

Abstract

The anaerobic conversion of choline to trimethylamine (TMA) by the human gut microbiota has been linked to multiple human diseases. The potential impact of this microbial metabolic activity on host health has inspired multiple efforts to identify small molecule inhibitors. Here, we use information about the structure and mechanism of the bacterial enzyme choline TMA-lyase (CutC) to develop a cyclic choline analog that inhibits the conversion of choline to TMA in bacterial whole cells and in a complex gut microbial community. In vitro biochemical assays and a crystal structure suggest that this analog is a competitive, mechanism-based inhibitor. This work demonstrates the utility of structure-based design to access inhibitors of radical enzymes from the human gut microbiota.
Keywords: Gut microbiota, choline, trimethylamine, small molecule inhibitor, cardiometabolic disease
The human gastrointestinal (GI) tract is colonized by trillions of microorganisms that have coevolved with the host. These organisms play an important role in maintaining host health by performing critical biological functions, including metabolizing inaccessible dietary components, synthesizing essential vitamins and nutrients, providing protection from invading pathogens, and priming the immune system.1,2 Gut microbial metabolic activities have also been linked to human disease. Indeed, metabolomics experiments have showed strong correlations between levels of human serum metabolites made or modified by gut microbes and various disease states.1,3
Anaerobic gut microbial choline metabolism is a disease-associated activity that has received much attention as a potential target for therapeutic intervention. While choline is an essential nutrient for the host, contributing to cell membrane function, methyl transfer events, and neurotransmission,4 gut anaerobes use choline as a source of carbon and energy, cleaving its C–N bond to generate acetaldehyde, which is further processed by alcohol dehydrogenase CutO and aldehyde oxidoreductase CutF to give ethanol and acetyl coenzyme A,5,6 and trimethylamine (TMA), a uniquely microbial metabolite (Figure 1A).7,8 While gut microbes generate TMA from multiple sources, including the nutrients carnitine and glycine betaine, choline is considered to be the major precursor.7 In the human body, TMA is further oxidized to trimethylamine-N-oxide (TMAO) by the liver enzyme flavin-dependent monooxygenase 3 (FMO3).9 Elevated levels of TMAO in human plasma have been linked to multiple diseases such as nonalcoholic fatty liver disease (NAFLD),10,11 cardiovascular disease,12−16 chronic kidney disease,17,18 type 2 diabetes,19 and artherosclerosis.20,21 If mutations in the gene encoding FMO3 result in a deficiency of this enzyme, TMA builds up in the body and its excessive excretion leads to a distinct odor. This inherited metabolic disorder is known as trimethylaminuria or “fish odor syndrome”.22,23 The correlation between TMA production by the gut microbiota and diseases, as well as its prevalence in the human gut microbiota, makes this metabolic activity a prominent target for manipulation and further study.
Figure 1.

Anaerobic choline metabolism is a disease-linked gut microbial activity and a promising target for inhibition. (A) Generation of TMA from choline by gut microbes and its subsequent processing in the human body. (B) Crystal structure of CutC (PDB = 5FAU) and (C) proposed mechanism for choline cleavage.
We have discovered and biochemically characterized the enzymes responsible for the key C–N bond cleavage reaction that converts choline to TMA: the glycyl radical enzyme (GRE) choline TMA-lyase (CutC) and its corresponding radical S-adenosylmethionine (SAM) activating protein (CutD).6,24−26 GREs are a family of oxygen-sensitive enzymes that use protein-based radical intermediates to catalyze chemically challenging reactions. Well characterized family members include pyruvate formate-lyase, class III (anaerobic) ribonucleotide reductase, benzylsuccinate synthase, and 4-hydroxyphenylacetate decarboxylase.27 The glycyl radical is installed by a dedicated partner enzyme that is a member of the radical SAM enzyme superfamily. A crystal structure of unactivated CutC with choline bound in the active site (Figure 1B), together with site-directed mutagenesis, supports a mechanism in which the glycyl radical of CutC abstracts a hydrogen atom from an adjacent cysteine (Cys489), generating a thiyl radical intermediate that initiates the reaction with choline by abstracting the pro-S hydrogen atom from C1. Deprotonation of the C1-OH by Glu491, followed by C–N bond cleavage via a “spin-center shift”, would then eliminate TMA. Hydrogen atom abstraction from Cys489 by the resulting acetaldehyde-centered radical would regenerate the thiyl radical and produce acetaldehyde. Proton transfer would then prepare the CutC active site for the next round of catalysis (Figure 1C).
Targeting microbial choline metabolism to reduce TMA generation (leading to lowered TMAO levels) has been proposed as a potential therapeutic approach for the treatment of cardiometabolic diseases. As CutC is the enzyme responsible for TMA generation, it may be an ideal therapeutic target. Hazen and co-workers reported the first CutC inhibitor, 3,3-dimethyl-1-butanol (DMB), replacing the nitrogen atom of choline by a carbon (Figure 2).21 They demonstrated that orally administered DMB reduced plasma TMAO levels in mice. They also observed that DMB treatment reduced foam cell formation induced by a high choline diet in an atherosclerosis mouse model. However, in our hands DMB poorly inhibits CutC activity in vitro and in bacterial cell culture, suggesting these effects may arise from interactions with a different target. Subsequently, Hazen et al. reported greatly improved rationally designed CutC inhibitors, including iodomethylcholine (IMC)28 and various N,N-dimethylprop-2-yn derivatives29−32 (Figure 2). Bhandari and co-workers recently described a pyridine derivative, trigonelline, that reduces TMA production from choline by inhibiting growth of choline metabolizing bacterial strains, like Citrobacter freundii, and also directly inhibits FMO3 (Figure 2).33 More recently, Boehringer-Ingelheim used a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF)-based high-throughput screen with whole cells of Clostridium hathewayi to identify new inhibitors of choline metabolism.34 This study revealed the ability of an inhibitor of choline transport to prevent TMA production (Figure 2).
Figure 2.

Previously reported small molecule inhibitors of anaerobic bacterial choline metabolism.
Concurrently, we reported an additional CutC inhibitor, betaine aldehyde (BA) (in vitro IC50 = 26 μM), which was identified by screening a focused library of choline analogs for activity toward the cut gene cluster-containing human gut isolate Escherichia coli MS 200-1.35 A cocrystal structure of BA bound to unactivated CutC showed the formation of a thiohemiacetal with Cys489 of CutC, suggesting this compound could to be a covalent, reversible inhibitor. We also found that BA inhibited TMA formation from choline in whole cell assays with multiple choline-degrading human gut bacteria (EC50 = 0.4–2.5 mM range).
Building on this proof of concept work, we sought to use structural information to design more potent CutC inhibitors. Examining the conformation of choline within the CutC active site, we noticed that the trimethylammonium group is oriented gauche to the hydroxyl, with an average dihedral angle of 61°. The aromatic ring of Phe395 may stabilize this conformation of choline by forming a face-on cation−π-like interaction with C2 (C2 to ring center distance: 3.8 Å), which would be expected to have a partial positive charge due to the adjacent quaternary ammonium.24 Overlaying choline- and BA-bound CutC crystal structures showed that BA binds within the active site in a similar conformation to choline, maintaining contacts with key active site residues.
Together, these structures suggested the possibility of building this conformation into an inhibitor scaffold by forming a ring between C1 and an N-methyl group, leading either to N-methyl-3-pyrrolidinol 1 or N-methyl-3-piperidinol 2 (Figure 3). These modifications could potentially improve inhibitor potency by constraining its conformation and thereby reducing the entropic penalty of binding.36,37 These compounds (synthesis detailed in the SI) were tested for inhibition of both anaerobic choline metabolism by E. coli MS 200-1 and CutC activity in vitro (Tables 1 and 2).
Figure 3.

Strategy for rigidifying choline analogs leading to N-methyl-3-pyrrolidinol 1 or N-methyl-3-piperidinol 2.
Table 1. Inhibitory Activity of Compounds 1–8.

| Compound | IC50 against E. coli MS 200–1 (μM) | In vitro IC50 against CutC (μM) |
|---|---|---|
| 1 | 6,300 ± 1,800 | 840 ± 100 |
| 2 | 950 ± 340 | 2,000 ± 300 |
| 3 | 1,600 ± 300 | 7,000 ± 2,000 |
| 4 | 760 ± 150 | 2,600 ± 1,100 |
| 5 | 27,000 ± 4,000 | >10,000 |
| 6 | 10 ± 4 | 2.9 ± 0.7 |
| 7 | 920 ± 80 | 420 ± 100 |
| 8 | 1,800 ± 600 | N/Aa |
N/A = not available.
Table 2. Inhibitory Activity of Compounds 9–14.
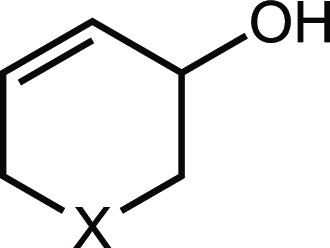
| Compound | X | IC50 against E. coli MS 200-1 (μM) | In vitro IC50 against CutC (μM) |
|---|---|---|---|
| 6 | N-Me | 10 ± 0.4 | 2.9 ± 0.7 |
| 9 | CH2 | 260 ± 110 | 710 ± 270 |
| 10 | O | 80 ± 22 | 2,900 ± 1,300 |
| 11 | N-nPr | 200 ± 60 | 140 ± 20 |
| 12 | N-cyclobutyl | 190 ± 60 | 200 ± 90 |
| 13 | N-Bn | 370 ± 70 | 100 ± 10 |
| 14 | N-Me2 | 7.6 ± 2.4 | 7.0 ± 1.0 |
| (S)-6 | N-Me | 9.8 ± 1.9 | 2.4 ± 0.3 |
| (R)-6 | N-Me | 31 ± 5 | 22 ± 3 |
Both 1 and 2 inhibited TMA formation from choline in vitro and in bacterial cell culture. We decided to prioritize compound 2 for further optimization as it was more active in whole cells. We found that moving the hydroxyl group to the 4-position (compound 3) led to a 3- and 2-fold loss in vitro and in whole cell activities, respectively. Expanding the size of the heterocyclic ring (compound 4) did not improve activity relative to 2. Oxidation of the alcohol to a ketone (5) led to a compound that was 20-fold less active toward whole cells, indicating that installing an electrophile would not improve activity. This loss of potency could also arise from the absence of a hydrogen atom on C3 if its abstraction is critical for activity.
We next introduced an alkene into the piperidine ring (compound 6), hypothesizing that this modification could affect potency by activating the C3 hydrogen atom for abstraction and/or altering the piperidine ring conformation. This modification improved inhibitory activity 100-fold both in whole cells and in vitro. The addition of a methyl group to this derivative at position 5 (compound 7) decreased activity. An acetylated analog (compound 8) was 100-fold less active than 6 in whole cells.
We then turned our attention toward the N-methyl group (Table 2), replacing it with both a carbon atom (compound 9) and an oxygen atom (compound 10). The methylene-containing derivative 9 was 25- and 230-fold less active than 6in vitro and in whole cells, respectively. While 10 possessed activity similar to that of 6 in the whole cell assay, it was 1,000-fold less active toward CutC in vitro, suggesting that it may interact with an additional, unknown target. These results highlight the importance of the amine for activity toward CutC and suggest this part of the molecule may be mimicking the trimethylammonium group of choline.
Next, we varied the substituent on the piperidine nitrogen atom. Introduction of bulkier substituents like n-propyl (compound 11), cyclobutyl (compound 12), and benzyl (compound 13) all reduced activity. We also tested the dimethylated quaternary ammonium analog 14, and we found this compound possessed similar activity to 6 in whole cells but was slightly less potent against CutC in vitro. Finally, we synthesized and tested both enantiomers of compound 6. While (S)-6 possessed similar activity to the racemic mixture, (R)-6 was at least 3-fold less active both in whole cells and in the CutC assay. Overall, this optimization effort revealed (S)-6 to be the most promising inhibitor, with a high ligand efficiency (0.98) and fit quality score (1.38).38
To gain further structural insights into the interactions between (S)-6 and CutC, we obtained a cocrystal structure of an unactivated enzyme–inhibitor complex. Following incubation of wild-type CutC with 10 mM (S)-6, CutC was crystallized using similar conditions to those reported previously.24 The resulting structure determined to 2.3-Å resolution revealed nonproteinogenic electron density within the CutC active site. Based on the observed electron density we modeled compound (S)-6 (Figure 4A), which fits well in the composite omit map density (Figure 4B). Overall, inhibitor (S)-6 bound in a similar fashion as choline, exploiting some of the same key binding interactions observed within the CutC-choline complex (Figure 4A,C). Of interest, the alcohol moiety of (S)-6 is within hydrogen bond distance (2.7 Å) of Glu491, placing the C3 hydrogen within 3.8 Å of Cys489. Similar to the CH–O bond interactions observed in the CutC–choline bound structure that are involved in securing the trimethylammonium moiety of choline in place, comparable CH–O interactions are observed between the N-methyl group, C2, and C6, which are adjacent to the tertiary amine of (S)-6, and residues Asp216, Tyr506, and Tyr208, respectively. Disruption of these interactions could provide a rationale for the decrease in activity observed for compounds 9–14 where the N-methyl functionality has been substituted. Perhaps unexpectedly, the two carbons that we anticipated would mimic the choline backbone (C2 and C3) are not located in the same region of the active site; instead, (S)-6 binds in a conformation amenable for π–π interaction between C4–C5 of (S)-6 and F395.39 This interaction may contribute to the boost in potency observed upon introducing this functional group. The similar binding orientation of (S)-6 compared to choline is also reflected in the observation that active site residues do not rearrange upon binding the different molecules (Figure 4D). Overall, the structures are very similar, with low RMSD values (0.28 Å over 792 residues out of 846).
Figure 4.
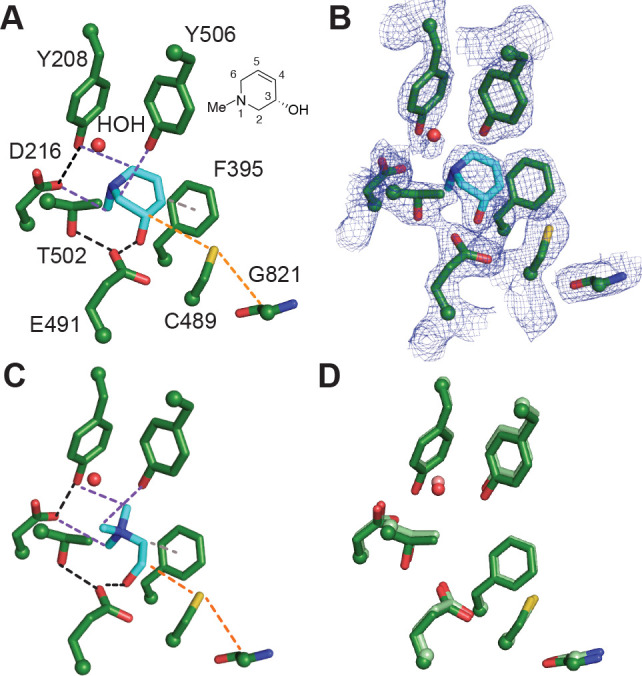
Binding of (S)-6 to CutC reveals key interactions important for recognition. (A) Crystal structure of unactivated CutC soaked with (S)-6 reveals interactions between bound inhibitor and CutC active site residues. Black dashes represent H-bond interactions, purple dashes represent CHO interactions, gray dashes represent a potential π–π interaction, and orange dashes represent the path of radical transfer. Chemical structure of (S)-6 with numbered atoms is shown next to the structure. (B) Simulated annealing composite omit map (2Fo – Fc, blue mesh) contoured at 1.5σ showing the location of the inhibitor. (C) Previously described crystal structure of unactivated CutC bound to choline (PDB 5FAU). Interactions between CutC and choline are color coded as described above with the exception of a cation−π interaction which is shown in gray dashes. (D) Comparison of choline-bound (protein residues are colored in light green) and (S)-6-bound (colored in green) CutC active sites. Choline and (S)-6 were omitted from the structure for clarity.
Together, our inhibitor optimization studies and structural data allow us to propose that cyclic compound 6 may be a mechanism-based inhibitor (Scheme 1). The similarity of its binding mode to that of choline, coupled with the proximity of the C3 hydrogen atom of (S)-6 to Cys489, suggests that this compound could undergo C–H abstraction to form an inhibitor-centered ketyl radical. Notably, such a radical would be stabilized by the adjacent alkene, which could derail catalysis and lead to inhibition. This proposal is consistent with the dramatic improvement in activity observed upon introducing the alkene into the cyclic scaffold. It is also consistent with improved potency for the (S) versus (R) enantiomer, as the (S) enantiomer should position the C3 hydrogen optimally for abstraction.24 To test this hypothesis, we synthesized an analog of 6 that is deuterated at C3 (15) and assayed it both in whole cells and in vitro (Table 3). Racemic 15 displays a 3- and 2-fold loss of activity in whole cells and in vitro against CutC in comparison to 6. This preliminary result provides support for our mechanistic hypothesis, as the loss in activity could potentially arise from a reduced rate of deuterium abstraction due to a kinetic isotope effect. An alternative mode of inhibition that we cannot yet rule out would be the oxidation of 6 to an enone followed by conjugate addition of C489 at C5.
Scheme 1. Potential Mechanism of CutC Inhibition by (S)-6.

Table 3. Inhibitory Activity of Compounds 6 and 15.
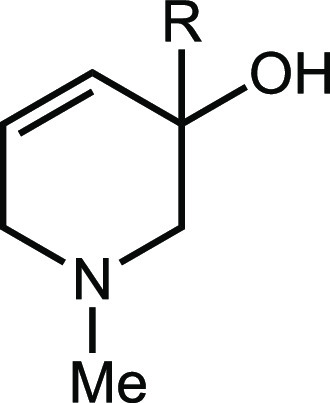
| Compounda | R | IC50 against E. coli MS 200-1 (μM) | IC50 against CutC (μM) |
|---|---|---|---|
| 6 | H | 10 | 3 |
| 15 | D | 30 | 6 |
Compounds 6 and 15 were assayed at the same time.
The cut genes are widely distributed among human gut bacteria and likely encode a major pathway for choline utilization in the GI tract. We therefore tested the potency of our best lead compound (S)-6 and our reference inhibitor BA for inhibition of TMA production from choline by additional Gram-negative (Proteus mirabilis, Escherichia coli MS69-1, Klebsiella sp. MS 92-3) and Gram-positive (Clostridium sporogenes) choline-metabolizing human gut isolates. Compound (S)-6 was effective against all strains analyzed, with IC50 values ranging from 5 to 97 μM, which is at least 20-fold more active than betaine aldehyde (Table 4). Finally, (S)-6 inhibited the conversion of choline to TMA by a human fecal suspension ex vivo with an EC50 of 60 μM, confirming its efficacy in a complex community more reminiscent of the gut microbiota (Table 4). Although (S)-6 possesses a somewhat modest biochemical IC50, the small shift between our in vitro and ex vivo IC50 values demonstrates its potential utility as a tool for in vivo studies.
Table 4. Activity of BA and (S)-6 toward Diverse Human Gut Bacteria and Human Fecal Suspension.
| Isolate | IC50 (μM) for BA | IC50 (μM) for (S)-6 |
|---|---|---|
| E. coli MS 69-1 | 465 | 22 |
| P. mirabilis | 2,488 | 92 |
| Klebsiella sp. | 212 | 4 |
| C. sporogenes | 1,000 | 12 |
| Fecal suspension | 1,273 | 60 |
In summary, we have discovered a cyclic choline analog that inhibits anaerobic choline metabolism by both Gram-negative and Gram-positive human gut isolates as well as a more complex gut community. Structural information and preliminary mechanistic studies suggest 6 may be a mechanism-based inhibitor that is processed by CutC to generate a stabilized radical intermediate. This finding adds another molecular scaffold to the growing arsenal of small molecules that inhibit gut microbial choline metabolism.
Acknowledgments
We acknowledge support from the Packard Fellowship for Science and Engineering and a Blavatnik Biomedical Accelerator Award (E.P.B.). C.L.D. is a Howard Hughes Medical Investigator. This work was supported by the National Institutes of Health grant R35 GM126982 (to C.L.D). M.O. is a Merck Fellow of the Life Sciences Research Foundation and a Postdoctoral Enrichment Program Grant recipient from the Burroughs Wellcome Fund. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30GM124165). The Pilatus 6M detector on the 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We acknowledge the laboratory of Federico Rey (University of Wisconsin Madison) for supplying the fecal sample.
Glossary
Abbreviations
- BA
betaine aldehyde
- CutC
glycyl radical enzyme choline TMA-lyase
- DMB
3,3-dimethyl-1-butanol
- FMO3
flavin-dependent monooxygenase 3
- GI
human gastrointestinal
- GRE
glycyl radical enzyme
- NAFLD
nonalcoholic fatty liver disease
- SAM
S-adenosylmethionine
- TMA
trimethylamine
- TMAO
trimethylamine-N-oxide.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00005.
Experimental data and procedures and crystallographic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sharon G.; Garg N.; Debelius J.; Knight R.; Dorrestein P. C.; Mazmanian S. K. Specialized Metabolites from the Microbiome in Health and Disease. Cell Metab. 2014, 20 (5), 719–730. 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N.; Balskus E. P. Exploring and Understanding the Biochemical Diversity of the Human Microbiota. Cell Chem. Bio. 2016, 23 (1), 18–30. 10.1016/j.chembiol.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Postler T. S.; Ghosh S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26 (1), 110–130. 10.1016/j.cmet.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel S. H.; da Costa K.-A. Choline: An Essential Nutrient for Public Health. Nutr. Rev. 2009, 67 (11), 615–623. 10.1111/j.1753-4887.2009.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin D. A. Ethanolamine Utilization in Bacterial Pathogens: Roles and Regulation. Nat. Rev. Microbiol. 2010, 8 (4), 290–295. 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S.; Balskus E. P. Microbial Conversion of Choline to Trimethylamine Requires a Glycyl Radical Enzyme. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (52), 21307–21312. 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain M.; Fornasini G.; Evans A. Trimethylamine: Metabolic, Pharmacokinetic and Safety Aspects. Curr. Drug Metab. 2005, 6 (3), 227–240. 10.2174/1389200054021807. [DOI] [PubMed] [Google Scholar]
- Zeisel S. H.; Wishnok J. S.; Blusztajn J. K. Formation of Methylamines from Ingested Choline and Lecithin. J. Pharmacol. Exp. Ther. 1983, 225 (2), 320–324. [PubMed] [Google Scholar]
- Krueger S. K.; Williams D. E. Mammalian Flavin-Containing Monooxygenases: Structure/Function, Genetic Polymorphisms and Role in Drug Metabolism. Pharmacol. Ther. 2005, 106 (3), 357–387. 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas M.-E.; Barton R. H.; Toye A.; Cloarec O.; Blancher C.; Rothwell A.; Fearnside J.; Tatoud R.; Blanc V.; Lindon J. C.; Mitchell S. C.; Holmes E.; McCarthy M. I.; Scott J.; Gauguier D.; Nicholson J. K. Metabolic Profiling Reveals a Contribution of Gut Microbiota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (33), 12511–12516. 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M. D.; Hamp T. J.; Reid R. W.; Fischer L. M.; Zeisel S. H.; Fodor A. A. Association Between Composition of the Human Gastrointestinal Microbiome and Development of Fatty Liver With Choline Deficiency. Gastroenterology 2011, 140 (3), 976–986. 10.1053/j.gastro.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Klipfell E.; Bennett B. J.; Koeth R.; Levison B. S.; DuGar B.; Feldstein A. E.; Britt E. B.; Fu X.; Chung Y.-M.; Wu Y.; Schauer P.; Smith J. D.; Allayee H.; Tang W. H. W.; DiDonato J. A.; Lusis A. J.; Hazen S. L. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472 (7341), 57–63. 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth R. A.; Wang Z.; Levison B. S.; Buffa J. A.; Org E.; Sheehy B. T.; Britt E. B.; Fu X.; Wu Y.; Li L.; Smith J. D.; DiDonato J. A.; Chen J.; Li H.; Wu G. D.; Lewis J. D.; Warrier M.; Brown J. M.; Krauss R. M.; Tang W. H. W.; Bushman F. D.; Lusis A. J.; Hazen S. L. Intestinal Microbiota Metabolism of l-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19 (5), 576–585. 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W. H. W.; Wang Z.; Levison B. S.; Koeth R. A.; Britt E. B.; Fu X.; Wu Y.; Hazen S. L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368 (17), 1575–1584. 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory J. C.; Buffa J. A.; Org E.; Wang Z.; Levison B. S.; Zhu W.; Wagner M. A.; Bennett B. J.; Li L.; DiDonato J. A.; Lusis A. J.; Hazen S. L. Transmission of Atherosclerosis Susceptibility with Gut Microbial Transplantation. J. Biol. Chem. 2015, 290 (9), 5647–5660. 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K.; Rey F. E. The Emerging Role of Gut Microbial Metabolism on Cardiovascular Disease. Curr. Opin. Microbiol. 2019, 50, 64–70. 10.1016/j.mib.2019.09.007. [DOI] [PubMed] [Google Scholar]
- Missailidis C.; Hällqvist J.; Qureshi A. R.; Barany P.; Heimbürger O.; Lindholm B.; Stenvinkel P.; Bergman P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS One 2016, 11 (1), e0141738 10.1371/journal.pone.0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.H. W.; Wang Z.; Kennedy D. J.; Wu Y.; Buffa J. A.; Agatisa-Boyle B.; Li X. S.; Levison B. S.; Hazen S. L. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116 (3), 448–455. 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J.; Ling A. V.; Manthena P. V.; Gearing M. E.; Graham M. J.; Crooke R. M.; Croce K. J.; Esquejo R. M.; Clish C. B.; Vicent D.; Biddinger S. B. Flavin-Containing Monooxygenase 3 as a Potential Player in Diabetes-Associated Atherosclerosis. Nat. Commun. 2015, 6 (1), 1–10. 10.1038/ncomms7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. J.; Vallim T. Q. de; Aguiar; Wang Z.; Shih D. M.; Meng Y.; Gregory J.; Allayee H.; Lee R.; Graham M.; Crooke R.; Edwards P. A.; Hazen S. L.; Lusis A. J. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013, 17 (1), 49–60. 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Roberts A. B.; Buffa J. A.; Levison B. S.; Zhu W.; Org E.; Gu X.; Huang Y.; Zamanian-Daryoush M.; Culley M. K.; DiDonato A. J.; Fu X.; Hazen J. E.; Krajcik D.; DiDonato J. A.; Lusis A. J.; Hazen S. L. Non-Lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163 (7), 1585–1595. 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treacy E. P.; Akerman B. R.; Chow L. M. L.; Youil R.; Lin C. B.; Bruce A. G.; Knight M.; Danks D. M.; Cashman J. R.; Forrest S. M. Mutations of the Flavin-Containing Monooxygenase Gene (FMO3) Cause Trimethylaminuria, a Defect in Detoxication. Hum. Mol. Genet. 1998, 7 (5), 839–845. 10.1093/hmg/7.5.839. [DOI] [PubMed] [Google Scholar]
- Mitchell S. C.; Smith R. L. Trimethylaminuria: The Fish Malodor Syndrome. Drug Metab. Dispos. 2001, 29 (4), 517–521. [PubMed] [Google Scholar]
- Bodea S.; Funk M. A.; Balskus E. P.; Drennan C. L. Molecular Basis of C–N Bond Cleavage by the Glycyl Radical Enzyme Choline Trimethylamine-Lyase. Cell Chem. Biol. 2016, 23 (10), 1206–1216. 10.1016/j.chembiol.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo A. M.; Bodea S.; Hamer H. A.; Marks J. A.; Haiser H. J.; Turnbaugh P. J.; Balskus E. P. Characterization and Detection of a Widely Distributed Gene Cluster That Predicts Anaerobic Choline Utilization by Human Gut Bacteria. mBio 2015, 6 (2), e00042-15 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S.; Marks J. A.; Balskus E. P. Characterization of Choline Trimethylamine-Lyase Expands the Chemistry of Glycyl Radical Enzymes. ACS Chem. Biol. 2014, 9 (7), 1408–1413. 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- Backman L. R. F.; Funk M. A.; Dawson C. D.; Drennan C. L. New Tricks for the Glycyl Radical Enzyme Family. Crit. Rev. Biochem. Mol. Biol. 2017, 52 (6), 674–695. 10.1080/10409238.2017.1373741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. B.; Gu X.; Buffa J. A.; Hurd A. G.; Wang Z.; Zhu W.; Gupta N.; Skye S. M.; Cody D. B.; Levison B. S.; Barrington W. T.; Russell M. W.; Reed J. M.; Duzan A.; Lang J. M.; Fu X.; Li L.; Myers A. J.; Rachakonda S.; DiDonato J. A.; Brown J. M.; Gogonea V.; Lusis A. J.; Garcia-Garcia J. C.; Hazen S. L. Development of a Gut Microbe–Targeted Nonlethal Therapeutic to Inhibit Thrombosis Potential. Nat. Med. 2018, 24 (9), 1407–1417. 10.1038/s41591-018-0128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen S. L.; Garcia-Garcia J. C.; Gerberick G. F.; Wos J. A.; Gu X.; Reilly M.; Sivik M. R.; Reed J. M.; Cody D. B.. Methods for Inhibiting Conversion of Choline to Trimethylamine (TMA). US20180000754A1, January 4, 2018.
- Garcia-Garcia J. C.; Gerberick G. F.; Reilly M.; Hazen S. L.; Gu X.. Methods for Inhibiting Conversion of Choline to Trimethylamine (Tma). US20190099384A1, April 4, 2019.
- Garcia-Garcia J. C.; Gerberick G. F.; Hazen S. L.; Gu X.. Methods for Inhibiting Conversion of Choline to Trimethylamine (TMA). US20190099390A1, April 4, 2019.
- Garcia-Garcia J. C.; Gerberick G. F.; Wos J. A.; Hazen S. L.; Gu X.. Methods for Inhibiting Conversion of Choline to Trimethylamine (TMA). US20190099389A1, April 4, 2019.
- Anwar S.; Bhandari U.; Panda B. P.; Dubey K.; Khan W.; Ahmad S. Trigonelline Inhibits Intestinal Microbial Metabolism of Choline and Its Associated Cardiovascular Risk. J. Pharm. Biomed. Anal. 2018, 159, 100–112. 10.1016/j.jpba.2018.06.027. [DOI] [PubMed] [Google Scholar]
- Winter M.; Bretschneider T.; Thamm S.; Kleiner C.; Grabowski D.; Chandler S.; Ries R.; Kley J. T.; Fowler D.; Bartlett C.; Binetti R.; Broadwater J.; Luippold A. H.; Bischoff D.; Büttner F. H. Chemical Derivatization Enables MALDI-TOF-Based High-Throughput Screening for Microbial Trimethylamine (TMA)-Lyase Inhibitors. SLAS DISCOVERY: Advancing Life Sciences R&D 2019, 24 (7), 766–777. 10.1177/2472555219838216. [DOI] [PubMed] [Google Scholar]
- Orman M.; Bodea S.; Funk M. A.; Campo A. M.; Bollenbach M.; Drennan C. L.; Balskus E. P. Structure-Guided Identification of a Small Molecule That Inhibits Anaerobic Choline Metabolism by Human Gut Bacteria. J. Am. Chem. Soc. 2019, 141 (1), 33–37. 10.1021/jacs.8b04883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett N. C.; Chaires J. B. Thermodynamic Studies for Drug Design and Screening. Expert Opin. Drug Discovery 2012, 7 (4), 299–314. 10.1517/17460441.2012.666235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. R.; Parrish J. C.; Fraser M. E.; Smith W. W.; Bartlett P. A.; James M. N. G. Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes. Biochemistry 1998, 37 (48), 16839–16845. 10.1021/bi9821364. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Tounge B. A.; Bembenek S. D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51 (8), 2432–2438. 10.1021/jm701255b. [DOI] [PubMed] [Google Scholar]
- Corne V.; Sarotti A. M.; Arellano C. R. de; Spanevello R. A.; Suárez A. G. Experimental and Theoretical Insights in the Alkene–Arene Intramolecular π-Stacking Interaction. Beilstein J. Org. Chem. 2016, 12 (1), 1616–1623. 10.3762/bjoc.12.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.