

Abstract

To derive new uricosuric agents, novel phenol derivatives were synthesized to overcome the disadvantages of benzbromarone (BBR), attributed by its structural features. Herein, we report the discovery of new phenol derivatives with a 1,1-dioxo-1,2-dihydro-3H-1,3-benzothiazole scaffold. The selected compound 11 (dotinurad, FYU-981) demonstrated remarkable inhibitory activity on uric acid uptake by primary human renal proximal tubule epithelial cells (RPTECs) and URAT1-mediated uric acid transport, with weak inhibitory activity against mitochondrial respiration. Dotinurad also displayed favorable pharmacokinetic profiles and higher potency in decreasing uric acid than BBR did in Cebus monkeys. Dotinurad has been approved as a new uricosuric medicine in Japan. Our strategy, which focuses on the structural features resulting in unfavorable effects, could be applied to the future developments of other drugs with disadvantages, particularly those having a bis-aryl ketone structure.
Keywords: Uricosurics, mitochondrial toxicity, benzbromarone, primary human renal proximal tubule epithelial cells, dotinurad
Hyperuricemia, a causative factor of gout, is one of the most studied diseases globally.1 Hyperuricemia is involved in diseases such as metabolic disorders, cardiovascular disease, and renal dysfunctions.2,3 There are two significant causes of hyperuricemia, which are overproduction of uric acid and a reduction in its renal elimination or both. Hence, medicines used against hyperuricemia are classified into two types based on their mechanisms of action: xanthine oxidoreductase inhibitors (XOIs) and uricosuric agents. Allopurinol, a representative XOI that decreases the blood level of uric acid by inhibiting uric acid synthesis from xanthine, has been used worldwide as the standard medicine for a long time. Recently, new XOIs, febuxostat and topiroxostat, were launched to provide treatment options for gout and hyperuricemia.4
Among uricosurics that increase urinary excretion of uric acid, probenecid and benzbromarone (BBR) have been used for a long time in some countries, and recently, a new uricosuric agent, lesinurad, was approved in the US and EU. These agents act by inhibiting urate reabsorption in the renal tubules.5,6 However, these medicines also possess disadvantages. Probenecid, which has also been used to maintain the blood concentration of other drugs such as penicillin, might cause drug–drug interactions due to nonselective inhibition of anion transporters.7 Because BBR has a highly potent hypouricemic effect in humans, it is used as one of the standard medicines in some countries such as Japan, Europe, and Brazil, especially when the effect of XOI is insufficient. However, following reports that fulminant hepatitis, a rare but life-threatening disorder, was associated with BBR administration, restriction of its use via warnings and withdrawal occurred in Japan and Europe, respectively.
Furthermore, BBR is an inhibitor of cytochrome P450 2C9 (CYP2C9), known as a polymorphic enzyme, and is metabolized by the enzyme. Metabolism by CYP2C9 may lead to drug–drug interaction and account for differences in metabolism among individuals.8,9 Lesinurad is only used in combination with XOIs and an inhibitor of CYP2C9 similar to BBR.10−12
Many studies have been carried out to investigate the mechanisms of BBR-induced hepatic toxicity13−19 including the involvement of metabolism, metabolites, and mitochondrial toxicity. Among these potential mechanisms, mitochondrial inhibitory activity (MIA) is one of the most promising theories for fulminant hepatitis.15,16 Based on these findings, to obtain compounds that possess potent pharmacological effects without mitochondrial toxicity, we modified BBR to overcome its disadvantages, which may be related to a structural flaw.
The association between the structure and mitochondrial toxicity of BBR and its analogs including amiodarone has been previously reported (Figure 1).17−19
Figure 1.

Ranking of benzbromarone (BBR) and its derivatives based on mitochondrial toxicity and structures of tolcapone and fenofibrate.
These studies reported that each of the structural parts (substituted benzoic acid and benzofuran) had no or minimal inhibitory activity on mitochondrial respiration. In contrast, substituted benzoyl-benzofuran analogs such as amiodarone, benzarone, and BBR (Figure 1), which are combined with these two units, had distinct inhibitory activities, and the bromo substitutions on the phenol ring enhance it. Because these derivatives have a bis-aryl ketone moiety known to be a radical stabilizing structure that is associated with some adverse effects,20 we assume the moiety affects the mitochondrial electron transfer system. Some medicines with the bis-aryl ketone such as tolcapone and fenofibrate (Figure 1) inhibit the mitochondrial function21,22 and are associated with the occurrence of hepatic toxicity. Therefore, this structure appears to have a risk of exerting mitochondrial toxicity. Based on the knowledge and the hypothesis stated above, we derived two modification strategies to avoid mitochondrial toxicity. The first is a conversion from bis-aryl ketone structures to aryl-benzamide structures, where the introduction of a nitrogen atom leads to a nonketone structure. At the same time, the other is the transformation of a bromine atom into other electron-withdrawing substituents. We also considered the structural features of BBR required to show urate uptake inhibition (UUI).
Because BBR has a plane structure and moderate acidity with a pKa value of 5.17, similar to that of uric acid (5.75), we assumed that BBR mimics uric acid. Therefore, electron-withdrawing groups such as bromide on phenol might play important roles in maintaining acidity, thereby allowing compounds to exert urate uptake inhibitory potency.
We prepared the new compounds, amide-linked indole and indoline derivatives with cyano-substituent as an electron-withdrawing group instead of bromine atoms on the phenol moiety, as alternatives to the acyl benzofuran structure (compounds 1 and 2a in Table 1).
Table 1. UUI and MIA of BBR, 1, and 2a.

UUI: Inhibitory activity of urate uptake into RPTECs.
MIA: Mitochondrial respiratory control ratio (RCR), IC50 (μM), or inhibition (%) at 100 μM.
To verify our strategy, these compounds were evaluated for their urate uptake inhibitory potency and mitochondrial toxicity. We established a novel method to evaluate the UUI of these compounds using primary human renal proximal tubule epithelial cells (RPTECs) (Method: Supporting Information). RPTECs play important roles in urate excretion and reabsorption. In fact, they can express many transporters including urate transporter 1 (URAT1), organic anion transporter 1 (OAT1), OAT3, multidrug resistance protein 4 (MRP4), ATP-binding cassette subfamily G member 2 (ABCG2), and glucose transporter 9 (GLUT9), which are involved in the transport of urate.23−25
In our experimental condition, BBR, known as a selective URAT1 inhibitor,26 inhibited uric acid uptake into RPTECs with an IC50 of single-digit micromolar and in a concentration-dependent manner. Hence, we believed that the urate uptake into RPTECs is mainly due to URAT1. This method will be reported in detail elsewhere. The mitochondrial respiratory control ratio (RCR) was used as an index of MIA. Both UUI (inhibitory activity of urate uptake into RPTECs, IC50) and MIA (RCR, IC50 (μM), or inhibition (%) at 100 μM) assays were performed for these compounds (Table 1).
The data indicated that the UUI potency levels of both indole 1 and indoline derivative 2a are the same as that of BBR. Although structural conversion to indole maintained high MIA, conversion to indoline lowered it. Hence, the aromatic indole ring might confer the same character as that of BBR with a bis-aryl ketone structure, while the indoline derivative alters the character owing to the bis-aryl ketone structure. These results suggested that modifying the bis-aryl ketone moiety is critical to avoiding mitochondrial toxicity. Because 2a was considered a seed compound, we prepared 2a-derivatives by adding another substituent on the phenol ring to investigate the compounds’ properties.
The synthetic procedures of 1 and 2 are shown in Scheme 1. The synthetic methods for substituted benzoic acid as intermediates have been reported earlier,27 and we have described some new benzoic acids in the Supporting Information. Acylation of indoline and 2,3-dimethylindole was accomplished with substituted or nonsubstituted 4-methoxy or 4-methoxymethoxy-3-cyanobenzoic acid and 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDC). The subsequent deprotection of phenol −OH yielded the desired analogs.
Scheme 1. Synthesis of Indole and Indoline Analogs.

Reagents and conditions: (a) EDC, HOBt in DMF, CHCl3; (b) R2 = MOM; HCl, 1,4-dioxane, THF or TFA, CH2Cl2, R2 = Me; LiCl, DMF, 35–91% over two steps.
The assay data of UUI and MIA, and the calculated LogP of these derivatives are shown in Table 2.
Table 2. UUI and MIA of Compounds 1 and 2 in Scheme 1.
| UUIa |
|||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 (μM) | % | MIAb IC50 (μM) or % | LogPc |
| 1 | 10.8 | 52.1 | 3.6 | 3.89 | |
| 2a | H | 2.5 | 67.5 | 43% | 2.70 |
| 2b | OMe | >100 | 21.7 | –10% | 2.57 |
| 2c | I | 5.3 | 65.1 | 53 μM | 4.05 |
| 2d | CN | 83.9 | 26.3 | 47% | 2.73 |
| 2e | SMe | >100 | 24.2 | 40 μM | 3.14 |
| 2f | tert-Butyl | 31.6 | 32.4 | 6.6 μM | 4.4 |
| 2g | Cyclopropyl | 8.8 | 67.8 | 24 μM | 3.43 |
| 2h | Cl | 14.7 | 38.2 | 31 μM | 3.26 |
| 2i | CF3 | >100 | 30.0 | 40% | 3.62 |
UUI potency seemed to be affected by substituent characters such as steric and electric properties including hydrophobicity. Although 2a, 2c, and 2g showed a relatively high activity, the bulky substituent (2f) had a lower potency compared to these three compounds presumably resulting from the disruption of the flatness of the molecules shared by uric acid. The electron donating (2b) and withdrawing (2d, 2i) groups affected pKa and their potencies. Furthermore, hydrophobicity seems to influence UUI potency; thus, UUI improved with increasing LogP values, except for one-substituted (2a) and bulky (2f) types.
As shown in Figure 2, a correlation was observed between MIA and LogP among the compounds in Table 2, in which MIA augmented with increasing LogP values. Although almost all compounds were close to the regression line (R2 = 0.54), compound 1 (indole derivative) showed an inferior character with stronger MIA than the other compounds. In contrast, 2c and 2i had weaker MIA for their LogP than the other compounds did.
Figure 2.

Correlation between the MIA (RCR, IC50 μM) and compounds’ LogP in Table 2. Compounds 2a, 2d, and 2i showed ca. 50% MIA at 100 μM; these IC50 values are assigned as 100 μM. Compound 2b showed no MIA at 100 μM; the IC50 value is assigned as 200 μM.
Although causes for the association between hydrophobicity and MIA are unclear, we speculate that it was dependent on membrane permeability and affinities to target proteins.
The association between UUI at 10 μM and MIA, which should ideally diverge from each other, is shown in Figure 3.
Figure 3.

Association between UUI (% inhibition at 10 μM) and MIA (RCR, IC50 μM) derived from Table 2 and Figure 2.
Although compounds with higher UUI tended to show higher MIA, 2a, 2c, and 2g showed promising in vitro character: high UUI and low MIA. Similar association was observed when UUI was evaluated at higher concentrations (30 or 100 μM). Therefore, the Cmax and urinary excretion rate (Exc.) of 2a, 2c, and 2g were then investigated as the pharmacokinetic (PK) screening compared to BBR (Table 3).
Table 3. Pharmacokinetic Profiles of 2a, 2c, 2g, and BBR.
| Compound | R1 | Cmaxa (μg/mL) | Exc.b (%) |
|---|---|---|---|
| 2a | H | 0.89 | 5.9 |
| 2c | I | 5.95 | 0.02 |
| 2g | Cyclopropyl | 1.36 | 0.35 |
| BBR | 4.42 | 0 |
Maximum concentration (Cmax).
Excretion rate in urine (%, 0–4 h) after oral administration of 3 mg/kg to rats.
To produce a pharmacological effect, special attention was given to the Exc. (%) of compounds based on the PK profiles of BBR in humans reported by some groups.28−33 Although BBR is typically eliminated from plasma at 48 h after administration and is excreted little in urine, a main metabolite, 6-OH-BBR,28,29 remains in the plasma and is excreted (1.2% of the dose) over 72 h in humans;30−32 this metabolites’ URAT1 inhibitory activity was only several times lower than that of BBR (6-OH-BBR IC50: 0.20 μM, BBR IC50: 0.0345 μM).31,32 As a long lasting effect of BBR in humans33 is consistent with the sustained urinary excretion of 6-OH-BBR, and URAT1 is present on the luminal side of epithelial cells of the renal tubule, it is suggested that 6-OH-BBR in urine contributes to the inhibition of uric acid reabsorption.32,33 Moreover, unlike BBR, the compounds in this study should not form active metabolites to avoid metabolic conversion by enzymes such as CYP2C9. Therefore, we considered that the excretion of unchanged drug with activity equivalent to that of BBR in urine is not only important to show activity, but also for the ideal property. Hence, to obtain candidate compounds, PK screening was performed with a target value of >1% of unchanged drug in 0–4 h urine in rat, based on the above 6-OH-BBR data.
Of the three compounds, only 2c showed a high Cmax, while its Exc. was low (0.02%). We concluded that the metabolic susceptibility of the core indoline structure with benzyl-CH2 was one of the causes of the inferior PK profiles. Therefore, we then investigated another 1,1-dioxo-1,2-dihydro-3H-1,3-benzothiazole derivative with −SO2– instead of benzyl-CH2– in the core that would be of metabolic resistance. Compound 7 of the new core structure with cyclopropyl substituent was prepared and compared with the indoline-type analog 2g and BBR regarding their UUI, MIA, PK, and CYP2C9 inhibitory assays (Table 4).
Table 4. Properties of Compound 7.

Compared to 2g, compound 7 showed satisfactory properties, with adequate UUI, weak MIA, and improved Cmax; furthermore, 7 had a higher Exc. than 2g did and appeared to have low CYP2C9 inhibitory activity (IC50 = 57 μM). Because 7 was considered a lead compound, we prepared 1,2-dihydro-3H-1,3-benzothiazole derivatives by converting the phenol substituents (Table 5). Although the synthetic method for these derivatives was previously described,27 additional information is added in the Supporting Information.
Table 5. UUI, MIA, and Pharmacokinetic Profiles of 1,2-Dihydro-3H-1,3-benzothiazole Derivatives.

| Compound | R1 | R2 | UUIa IC50 (μM) | MIAb IC50 (μM) or % | Cmaxc (μg/mL) | Exc.d (%) |
|---|---|---|---|---|---|---|
| 8 | CN | CF3 | >100 | 12% | ||
| 9* | CN | CF3 | 13.1 | 55 μM | 3.15 | 0.1 |
| 10 | CN | Cl | >100 | 20% | 1.35 | 23 |
| 11 | Cl | Cl | 6.8 | 27 μM | 9.14 | 1.1 |
| 12 | CN | iPr | 83.3 | 12 μM | 4.46 | 0.1 |
| 13 | CN | Et | >100 | 37 μM | 4.24 | 1.7 |
| 14 | CN | ethynyl | 3.2 | 13% | 1.58 | 12.4 |
| 15 | CN | SMe | 7.1 | 21% | 1.08 | 9.4 |
| 16 | CF3 | Cl | 2.6 | 18 μM | 6.41 | 0.1 |
| 17 | F | Cl | 33.9 | 63 μM | 7.70 | 4.3 |
| 18 | Cl | SMe | 5.7 | 59 μM | 4.64 | 0.2 |
| 19 | CF3 | OMe | 17.8 | 47% | 3.99 | 0 |
| 20 | Cl | OMe | 4.3 | 49% | 1.06 | 0.1 |
Many compounds showed potent UUI with single-digit μM (IC50) activity, whereas 8 and 10, having two electron-withdrawing substituents, had low UUI (Table 5). However, compared to 8 (core; −SO2−), 9 (core; −S−) maintained its UUI potency as the substituent effect may be partially due to the hydrophobicity of compounds. Among compounds with high potency, 11, 14, and 15 showed Exc. > 1%, and 11 had the highest Cmax.
The URAT1 assay was performed for some compounds to confirm the mechanism of action (Table 6). These compounds showed URAT1 inhibitory activity (0.1 μM) along with UUI in RPTECs (10 μM). A positive correlation was observed between the two assays as shown in the analysis of the 7 compounds in Table 6 (R2 = 0.59; Figure 4) and, furthermore, in a dose-dependency test of 11 (R2 = 0.87; Figure 4-1 in the Supporting Information). Thus, urate uptake into RPTECs seems to be mainly mediated by URAT1, as mentioned above.
Table 6. Urate Transporter 1 Inhibition and UUI of 1,2-Dihydro-3H-1,3-benzothiazole Derivatives and BBR.
Figure 4.

Correlation between the RPTEC and URAT1 assays among the compounds in Table 6.
The dichloro-substituted derivative 11 appeared to possess highly potent UUI and URAT1 inhibitory activities at least equivalent to that of BBR, low MIA, and high Cmax with moderate urinary excretion rate (>1%, 0–4 h). Although 11 showed weak CYP2C9 inhibitory activity (IC50 = 5.7 μM), its activity was thought to be sufficiently separated from uricosuric activity based on its PK profile. As 11 (dotinurad) had a favorable balance of these parameters, it was selected for further development. The results of the additional in vitro and in vivo assay using rats and Cebus monkeys are shown in Table 7 and Figure 5.
Table 7. Properties of 11 and BBR.

| Rat | Cmax or C0a (ng/mL) | AUC0-infb (ng·h/mL) | BA.c |
|---|---|---|---|
| 11 | 1085.8 ± 100.4 | 5744.7 ± 188.8 | 86.8% |
| 0.3 mg/kg p.o. | |||
| 11 | 2401.3 ± 65.6 | 6619.5 ± 2041.8 | |
| 0.3 mg/kg i.v. |
| Cebus monkeys | AUC0–24ha (μg·h/mL) | FEUAd (%) | Difference of ΔPUAefrom control (mg/dL) |
|---|---|---|---|
| 11 | 108.55 ± 52.94 | 16.5 ± 4.2 | 0.97 |
| 5 mg/kg p.o. | |||
| BBR | 95.24 ± 28.12 | 11.5 ± 7.9 | 0.46 |
| 30 mg/kg p.o. |
| In vitro evaluation | URAT1 inhibition IC50 (nM) |
|---|---|
| 11 | 37.2 |
| BBR | 190 |
Maximum concentration (Cmax or C0).
Area under the blood concentration–time curve (AUC); inf: infinity.
BA.:Bioavailability.
FEUA: Fractional excretion of urate at 0–4 h; FEUA value of control: 8.9 ± 4.0%.
ΔPUA: Changes in plasma urate level at 0–8 h; value of control: 1.65 ± 0.78 mg/dL.
Figure 5.
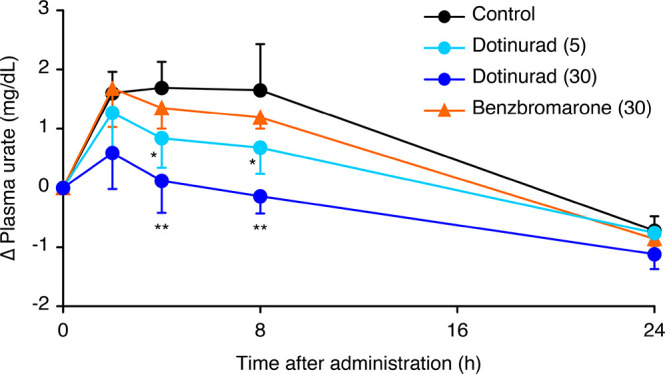
Changes in plasma urate level compared with the predosing value in Cebus monkeys. 11 (dotiurad); 5 and 30 mg/kg, BBR; 30 mg/kg p.o. Values are the means + SD of five animals. The average predosing value of control: 2.98 mg/dL. * P < 0.05, ** P < 0.01, significantly different from control at each time point by Dunnett’s test.
A detailed URAT1 assay of 11 showed an IC50 of 37.2 nM, indicating slightly higher potency than that of BBR (original data, IC50 = 190 nM), similar to the equipotent inhibition in the RPTEC assay. Compound 11 had excellent PK profiles in both animals. In Cebus monkeys, which could be an effective model for extrapolating the uricosuric effect in humans, 5 mg/kg of 11 showed a potent uricosuric effect with the fractional excretion of urate at 0–4 h (FEUA; 16.5% vs 11.5% of 30 mg/kg of BBR). Moreover, 11 decreased the plasma urate level more potently than did BBR in Cebus monkeys. This potent effect has not been reported with conventional medicines.
Because compound 11 only weakly inhibited ABCG2 and OAT1/3, it was shown to be a selective urate reabsorption inhibitor (SURI), which was defined as a potent URAT1 inhibitor with minimal effect on urate secretion transporters.34
The major metabolites of 11 in humans are glucuronide and sulfate of phenols, with excretion rates in urine during 0–72 h of 44.3% and 20.0%, respectively, in a mass balance test.35 The formation of reactive metabolites with potential to cause hepatotoxicity, as reported for BBR,13 was not observed. These results suggested that 11 had a safe metabolic pathway.
In conclusion, this series of studies led to the discovery of compound 11. We proceeded to clinical trials with 11 and obtained excellent results. The registration of 11 as a new medicine (dotinurad: FYU-981) has been approved in Japan.
We obtained new medicine by focusing on the negative aspects of existing drugs. The method used to avoid chemical structures that may cause unfavorable effects could be applied to other drugs with disadvantages.
Acknowledgments
We thank Koichi Omura for his contribution to the analytical work.
Glossary
Abbreviations
- DMF
N,N-dimethylformamide
- MOM
methoxymethyl
- EDC
1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride
- m-CPBA
m-chloroperbenzoic acid
- TFA
trifluoroacetic acid
- CYP2C9
cytochrome P450 2C9
- URAT1
urate transporter 1
- ABCG2
ATP-binding cassette subfamily G member 2
- OAT1/3
organic anion transporter 1/3
- MRP4
multidrug resistance protein 4
- GLUT9
glucose transporter 9
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00176.
Author Contributions
The manuscript was written through the contributions of all authors.
No external funding was received for this study.
The authors declare no competing financial interest.
Supplementary Material
References
- Benn C. L.; Dua P.; Gurrell R.; Loudon P.; Pike A.; Storer R. I.; Vangjeli C. Physiology of Hyperuricemia and Urate-Lowering Treatments. Front. Med. 2018, 5, 160. 10.3389/fmed.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbay M.; Afsar B.; Covic A. Uric Acid as a Cardiometabolic Risk Factor: To Be or Not to Be. Contrib. Nephrol. 2011, 171, 62–67. 10.1159/000327160. [DOI] [PubMed] [Google Scholar]
- Lipkowitz M. S. Regulation of Uric Acid Excretion by the Kidney. Curr. Rheumatol. Rep. 2012, 14, 179–188. 10.1007/s11926-012-0240-z. [DOI] [PubMed] [Google Scholar]
- Diaz-Torné C.; Perez-Herrero N.; Perez-Ruiz F. New Medications in Development for the Treatment of Hyperuricemia of Gout. Curr. Opin. Rheumatol. 2015, 27, 164–169. 10.1097/BOR.0000000000000146. [DOI] [PubMed] [Google Scholar]
- Diamond H. S.; Paolino J. S. Evidence for a Postsecretory Reabsorptive Site for Uric Acid in Man. J. Clin. Invest. 1973, 52, 1491–1499. 10.1172/JCI107323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman W.; Locovei S.; Dahl G. Probenecid, a Gout Remedy, Inhibits Pannexin 1 Channels. Am. J. Physiol. Cell Physiol. 2008, 295, C761–C767. 10.1152/ajpcell.00227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizaki R.; Nishijima T.; Aoki T.; Teruya K.; Kikuchi Y.; Oka S.; Gatanaga H. High-Dose Oral Amoxicillin Plus Probenecid Is Highly Effective for Syphilis in Patients with HIV Infection. Clin. Infect. Dis. 2015, 61, 177–183. 10.1093/cid/civ270. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Graham G. G.; Williams K. M.; Day R. O. A Benefit-Risk Assessment of Benzbromarone in the Treatment of Gout. Was Its Withdrawal from the Market in the Best Interest of Patients?. Drug Saf. 2008, 31, 643–665. 10.2165/00002018-200831080-00002. [DOI] [PubMed] [Google Scholar]
- Sistonen J.; Fuselli S.; Palo J. U.; Chauhan N.; Padh H.; Sajantila A. Pharmacogenetic Variation at CYP2C9, CYP2C19, and CYP2D6 at Global and Microgeographic Scales. Pharmacogenet. Genomics 2009, 19, 170–179. 10.1097/FPC.0b013e32831ebb30. [DOI] [PubMed] [Google Scholar]
- Dalbeth N.; Jones G.; Terkeltaub R.; Khanna D.; Kopicko J.; Bhakta N.; Adler S.; Fung M.; Storgard C.; Baumgartner S.; Perez-Ruiz F. Lesinurad, a Selective Uric Acid Reabsorption Inhibitor, in Combination with Febuxostat in Patients with Tophaceous Gout: Findings of a Phase III Clinical Trial. Arthritis Rheumatol. 2017, 69, 1903–1913. 10.1002/art.40159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abhishek A. New Urate-Lowing Therapies. Curr. Opin. Rheumatol. 2018, 30, 177–182. 10.1097/BOR.0000000000000476. [DOI] [PubMed] [Google Scholar]
- Dean L.Lesinurad Therapy and CYP2C9 Genotype, Medical Genetics Summaries [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK537366/ (accessed 2020-03-13). Created: 2019-02-11.
- McDonald M. G.; Rettie A. E. Sequential Metabolism and Bioactivation of the Hepatotoxin Benzbromarone: Formation of Glutathione Adducts from a Catechol Intermediate. Chem. Res. Toxicol. 2007, 20, 1833–1842. 10.1021/tx7001228. [DOI] [PubMed] [Google Scholar]
- Ohe T.; Umezawa R.; Kitagawara Y.; Yasuda D.; Takahashi K.; Nakamura S.; Abe A.; Sekine S.; Ito K.; Okunushi K.; Morio H.; Furihata T.; Anzai N.; Mashino T. Synthesis of Novel Benzbromarone Derivatives Designed to Avoid Metabolic Activation. Bioorg. Med. Chem. Lett. 2018, 28, 3708–3711. 10.1016/j.bmcl.2018.10.023. [DOI] [PubMed] [Google Scholar]
- Ai Q.; Jing Y.; Jiang R.; Lin L.; Dai J.; Che Q.; Zhou D.; Jia M.; Wan J.; Zhang L. Rotenone, a Mitochondrial Respiratory Complex I Inhibitor, Ameliorates Lipopolysaccharide/D-Galactosamine-Induced Fulminant Hepatitis in Mice. Int. Immunopharmacol. 2014, 21, 200–207. 10.1016/j.intimp.2014.04.028. [DOI] [PubMed] [Google Scholar]
- Meyer J. N.; Leuthner T. C.; Luz A. L. Mitochondrial Fusion, Fission, and Mitochondrial Toxicity. Toxicology 2017, 391, 42–53. 10.1016/j.tox.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaniol M.; Bracher R.; Ha H. R.; Follath F.; Krähenbühl S. Toxicity of Amiodarone and Amiodarone Analogues on Isolated Rat Liver Mitochondria. J. Hepatol. 2001, 35, 628–636. 10.1016/S0168-8278(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Kaufmann P.; Török M.; Hänni A.; Roberts P.; Gasser R.; Krähenbühl S. Mechanisms of Benzarone and Benzbromarone-Induced Hepatic Toxicity. Hepatology 2005, 41, 925–935. 10.1002/hep.20634. [DOI] [PubMed] [Google Scholar]
- Waldhauser K. M.; Török M.; Ha H. R.; Thomet U.; Konrad D.; Brecht K.; Follath F.; Krähenbühl S. Hepatocellular, Toxicity and Pharmacological Effect of Amiodarone and Amiodarone Derivatives. J. Pharmacol. Exp. Ther. 2006, 319, 1413–1423. 10.1124/jpet.106.108993. [DOI] [PubMed] [Google Scholar]
- Barratt M. D. Structure-Activity Relationships, and Prediction of the Phototoxicity and Phototoxic Potential of New Drugs. ATLA, Altern. Lab. Anim. 2004, 32, 511–524. 10.1177/026119290403200506. [DOI] [PubMed] [Google Scholar]
- Brunmair B.; Lest A.; Staniek K.; Gras F.; Scharf N.; Roden M.; Nohl H.; Waldhäusl W.; Fürnsinn C. Fenofibrate Impairs Rat Mitochondrial Function by Inhibition of Respiratory Complex I. J. Pharmacol. Exp. Ther. 2004, 311, 109–114. 10.1124/jpet.104.068312. [DOI] [PubMed] [Google Scholar]
- Grünig D.; Felser A.; Bouitbir J.; Krähenbühl S. The Catechol-O-Methyltransferase Inhibitors Tolcapone and Entacapone Uncouple and Inhibit the Mitochondrial Respiratory Chain in HepaRG Cells. Toxicol. In Vitro 2017, 42, 337–347. 10.1016/j.tiv.2017.05.013. [DOI] [PubMed] [Google Scholar]
- Xu L.; Shi Y.; Zhuang S.; Liu N. Recent Advances on Uric Acid Transporters. Oncotarget 2017, 8, 100852–100862. 10.18632/oncotarget.20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J.; Zhang X.; Fu C.; Yang Q.; Xie Y.; Zhang Z.; Ye Z. Impaired Na+-K+-ATPase Signaling in Renal Proximal Tubule Contributes to Hyperuricemia-induced Renal Tubular Injury. Exp. Mol. Med. 2018, 50, e452 10.1038/emm.2017.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonagh E. M.; Thorn C. F.; Callaghan J. T.; Altman R. B.; Klein T. E. PharmGKB summary: Uric Acid-Lowering Drugs Pathway, Pharmacodynamics. Pharmacogenet. Genomics 2014, 24, 464–476. 10.1097/FPC.0000000000000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P. K.; Ostertag T. M.; Miner J. N. Mechanism of High Affinity Inhibition of the Human Urate Transporter URAT1. Sci. Rep. 2016, 6, 34995. 10.1038/srep34995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobashi S.; Uda J.; Miyata S.; Inoue T.; Ashizawa N.; Matsumoto K.; Taniguchi T.; Iwanaga T.; Nagata O.. Novel Phenol Derivative. US 8367843 B2, February 5, 2013.
- Walter-Sack I.; de Vries J. X.; von Bubnoff A.; Pfleilschifter V.; Raedsch R. Biotransformation and Uric Acid Lowering Effect of Benzbromarone in Patients with Liver Cirrhosis - Evidence for Active Benzbromarone Metabolites?. Eur. J. Med. Res. 1995, 1, 16–20. [PubMed] [Google Scholar]
- Walter-Sack I.; de Vries J. X.; Ittensohn A.; Raedsch R. Biliary Excretion of Benzbromarone and Its Hydroxylated Main Metabolites in Humans. Eur. J. Med. Res. 1998, 3, 45–49. [PubMed] [Google Scholar]
- Oikawa T.; Kunishima C.; Adachi Y.; Sato Y.; Okamura Y.; Tanaka N.; Hosoda T. Metabolism Study of Benzbromarone: In vivo Metabolism and Pharmacokinetics in Healthy Volunteers. J. New Rem. & Clin. 2004, 53, 682–691. [Google Scholar]
- Oikawa T.; Kunishima C.; Matsumoto K.; Ishikawa S.; Endou H. The Inhibitory Effect of Benzbromarone and 6-Hydroxybenzbromarone on Urate Transporter (URAT1). J. New Rem. & Clin. 2005, 54, 645–650. [Google Scholar]
- Endou H.; Oikawa T.. Medicinal Compositions Containing 6-Hydroxybenzbromarone or Salts Thereof. US 7521570 B2, April 21, 2009.
- Jain A. K.; Ryan J. R.; McMahon F. G.; Noveck R. J. Effect of Single Oral Doses of Benzbromarone on Serum and Urinary Uric Acid. Arthritis Rheum. 1974, 17, 149–157. 10.1002/art.1780170207. [DOI] [PubMed] [Google Scholar]
- Taniguchi T.; Ashizawa N.; Matsumoto K.; Saito R.; Motoki K.; Sakai M.; Chikamatsu N.; Hagihara C.; Hashiba M.; Iwanaga T. Pharmacological Evaluation of Dotinurad, a Selective Urate Reabsorption Inhibitor. J. Pharmacol. Exp. Ther. 2019, 371, 162–170. 10.1124/jpet.119.259341. [DOI] [PubMed] [Google Scholar]
- Omura K.; Miyata K.; Kobashi S.; Ito A.; Fushimi M.; Uda J.; Sasaki T.; Iwanaga T.; Ohashi T. Ideal Pharmacokinetic Profile of Dotinurad as a Selective Urate Reabsorption Inhibitor. Drug Metab. Pharmacokinet. 2020, 35, 313–320. 10.1016/j.dmpk.2020.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.