

Abstract
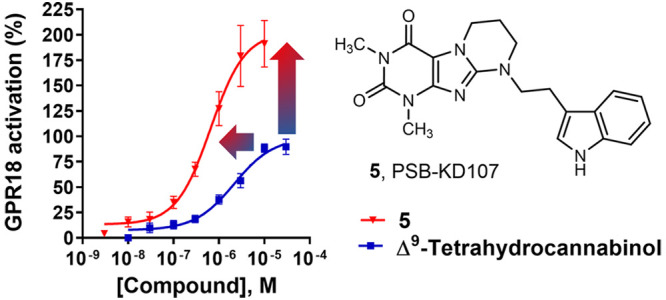
GPR18 is a rhodopsin-like orphan G-protein-coupled receptor (GPCR) that is activated by the natural cannabinoid (CB) Δ9-tetrahydrocannabinol (THC). It is highly expressed in immune cells and represents a promising new drug target. However, THC is much more potent in activating CB receptors than GPR18, and several other proposed lipidic agonists for GPR18 have not been independently confirmed. Herein we describe the first non-lipid-like agonists for GPR18 based on a tricyclic xanthine-derived scaffold, along with initial structure–activity relationships. PSB-KD107 (5) and PSB-KD477 (16) displayed significantly higher potency and efficacy than THC, determined in a GPR18-dependent β-arrestin recruitment assay, and were found to be selective versus the CB-sensitive receptors CB1, CB2, and GPR55. Structure–activity relationships were steep, and indole substitution was crucial for biological activity. These first selective agonists, which are structurally distinct from the lipidic agonist(s), will allow target validation studies and may eventually contribute to the deorphanization of GPR18.
Keywords: Agonist, β-Arrestin, GPR18, Orphan receptor, Tetrahydrocannabinol, Xanthine
First described in 1997, GPR18 is an orphan G-protein-coupled receptor (GPCR) that is located in the δ-branch of class-A, rhodopsin-like GPCRs.1 The receptor is predominantly expressed in cells and tissues of the immune system including the spleen, thymus, lymphocytes, pro-inflammatory macrophages,2−9 and microglia.10,11 Furthermore, expression in the testis5,12 and cancer cells13,14 has been reported. Recent findings showed that GPR18 is also expressed in the eye and upregulated upon corneal damage, where it contributes to wound healing.15,16 Thus GPR18 expression suggests its involvement in immunomodulatory and inflammatory processes, wound healing, and cancer progression, which makes it a promising new drug target.
In the past years, various lipidic and lipid-like physiological compounds have been proposed as agonists for GPR18. In 2006, Kohno et al. suggested N-arachidonoylglycine (Figure 1, NAGly, 1), a metabolite of anandamide, to act as an endogenous ligand of GPR18 based on its effects in Ca2+ mobilization and cAMP accumulation assays in several cell lines expressing GPR18.2 The cAMP-mediated signal by NAGly was abolished by pertussis toxin pretreatment, implying that NAGly activates GPR18 via Gαi/0 proteins. These findings were supported by McHugh et al., who performed experiments using the microglial cell line BV-2 and the endometrial cell line HEC1b.11,17 According to their results, GPR18 appeared to be activated by abnormal cannabidiol (2) and Δ9-tetrahydrocannabinol (THC, 3), both of which promoted cell migration.17,18 These results suggested that GPR18 might be a novel type of cannabinoid (CB) receptor.19,20 Console-Bram et al. supported these results with their findings that NAGly and THC produced increases in intracellular Ca2+ levels and induced mitogen-activated protein kinase (MAPK) activation. However, these authors reported that only THC, and not NAGly, was able to modulate GPR18 activity in β-arrestin recruitment assays.21 Similar results were obtained by Yin et al.22 and Rempel et al.,23,24 who did not detect GPR18 activation in β-arrestin recruitment assays upon stimulation with NAGly. In fact, several groups failed to observe GPR18 activation by NAGly in a number of assays, including those measuring G-protein-dependent signaling.22,25,26 Lu et al. reported the lack of GPR18 modulation by NAGly using electrophysiological experiments.25 Finlay et al. performed a thoroughly controlled study reporting that in several functional assays under various conditions, GPR18-dependent effects on Gαi/0 or Gs signaling by NAGly were observed in neither Chinese hamster ovary (CHO) nor human embryonic kidney (HEK) cells recombinantly expressing GPR18.26 These findings are in agreement with results from our laboratory24,27 (and unpublished results).
Figure 1.

Structures of proposed GPR18 agonists.
Besides NAGly, the polyunsaturated fatty acid metabolite resolvin D2 (RvD2, 4) was proposed to act as an endogenous GPR18 agonist promoting the resolution of bacterial infections via GPR18.3 However, this finding still awaits independent confirmation, and in our hands, 4 was not able to activate GPR18 (see Table 1).
Table 1. Agonistic Potencies of Xanthine Derivatives at GPR18 and GPR55 Determined in β-Arrestin Recruitment Assays.

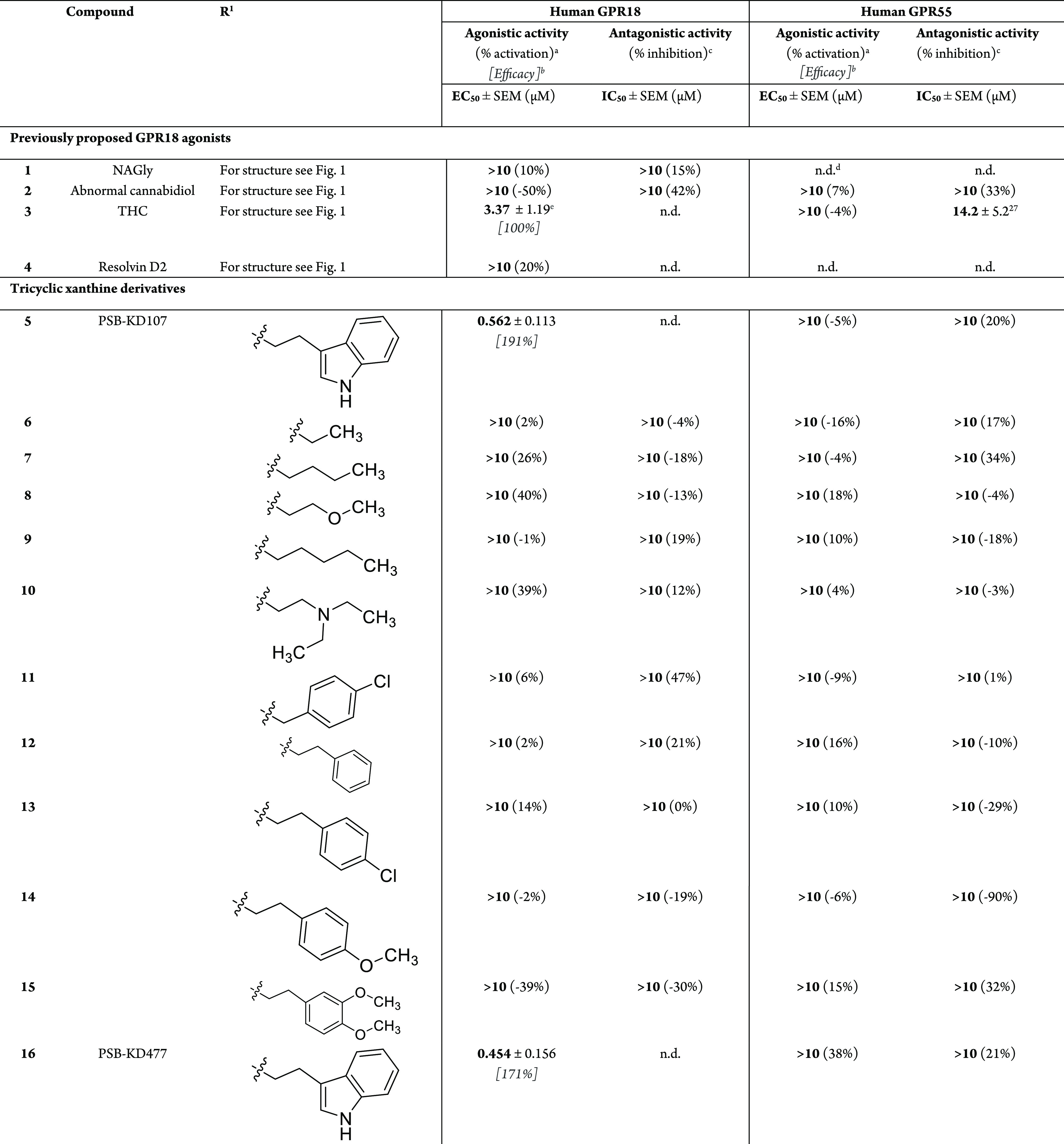

Compounds were tested at a concentration of 10 μM. Effects were normalized to the signal induced by 10 μM THC (GPR18) or 1 μM LPI (GPR55).
Efficacy relative to the maximal effect of the standard agonist (GPR18, 30 μM THC; GPR55, 10 μM LPI) set at 100%.
Percent inhibition of agonist effect (GPR18, 10 μM THC; GPR55, 1 μM LPI) by test compound at 10 μM.
n.d., not determined.
Literature value: EC50 = 4.61 ± 0.50.27
Up to now, the only published GPR18 agonist that has been functional in our hands is THC. It allowed us to develop the first GPR18 antagonists, lipophilic heterocyclic compounds that were characterized to be competitive versus THC.23,24,28 However, THC is lacking GPR18 selectivity due to its interaction with both CB receptor subtypes, CB1 and CB2, and with the lipid-activated orphan receptor GPR55.27
Contradictory reports and inconsistent results on GPR18 and its proposed ligands severely hamper the current GPR18 research. Moreover, the lack of selectivity of the confirmed agonist THC complicates the evaluation of GPR18 in biological systems. Thus, potent and selective agonists are urgently needed as tool compounds to explore the (patho)physiological roles of GPR18 and to allow its validation as a drug target.
Here we describe the discovery and initial structure–activity relationships (SARs) of a novel class of GPR18 agonists that are derived from a non-lipid-like heterotricyclic xanthine scaffold.
Results and Discussion
To identify GPR18 agonists, we screened sublibraries of our proprietary compound collection (https://www.pharmchem1.uni-bonn.de/www-en/pharmchem1-en/mueller-laboratory/compound-library) using a β-arrestin recruitment assay that is based on enzyme complementation technology.29 GPR18 and β-arrestin-2 were tagged with complementary parts of β-galactosidase. Upon β-arrestin recruitment to the tagged receptor, the enzyme is completed and can hydrolyze an added substrate, resulting in a luminescence signal. This assay thus detects GPCR activation regardless of the G-protein signaling pathway and is specific for the investigated GPCR.29 Artifacts, such as false-positive signals, are rare and can be controlled by performing the same assay using cell lines that express different GPCRs. Our GPR18 agonist screening campaign provided a single hit, the tricyclic xanthine derivative 5 (Figure 2).
Figure 2.

Screening of a compound sublibrary for GPR18 agonistic activity using a β-arrestin enzyme complementation assay. The human GPR18 was recombinantly expressed in CHO cells. Compound 5 was identified as a hit. Data were normalized to the effect observed for GPR18 activation by 10 μM Δ9-THC (corresponding to its EC80) set at 100%.
Xanthine 5 contains an annelated tetrahydropyrimidine ring that is substituted by an indolylethyl residue (Table 1). This compound had previously been identified as a moderately potent antagonist for the human adenosine A2A receptor (Ki = 4.56 μM).30 We subsequently determined the concentration-dependent activation of GPR18 by 5 and calculated an EC50 value of 0.562 ± 0.113 μM (Figure 3).
Figure 3.

Concentration–response curves of hit compound 5 (EC50 = 0.562 ± 0.113 μM) and Δ9-THC (EC50 = 3.37 ± 1.19 μM) determined in a β-arrestin enzyme complementation assay using CHO cells stably expressing the human GPR18. Data were normalized to the maximum effect of Δ9-THC (set at 100%). Data points shown are mean values of three independent experiments, each performed in duplicate.
We observed a significantly higher maximal effect for agonist 5 as compared with the standard agonist THC, namely, 191% of THC efficacy (= 100%). This means that THC behaves as a partial agonist in comparison with the new agonist 5. Thus 5 shows superior potency and efficacy.
To confirm that the effect observed for 5 was mediated by GPR18 activation and to study its selectivity, the compound was additionally tested at the related orphan GPCR GPR55 using the same assay system. GPR55 was chosen because it shares with GPR18 the ability to interact with cannabinoids. Compound 5, tested at a high concentration of 10 μM, neither induced β-arrestin recruitment in CHO cells stably expressing GPR55 nor blocked GPR55 activation by its agonist lysophosphatidylinositol (LPI) (Table 1). This is a strong indication that 5 is selective for GPR18 over GPR55. However, because we measured β2-arrestin recruitment only, which is well described for GPR55,31 and not β1-arrestin recruitment, it cannot be fully excluded at present that the compound might interact with GPR55 when differently assayed. The same applies to a potential bias toward G-protein signaling.32−34
When tested in radioligand binding studies at CB receptors, GPR18 agonist 5, tested at a high concentration of 10 μM, showed no significant displacement of the CB receptor-specific radioligand at either CB1 or at CB2 receptors. It also did not show any increase in radioligand binding that could have been indicative of positive allosteric CB receptor modulation. This implies that 5 is also selective versus both CB receptor subtypes, although an allosteric modulation of the CB receptor function cannot be fully excluded.
Encouraged by these findings, we further studied the SARs of this new class of GPR18 agonists.
Chemistry
Tricyclic xanthines 5–9, 11–15, 18, and 19 were synthesized as previously described.30,35−37 The new compounds 10, 16, and 17 were obtained according to the following procedure (Scheme 1): Theophylline (I) was brominated with HBr in the presence of NaOCl3, yielding 8-bromotheophylline (II) according to a previously described procedure.35 Compound II was then alkylated at N7 with 1-bromo-3-chloropropane and N,N-diisopropylethylamine (DIPEA) as a base, resulting in 8-bromo-7-(3-chloropropyl)-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (III). Xanthine III was then cyclized by the reaction with 2-aminoethanol, yielding the tricyclic compound 9-(2-hydoxyethyl)-1,3-dimethylpyrimido[2,1-f]purine-2,4-dione (IV), which was subsequently brominated using PBr3 yielding V. The final product 10 was obtained by the nucleophilic substitution of V with excess diethylamine. For the synthesis of products 16 and 17, 8-bromotheophylline (II) was alkylated either with 1,4-dibromobutane or with 1,2-dibromoethane using benzyltriethylammonium chloride (BTEAC) as a phase-transfer catalyst, resulting in 7-(2-bromoethyl)-8-bromotheophylline (VI) or 7-(4-bromobutyl)-8-bromotheophylline (VII), respectively. Compounds VI and VII were then cyclized with 3-(2-aminoethyl)indole in dimethylformamide (DMF) or 2-methoxyethanol, yielding diazepino- and imidazo-[2,1-f]purine-2,4-diones 16 and 17. The structures of the synthesized compounds were confirmed by MS, UV, IR, and 1H NMR spectra. The purity of all compounds was confirmed to be >95%.
Scheme 1. Synthesis of Tricyclic Xanthine Derivatives 11, 16, and 17.

Reagents and conditions: (a) HBr, NaClO3, CH3COOH, 60°C; (b) 1-bromo-3-chloropropane, benzyltriethylammonium chloride (BTEAC), K2CO3, acetone, reflux; (c) 2-aminoethanol, reflux; (d) PBr3, CHCl3, reflux; (e) diethylamine, propanol, reflux; (f) 1,4-dibromobutane for 16, 1,2-dibromoethane for 17, BTEAC, K2CO3, acetone, reflux; (g) 3-(2-aminoethyl)-indole, 2-methoxyethanol, reflux.
Structure–Activity Relationships
To study initial SARs, we modified the indolylethyl substituent on the xanthine core. Any replacement of the indolyl residue with other aromatic or aliphatic substituents (see 6–15 and 18) abolished the GPR18 activity. (See Table 1.) This indicates the importance of the indole residue attached to the tricyclic xanthine scaffold for GPR18 agonistic activity.
Previously, we had reported on several natural products bearing indole moieties, which were able to inhibit THC-induced GPR18 activation.38,39 In fact, these compounds had been the first antagonists described for GPR18. In light of the present findings, the observed requirement of an indole moiety not only for agonists but also for various antagonists suggests that the reported antagonists and the newly discovered agonists may share the same binding site on GPR18.
Next, we studied the tricyclic xanthine core itself. Extension of the annelated six-membered tetrahydropyrimidine ring present in agonist 5 to a seven-membered ring appeared to slightly increase the activity (16, PSB-KD477, EC50 0.454 ± 0.156 μM). However, decreasing the ring size to a five-membered imidazolidine ring significantly reduced the activity by about 10-fold (17, EC50 = 5.68 ± 1.54 μM).
Agonist 16 was also somewhat more potent at GPR55 (38% activation at 10 μM) and at the CB2 receptor (46% inhibition of radioligand binding at 10 μM) than lead structure 5. (See Table 1 and Table S1.) Therefore, we continued modifying the more selective derivative, the six-ring-annelated xanthine derivative 5.
In a subsequent step, we investigated the substituents in the N1- and N3-positions of the xanthine core. Replacement of the methyl groups in agonist 5 (which are also present in the naturally occurring xanthine alkaloids caffeine and theophylline) by the longer propyl residues (compound 19) drastically reduced the GPR18 activity. These steep SARs indicate limited space in the binding pocket of GPR18.
None of the compounds evaluated in this study, including those that showed no GPR18-agonistic activity, inhibited THC-induced β-arrestin recruitment in GPR18-expressing cells (Table 1). This shows that the investigated tricyclic compounds that failed to activate GPR18 also did not block the receptor.
Finally, we characterized the new agonist 5 by trying to block its effect by the THC-competitive GPR18 antagonist PSB-CB-27 (20; see Figure 4).24 In our previous study, we had shown that 20 was able to completely block THC-induced GPR18 activation in a β-arrestin recruitment assay, displaying an IC50 value of 0.650 μM. Agonist 5 was employed at a concentration of 1 μM, which corresponds to its EC80 value, and inhibition of its effect by GPR18 antagonist 20 was studied (Figure 4). Surprisingly, antagonist 20 was not able to fully inhibit GPR18 activation induced by the tricyclic xanthine agonist 5 (IC50 value of 0.944 μM, 65% maximal inhibition; see Figure 4).
Figure 4.

Concentration-dependent inhibition of GPR18 activation by an EC80 concentration of the GPR18 agonist THC (blue curve) and the GPR18 agonist 5 (red curve). IC50 values were 0.650 μM (vs THC, complete inhibition) and 0.944 μM (vs 5, 64% maximal inhibition), determined in a β-arrestin enzyme complementation assay using CHO cells stably expressing the human GPR18 receptor. Data points are means of three independent experiments, each performed in duplicate.
These results may be indicative of different binding sites or receptor conformations to which the structurally very different GPR18 agonists, the lipid-like THC, on the one hand, and the heterotricyclic xanthine derivative 5, on the other hand, are binding. Whereas the lipophilic antagonist 20 appears to bind to the same binding site as the lipid-like agonist THC, this is probably not the case for the xanthine-based agonist.
To confirm this hypothesis, we studied the concentration-dependent activation of GPR18 by xanthine agonist 5 in the absence and in the presence of different concentrations of the lipidic antagonist 20. In fact, increasing concentrations of 20 led to a lowering of the maximal effect induced by agonist 5. No significant rightward shift of the concentration–response curve could be observed (Figure 5), and the EC50 values were not significantly different from each other. (See Table S2.) This is consonant with an allosteric mechanism of inhibition for antagonist 20 versus the xanthine-type agonist 5. In contrast, antagonist 20 had previously been shown to block GPR18 activation by the lipidic GPR18 agonist THC in a competitive manner.24
Figure 5.

Activation of GPR18 by 5 in the absence and presence of different concentrations of 20. EC50 values and maximum effects are collected in Table S2. Determined by the β-arrestin enzyme complementation assay using CHO stably expressing the human GPR18 receptor. Data points shown are means of three independent experiments performed in duplicate.
As a next step, we studied the effects of combining the lipid-like “partial” GPR18 agonist THC with the xanthine-type “full” agonist 5 in the β-arrestin recruitment assay. In Figure 6A, the effects of fixed concentrations of xanthine agonist 5 on the concentration–response curve of THC are shown. In Figure 6B, results of the reverse experiment are shown, in which the effects of fixed concentrations of THC on concentration-dependent GPR18 activation by xanthine agonist 5 were studied. Xanthine agonist 5, used at its EC50 value of 1 μM, was able to increase the basal response of low concentrations of THC. However, at high concentrations of the xanthine agonist 5, at which it showed maximal GPR18 activation, its effect was inhibited by THC in a concentration-dependent manner (Figure 6A). This behavior is typical for partial agonists. Along the same lines, concentration-dependent activation of GPR18 by the xanthine agonist 5 was partly inhibited by 3 μM of THC and inhibited by >70% at a high concentration of 10 μM THC (Figure 6B). Unfortunately, we could not add even higher concentrations of THC due to its limited water solubility. These observations clearly indicate that THC is a partial agonist at GPR18 in comparison with the novel xanthine-derived agonist 5, and THC can even act as a GPR18 antagonist in the presence of the more efficacious agonist. These results highlight the superior properties of the newly discovered GPR18 agonist scaffold. It is interesting to note in this context that THC also behaves as a partial agonist at both CB receptor subtypes.40−44
Figure 6.

(A) Effects of fixed concentrations of xanthine agonist 5 on the concentration–response curve of THC. (B) Effects of fixed concentrations of THC on the concentration–response curve of xanthine-derived agonist 5. Activation of the human GPR18 expressed in CHO cells was determined in β-arrestin recruitment assays. Data points shown are means of three independent experiments performed in duplicate.
In conclusion, we discovered a novel class of surrogate agonists for the orphan GPCR GPR18 and investigated preliminary SARs, which were found to be steep. The new scaffold is characterized by a tricyclic dimethylxanthine core substituted by an indole ring attached via an ethylene bridge. Agonist 5, which showed a submicromolar EC50 value, was more potent and much more efficacious that the standard GPR18 agonist THC. These new GPR18 agonists represent suitable lead structures for further optimization to obtain potent and selective tool compounds. They will likely contribute to a breakthrough in the field of GPR18 research, allowing future target validation studies.
Acknowledgments
We thank Marion Schneider for the expert measurement of LCMS and NMR spectra.
Glossary
Abbreviations
- Abn-CBD
abnormal cannabidiol
- BTEAC
benzyltriethylammonium chloride
- BV-2
mouse microglial cell line BV-2
- CB
cannabinoid receptor
- CB1
cannabinoid receptor type 1
- CB2
cannabinoid receptor type 2
- CHO
Chinese hamster ovary
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- GPCR
G-protein-coupled receptor
- GPR18
G-protein-coupled receptor 18
- GPR55
G-protein-coupled receptor 55
- HEC-1B
human endometrial cell line 1-B
- HEK
human embryonic kidney
- LPI
lysophosphatidylinositol
- MAPK
mitogen-activated protein kinase
- NAGly
N-arachidonoylglycine
- RvD2
resolvin D2
- SAR
structure–activity relationship
- THC
Δ9-tetrahydrocannabinol
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00208.
Synthetic procedures, analytical data, assay procedures, and additional selectivity data (PDF)
Author Contributions
∥ C.T.S. and A.B.M. contributed equally. The manuscript was mainly written by C.E.M. and A.B.M. with contributions from all authors. All authors (C.T.S., A.B.M., A.D., K.K.-K., and C.E.M.) have given approval to the final version of the manuscript.
C.T.S., A.B.M., and C.E.M. were supported by the Deutsche Forschungsgemeinschaft (DFG) within the Research Training Group GRK 1873 “Pharmacology of 7TM-receptors and downstream signaling pathways” and the BMBF-funded Bonn International Graduate School Drug Sciences (BIGS DrugS). K.K.-K. was supported by the Jagiellonian University statutory funds (N42/DBS/000039). K.K.-K. and C.E.M. were supported by the EU COST Action ERNEST CA18133. C.T.S. received a BAYER Ph.D. fellowship through BIGS DrugS. A.B.M. was funded by the Ministry of Finance of Indonesia in the scheme of the Indonesia Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan (LPDP).
The authors declare no competing financial interest.
Supplementary Material
References
- Fredriksson R.; Lagerström M. C.; Lundin L.-G.; Schiöth H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Kohno M.; Hasegawa H.; Inoue A.; Muraoka M.; Miyazaki T.; Oka K.; Yasukawa M. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem. Biophys. Res. Commun. 2006, 347, 827–832. 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Chiang N.; Dalli J.; Colas R. A.; Serhan C. N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203. 10.1084/jem.20150225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Sumida H.; Cyster J. G. GPR18 is required for a normal CD8αα intestinal intraepithelial lymphocyte compartment. J. Exp. Med. 2014, 211, 2351–2359. 10.1084/jem.20140646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz I.; Muraoka A.; Yang Y. K.; Samuelson L. C.; Zimmerman E. M.; Cook H.; Yamada T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–6. 10.1006/geno.1997.4752. [DOI] [PubMed] [Google Scholar]
- Regard J. B.; Sato I. T.; Coughlin S. R. Anatomical profiling of G protein-coupled receptor expression. Cell 2008, 135, 561–571. 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A. M.; Callahan D. J.; Richner J. M.; Choi J.; DiPersio J. F.; Diamond M. S.; Bhattacharya D. GPR18 controls reconstitution of mouse small intestine intraepithelial lymphocytes following bone marrow transplantation. PLoS One 2015, 10, e0133854 10.1371/journal.pone.0133854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi R.; Inoue K.; Kambe Y.; Miyata A. N-arachidonoyl glycine induces macrophage apoptosis via GPR18. Biochem. Biophys. Res. Commun. 2012, 418, 366–71. 10.1016/j.bbrc.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Jablonski K. A.; Amici S. A.; Webb L. M.; Ruiz-Rosado J. d. D.; Popovich P. G.; Partida-Sanchez S.; Guerau-De-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS One 2015, 10, e0145342 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D. GPR18 in microglia: implications for the CNS and endocannabinoid system signalling. Br. J. Pharmacol. 2012, 167, 1575–82. 10.1111/j.1476-5381.2012.02019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D.; Wager-Miller J.; Page J.; Bradshaw H. B. siRNA knockdown of GPR18 receptors in BV-2 microglia attenuates N-arachidonoyl glycine-induced cell migration. J. Mol. Signaling 2014, 7, 10. 10.1186/1750-2187-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegel C.; Vogel F.; Hofreuter A.; Wojcik S.; Schoeder C.; Kiec-Kononowicz K.; Brockmeyer N. H.; Müller C. E.; Becker C.; Altmüller J.; Hatt H.; Gisselmann G. Characterization of non-olfactory GPCRs in human sperm with a focus on GPR18. Sci. Rep. 2016, 6, 32255. 10.1038/srep32255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y.; Verdegaal E. M. E.; Siderius M.; Bebelman J. P.; Smit M. J.; Leurs R.; Willemze R.; Tensen C. P.; Osanto S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigm. Cell Melanoma Res. 2011, 24, 207–218. 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- Qiao G.-J.; Chen L.; Wu J.-C.; Li Z.-R. Identification of an eight-gene signature for survival prediction for patients with hepatocellular carcinoma based on integrated bioinformatics analysis. PeerJ 2019, 7, e6548 10.7717/peerj.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murataeva N.; Daily L.; Taylor X.; Dhopeshwarkar A.; Hu S. S.-J.; Miller S.; McHugh D.; Oehler O.; Li S.; Bonanno J. A.; Mackie K.; Straiker A. Evidence for a GPR18 role in chemotaxis, proliferation, and the course of wound closure in the cornea. Cornea 2019, 38, 905–913. 10.1097/ICO.0000000000001934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell M. D.; Hu S. S.-J.; Viswanathan S.; Bradshaw H.; Kelly M. E. M.; Straiker A. A GPR18-based signalling system regulates IOP in murine eye. Br. J. Pharmacol. 2013, 169, 834–843. 10.1111/bph.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D.; Hu S. S.; Rimmerman N.; Juknat A.; Vogel Z.; Walker J. M.; Bradshaw H. B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D.; Page J.; Dunn E.; Bradshaw H. B. Δ9-Tetrahydrocannabinol and N-arachidonyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br. J. Pharmacol. 2012, 165, 2414–24. 10.1111/j.1476-5381.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander S. P. So what do we call GPR18 now?. Br. J. Pharmacol. 2012, 165, 2411–3. 10.1111/j.1476-5381.2011.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander S. P.; Christopoulos A.; Davenport A. P.; Kelly E.; Marrion N. V.; Peters J. A.; Faccenda E.; Harding S. D.; Pawson A. J.; Sharman J. L.; Southan C.; Davies J. A.; The consice guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174, S17–S129. 10.1111/bph.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Console-Bram L.; Brailoiu E.; Brailoiu G. C.; Sharir H.; Abood M. E. Activation of GPR18 by cannabinoid compounds: a tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. 10.1111/bph.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Chu A.; Li W.; Wang B.; Shelton F.; Otero F.; Nguyen D. G.; Caldwell J. S.; Chen Y. A. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–38. 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel V.; Atzler K.; Behrenswerth A.; Karcz T.; Schoeder C.; Hinz S.; Kaleta M.; Thimm D.; Kiec-Kononowicz K.; Müller C. E. Bicyclic imidazole-4-one derivatives: a new class of antagonists for the orphan G protein-coupled receptors GPR18 and GPR55. MedChemComm 2014, 5, 632–649. 10.1039/C3MD00394A. [DOI] [Google Scholar]
- Schoeder C. T.; Kaleta M.; Mahardhika A. B.; Olejarz-Maciej A.; Łażewska D.; Kieć-Kononowicz K.; Müller C. E. Structure-activity relationships of imidazothiazinones and analogs as antagonists of the cannabinoid-activated orphan G protein-coupled receptor GPR18. Eur. J. Med. Chem. 2018, 155, 381–397. 10.1016/j.ejmech.2018.05.050. [DOI] [PubMed] [Google Scholar]
- Lu V. B.; Puhl H. L.; Ikeda S. R. N-Arachidonyl glycine does not activate G-protein-coupled receptor 18 signaling via canonical pathways. Mol. Pharmacol. 2013, 83, 267. 10.1124/mol.112.081182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay D. B.; Joseph W. R.; Grimsey N. L.; Glass M. GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N-arachidonoyl glycine. PeerJ 2016, 4, e1835 10.7717/peerj.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel V.; Volz N.; Gläser F.; Nieger M.; Bräse S.; Müller C. E. Antagonists for the orphan G-protein-coupled receptor GPR55 based on a coumarin scaffold. J. Med. Chem. 2013, 56, 4798–810. 10.1021/jm4005175. [DOI] [PubMed] [Google Scholar]
- Neumann A.; Engel V.; Mahardhika A. B.; Schoeder C. T.; Namasivayam V.; Kiec-Kononowicz K.; Müller C. E. Computational Investigations on the Binding Mode of Ligands for the Cannabinoid-Activated G Protein-Coupled Receptor GPR18. Biomolecules 2020, 10, 686. 10.3390/biom10050686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassoni D. L.; Raab W. J.; Achacoso P. L.; Loh C. Y.; Wehrman T. S.. Measurements of β-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. In Receptor Binding Techniques; Davenport A. P., Ed.; Humana Press: Totowa, NJ, 2012; pp 181–203. [DOI] [PubMed] [Google Scholar]
- Drabczyńska A.; Müller C. E.; Schiedel A.; Schumacher B.; Karolak-Wojciechowska J.; Fruziński A.; Zobnina W.; Yuzlenko O.; Kieć-Kononowicz K. Phenylethyl-substituted pyrimido[2,1-f]purinediones and related compounds: Structure-activity relationships as adenosine A1 and A2A receptor ligands. Bioorg. Med. Chem. 2007, 15, 6956–6974. 10.1016/j.bmc.2007.07.051. [DOI] [PubMed] [Google Scholar]
- Heynen-Genel S.; Dahl R.; Shi S.; Milan L.; Hariharan S.; Bravo Y.; Sergienko E.; Hedrick M.; Dad S.; Stonich D.; Su Y.; Vicchiarelli M.; Mangravita-Novo A.; Smith L. H.; Chung T. D.; Sharir H.; Barak L. S.; Abood M. E.. Screening for selective ligands for GPR55-agonists. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information (US): 2011. [PubMed]
- Pertwee R. G.; Howlett A. C.; Abood M. E.; Alexander S. P.; Di Marzo V.; Elphick M. R.; Greasley P. J.; Hansen H. S.; Kunos G.; Mackie K.; Mechoulam R.; Ross R. A. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A.; Zhao P.; Sharir H.; Bai Y.; Caron M. G.; Barak L. S.; Abood M. E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284, 29817–29827. 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhouri L.; Cook C. D.; Al-Huniti M. H.; Console-Bram L. M.; Hurst D. P.; Spano M. B. S.; Nasrallah D. J.; Caron M. G.; Barak L. S.; Reggio P. H.; Abood M. E.; Croatt M. P. Design, synthesis and biological evaluation of GPR55 agonists. Bioorg. Med. Chem. 2017, 25, 4355–4367. 10.1016/j.bmc.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabczyńska A.; Müller C. E.; Lacher S. K.; Schumacher B.; Karolak-Wojciechowska J.; Nasal A.; Kawczak P.; Yuzlenko O.; Pȩkala E.; Kieć-Kononowicz K. Synthesis and biological activity of tricyclic aryloimidazo-, pyrimido-, and diazepinopurinediones. Bioorg. Med. Chem. 2006, 14, 7258–7281. 10.1016/j.bmc.2006.06.052. [DOI] [PubMed] [Google Scholar]
- Kieć-Kononowicz K.; Drabczyńska A.; Pȩkala E.; Michalak B.; Müller C. E.; Schumacher B.; Karolak-Wojciechowska J.; Duddeck H.; Rockitt S.; Wartchow R. New developments in A1 and A2 adenosine receptor antagonists. Pure Appl. Chem. 2001, 73, 1411. 10.1351/pac200173091411. [DOI] [Google Scholar]
- Drabczyńska A.; Karcz T.; Szymańska E.; Köse M.; Müller C. E.; Paskaleva M.; Karolak-Wojciechowska J.; Handzlik J.; Yuzlenko O.; Kieć-Kononowicz K. Synthesis, biological activity and molecular modelling studies of tricyclic alkylimidazo-, pyrimido- and diazepinopurinediones. Purinergic Signalling 2013, 9, 395–414. 10.1007/s11302-013-9358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazir M.; Harms H.; Loef I.; Kehraus S.; El Maddah F.; Arslan I.; Rempel V.; Müller C. E.; König G. M. GPR18 inhibiting Amauromine and the novel triterpene glycoside Auxarthonoside from the sponge-derived fungus Auxarthron reticulatum. Planta Med. 2015, 81, 1141–5. 10.1055/s-0035-1545979. [DOI] [PubMed] [Google Scholar]
- Harms H.; Rempel V.; Kehraus S.; Kaiser M.; Hufendiek P.; Müller C. E.; König G. M. Indoloditerpenes from a marine-derived fungal strain of Dichotomomyces cejpii with antagonistic activity at GPR18 and cannabinoid receptors. J. Nat. Prod. 2014, 77, 673–7. 10.1021/np400850g. [DOI] [PubMed] [Google Scholar]
- Hess C.; Schoeder C. T.; Pillaiyar T.; Madea B.; Müller C. E. Pharmacological evaluation of synthetic cannabinoids identified as constituents of spice. Forensic Toxicol. 2016, 34, 329–343. 10.1007/s11419-016-0320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel V.; Volz N.; Hinz S.; Karcz T.; Meliciani I.; Nieger M.; Wenzel W.; Bräse S.; Müller C. E. 7-Alkyl-3-benzylcoumarins: A versatile scaffold for the development of potent and selective cannabinoid receptor agonists and antagonists. J. Med. Chem. 2012, 55, 7967–7977. 10.1021/jm3008213. [DOI] [PubMed] [Google Scholar]
- Banister S. D.; Arnold J. C.; Connor M.; Glass M.; McGregor I. S. Dark Classics in Chemical Neuroscience: Δ9-Tetrahydrocannabinol. ACS Chem. Neurosci. 2019, 10, 2160–2175. 10.1021/acschemneuro.8b00651. [DOI] [PubMed] [Google Scholar]
- Banister S. D.; Connor M.. The Chemistry and Pharmacology of Synthetic Cannabinoid Receptor Agonists as New Psychoactive Substances: Origins. In New Psychoactive Substances: Pharmacology, Clinical, Forensic and Analytical Toxicology; Maurer H. H., Brandt S. D., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp 165–190. [DOI] [PubMed] [Google Scholar]
- Müller C. E. Fortschritte in der Cannabis-Forschung aus pharmazeutisch-chemischer Sicht (Progress in cannabis research from a pharmaceutical chemist’s point of view). Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz 2019, 62, 818–824. 10.1007/s00103-019-02964-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.