10.1002/prot.25073. [DOI] [PubMed] [Google Scholar]
- Bourne Y.; Radić Z.; Sulzenbacher G.; Kim E.; Taylor P.; Marchot P. Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 2006, 281, 29256–29267. 10.1074/jbc.M603018200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.