

All cells need to coordinate their growth and division, and small GTPases that are conserved throughout life play a key role in this regulation. One of these, Era, provides an essential function in the assembly of the 30S ribosomal subunit in Escherichia coli, but its role in regulating E. coli cell division is much less well understood. Here, we characterize a novel dominant negative mutant of Era (Era647) that uncouples these two activities when overproduced; it inhibits cell division by disrupting assembly of the Z ring, without significantly affecting ribosome production. The unique properties of this mutant should help to elucidate how Era regulates cell division and coordinates this process with ribosome biogenesis.
KEYWORDS: Era, GTPases, cell cycle checkpoints, cytokinesis, FtsZ
ABSTRACT
Cell growth and division are coordinated, ensuring homeostasis under any given growth condition, with division occurring as cell mass doubles. The signals and controlling circuit(s) between growth and division are not well understood; however, it is known in Escherichia coli that the essential GTPase Era, which is growth rate regulated, coordinates the two functions and may be a checkpoint regulator of both. We have isolated a mutant of Era that separates its effect on growth and division. When overproduced, the mutant protein Era647 is dominant to wild-type Era and blocks division, causing cells to filament. Multicopy suppressors that prevent the filamentation phenotype of Era647 either increase the expression of FtsZ or decrease the expression of the Era647 protein. Excess Era647 induces complete delocalization of Z rings, providing an explanation for why Era647 induces filamentation, but this effect is probably not due to direct interaction between Era647 and FtsZ. The hypermorphic ftsZ* allele at the native locus can suppress the effects of Era647 overproduction, indicating that extra FtsZ is not required for the suppression, but another hypermorphic allele that accelerates cell division through periplasmic signaling, ftsL*, cannot. Together, these results suggest that Era647 blocks cell division by destabilizing the Z ring.
IMPORTANCE All cells need to coordinate their growth and division, and small GTPases that are conserved throughout life play a key role in this regulation. One of these, Era, provides an essential function in the assembly of the 30S ribosomal subunit in Escherichia coli, but its role in regulating E. coli cell division is much less well understood. Here, we characterize a novel dominant negative mutant of Era (Era647) that uncouples these two activities when overproduced; it inhibits cell division by disrupting assembly of the Z ring, without significantly affecting ribosome production. The unique properties of this mutant should help to elucidate how Era regulates cell division and coordinates this process with ribosome biogenesis.
INTRODUCTION
GTPases are recognized as molecular switches whose conformation and activities change depending upon their GDP- or GTP-bound states. The era gene of Escherichia coli is located downstream of rnc in the multifunctional rnc operon (rnc-era-recO) and encodes a GTPase (∼35 kDa) essential for survival (1–3). The rnc gene encodes RNase III, a double-strand specific RNA endonuclease, which inhibits expression of the operon by processing the leader RNA transcript, causing mRNA degradation (4, 5). Independent of RNase III regulation, the translation rate of the two genes is coordinately controlled by growth rate. Cells grown in minimal medium produce ∼300 molecules of Era per cell, which is 5-fold less than that of cells grown in a rich medium (4, 6).
Era is highly conserved both in prokaryotes and in eukaryotes, including humans (7). Homologues of Era are present in every bacterial species except the Chlamydiales and Planctomycetes (7, 8). It has been shown that human Era plays a role in controlling cell growth and apoptosis (7, 9–11). Crystal structures of Era reveal an N-terminal GTPase domain and a C-terminal RNA-binding KH domain (Fig. 1) (12–14).
FIG 1.

The structure of the Era protein and location of the Era647 mutation. (A) A comparison of amino acid sequences among Era homologs from Aquifex aeolicus, Thermus thermophilus, and E. coli (PDB entries 3IEV, 1WF3, and 1EGA, respectively) is shown along with structural features. Identical residues shared among all three homologs are shaded dark green, similar residues shared among all three homologs are shaded light green, and identical residues shared with two homologs are shaded blue. Residues 131 to 137 changed in Era647 are highlighted with a blue box and magenta letters below; the deleted glutamate is shown as a dash. (B) The location of residues 131 to 137 near α helix 4 of the GTPase domain is indicated on the atomic structure of E. coli Era in complex with GDP (PDB entry 3IEU). The ribbon diagram represents the protein (helices as cylinders, strands as arrows, and loops as tubes), and the stick model in green represents the nucleotide. The GTPase and KH domains are colored in gray and light orange, respectively, and residues 131 to 137 are highlighted in red.
Biochemical, genetic, and physical evidence shows that Era is important in ribosome biogenesis and bacterial growth. The KH domain of Era binds 16S rRNA in vivo (15–18). Like many mutants with structural ribosome defects, the Era E200K mutant is cold sensitive for growth. The cold sensitivity of Era E200K can be suppressed by overexpression of the ksgA gene (19), which encodes the 16S rRNA adenosine dimethyl-transferase KsgA. Era binds to the 30S ribosome subunit and to the 16S rRNA (12, 13, 20). The GTP-bound form of Era (Era-GTP) binds specifically to the 10-nucleotide residues GAUCACCUCC containing the CCUCC anti-Shine-Dalgarno sequence near the 3′ end of the 16S rRNA (12). Here, Era also interacts with helix 45, which is the helix that KsgA binds for the methylation of 16S rRNA. Helix 45 and the following 10 nucleotides bound by Era are highly conserved in all three kingdoms of life. Binding of Era-GTP to this helix and the 10 nucleotides beyond it stimulates hydrolysis of bound GTP. Era-RNA structural studies show that only the GTP form of the protein can bind the rRNA and that the hydrolysis of GTP to GDP results in a major rearrangement of the structure of Era, releasing Era-GDP from the 16S rRNA (12, 13). Era stimulates processing of the 16S precursor rRNA to its mature form (21) and aids global ribosome biogenesis to the final maturation of the 30S particle (12, 13, 22, 23). Era may function at a terminal stage of 30S biogenesis as the checkpoint for the final activation of the mature 30S subunit and the initiation of mRNA translation. Overproduction of Era also enhances 16S rRNA processing as well as 70S ribosome assembly in the absence of the YbeY endoribonuclease (24).
The cytosol contains most of the cellular Era, as would be predicted for a protein modulating the biogenesis of each 30S ribosome subunit. However, about 20% of Era in a population of cells is associated with the inner membrane fraction (25), and purified Era-GDP has been demonstrated to bind specifically to isolated cytoplasmic membranes depleted of Era (26). Membrane binding is dependent upon the C-terminal KH domain of Era and on a site that overlaps the RNA-binding region (15, 16). This finding suggests that Era may cycle between a 16S rRNA-bound form of Era-GTP and a membrane-bound form of Era-GDP. Immunogold electron microscopy has been used to locate Era in patches along the membrane at mid- and quarter-cell positions corresponding to potential cell division sites (27).
In E. coli, a number of Era mutants in the highly conserved regions of both the N-terminal GTPase and the C-terminal KH domains have been isolated (2, 6, 18, 28–30). Characterization of these mutants revealed that both domains are essential for viability. Nonlethal mutations, like the era1 point mutation in the G1 segment of the Era GTP-binding domain (P17R), cause both reduced cell growth, especially at low temperatures, and delayed cell division. Increasing the expression of the era1 mutant on a multicopy plasmid alleviated its growth and division defects (6). Unlike known cell division mutants, the era1 mutant does not form the characteristic long filaments but instead arrests growth just prior to division, at a two-cell length stage. Simply reducing expression of wild-type era exerts a similar checkpoint phenotype in both E. coli and Bacillus subtilis; as Era levels are reduced, the growth rate is reduced in parallel, and an increasing fraction of cells exist at the two-cell stage (6, 31). Thus, under either era+ or era1 conditions, a critical level of GTPase activity seems to be required for growth and division. Until that threshold level is reached, the cells exist in a growth arrested, predivisional state (6).
Generally, when translation is inhibited, there is a subsequent block in cell growth and division. Thus, it is difficult to know whether Era, which affects ribosome biogenesis activity, is directly involved in cell division. Here, in examining this problem, we have isolated and identified a dominant mutant allele of Era that, when overproduced, maintains ribosome function and growth but is defective for cell division, thus separating the two functions of Era. We show that this defect is at the level of Z ring assembly, which can be corrected by increasing cellular levels of FtsZ or by a hypermorphic mutant of FtsZ.
RESULTS AND DISCUSSION
Identification of a dominant negative Era protein that blocks cell division.
Era has been shown to coordinate cell growth with division, and era mutations have been identified that block cell growth and division. A primary effect of these mutations is to inhibit 30S ribosome subunit assembly and subsequent translation. Because a ribosome defect would itself block cell growth and division, it has been unclear whether Era had a direct effect on the division process itself. Until we isolated the mutant era647, no era mutation existed with an effect on cell division that did not also block cell cycle progression.
The era647 mutation was initially created on plasmid pHKP60 carrying the era+ gene expressed from the arabinose-inducible pBAD promoter. Substitution mutants covering six to seven residues within the N-terminal GTP binding domain were created in a search for Era defective mutants. One of these mutants, in which 7 residues from 131 to 137 in the GTPase domain were replaced with 6 other residues, had defects in the presence of arabinose that caused cell filamentation at temperatures tested between 32°C and 42°C as described below. The mutation was given the name “6 for 7” (era647) because of the amino acid changes that may alter the Era structure of α helix 4 between the G4 and G5 motifs (Fig. 1). On LB agar in the presence of arabinose, colonies of strain W3110/pBADera647, expressing the mutant Era, had a flattened, “fried egg” phenotype (data not shown). This distinct colony phenotype enabled their selection.
Because the pBADera647 plasmid pHKP67 caused reduced growth even without adding arabinose, we constructed two new strains in which either wild-type era and/or era647 were placed on the bacterial chromosome in single copy within the pL operon of a defective λ prophage. In this configuration, the era alleles are under tight control by the heat labile bacteriophage λ cI repressor, allowing expression of the pL operon at 42°C but maintaining repression of pL at 32°C. Another copy of the wild-type era gene remained at its native site in the rnc operon on the chromosome of E. coli. Both new strains formed normal colonies at 32°C when pL is repressed. Inducing wild-type era expression at 42°C from pL did not alter colony formation; however, turning on era647 expression at 42°C prevented colony formation (Fig. 2A). This lethal effect was observed on LB or M63 minimal glucose agar plates (data not shown). Phase-contrast microscopy revealed that turning on expression of the era647 mutant relative to wild-type era at 42°C caused cells to filament without any sign of a septum being formed (Fig. 2B).
FIG 2.
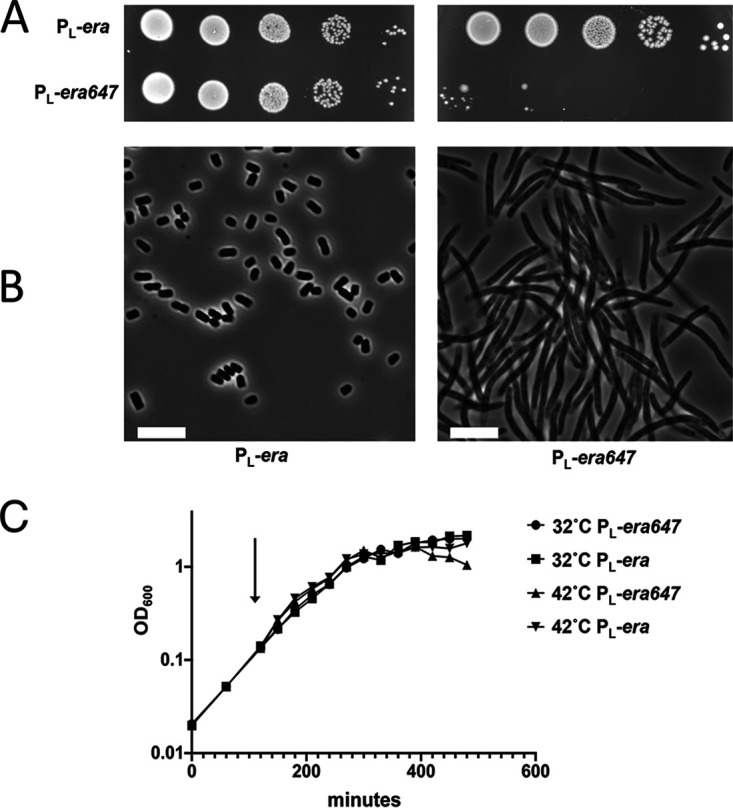
Phenotypic analysis of pL expression for strains carrying era (TUC540) and era647 (TUC05). (A) Colony viabilities were tested on LB agar at 32°C (left side) and 42°C (right side) by spotting 10-fold serial dilutions of cultures. (B) Phase-contrast microscopic analysis of cells grown for 2 hours in exponential phase at 42°C in LB culture. (C) Growth curves for the two strains grown in LB broth at 32°C or shifted from 32°C to 42°C. Cells were diluted 1:200 and incubated at 32°C for 2 h and then split into 32°C and 42°C cultures (arrow) and grown to stationary phase. Scale bars, 5 μm.
Effects of replacing wild-type era with era647 at the native chromosomal locus.
We PCR amplified a 519-bp segment of era carrying the era647 allele and inserted this segment with the mutation into the era gene within the genomic rnc operon by recombineering (see Materials and Methods). Colonies formed by this era647 mutant strain were normal at 42°C and 37°C, but they were nearly nonviable at 25°C (see Fig. S1 in the supplemental material). This finding is similar to the behavior of cells with the era1 allele, which, as mentioned earlier, confers cold sensitivity when at the native rnc locus but corrects the cold sensitivity when in multicopy (6). Thus, native expression of the era647 allele has a phenotype similar to the era1 allele, whereas the phenotypes of high-level expression are distinct.
Multicopy suppression of era647-mediated cell division inhibition.
A library of E. coli genes cloned into the BamHI site on pBR322 was used to isolate multicopy suppressors of the thermosensitive phenotype of era647 under pL control (see Materials and Methods). Two classes of suppressors were found that restored colony formation at 42°C.
The first class included clones of either the nusB gene or the rof (yaeO) gene, both of which partially suppressed the filamentation defect of Era647 (Fig. 3). The NusB protein is known to modify the RNA polymerase elongation complex during transcription (32, 33), and the Rof product is known to inhibit the transcription termination factor Rho (34). Because Rof suppresses era647, we also tested a rho mutation (TUC626) and found that it also suppresses the thermosensitive phenotype of era647 (data not shown). Since we suspected that suppression might occur by lowering the levels of Era647 expression from pL, we measured the effect of the nusB and rof plasmid clones on pL operon expression. To do this, we placed the lacZ gene in exactly the same position as era in the pL operon (see Fig. S2A in the supplemental material) and used the expression of β-galactosidase as a comparative reporter for Era expression from pL. Multicopy nusB or rof expression reduced the level of β-galactosidase expressed from lacZ (Fig. S2B), perhaps by interfering with antitermination mechanisms. To explore a potential transcription termination-independent mechanism, we asked if era647-mediated toxicity could be suppressed by expressing the lambda Cro repressor, which is known to reduce transcription from the pL promoter by 2- to 4-fold (35). We found that excess Cro did suppress the toxicity (data not shown). We conclude that this class of suppressors reduces the expression of era647 from pL to a level that reduces toxicity and allows colony formation.
FIG 3.
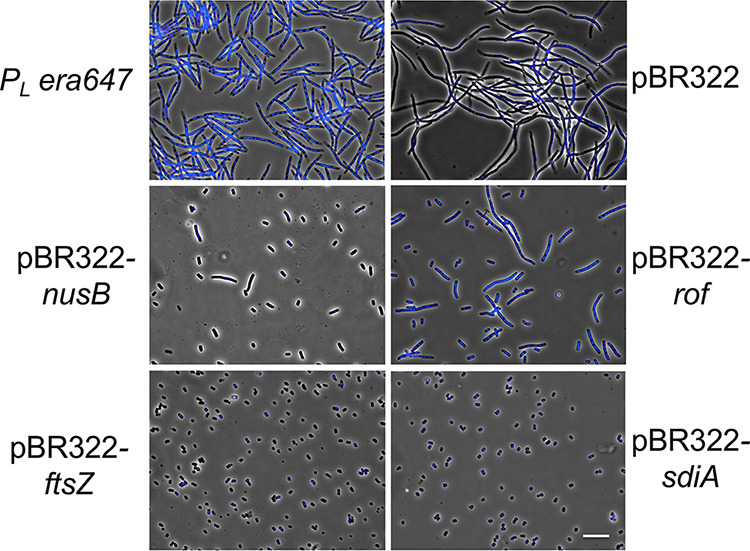
Multicopy suppressors of Era647-mediated cell filamentation. Shown are phase contrast-fluorescence images of TUC05 cells grown at 32°C to an OD600 of 0.1 and 0.2 and then induced to express era647 from the lambda pL promoter at 42°C for 3 h. The top left image shows TUC05 cells. The top right image shows TUC05 with plasmid pBR322. The other images show TUC05 with clones expressing the genes shown. Cells were also stained with DAPI to show DNA. Scale bar, 5 μm.
The second class of multicopy suppressors of era647 included plasmids that carry the ftsZ gene, which would increase the level of the tubulin-like, major cell division protein FtsZ. In addition to clones carrying the ftsZ gene, clones carrying the sdiA gene also suppressed the era647 temperature-sensitive defect. Multicopy sdiA is known to stimulate the transcription of the ftsQAZ operon and increase FtsZ protein levels (36). Thus, both ftsZ and sdiA clones suppress the temperature-sensitive defect of the ftsZ84 allele and the toxic effect of Era647-induced cell filamentation.
Both ftsZ and sdiA in multicopy completely prevented Era647-induced cell filamentation compared with nusB or rof plasmids (Fig. 3). Neither FtsZ nor SdiA changed the level of expression of genes under pL control (Fig. S2B), but both suppressed the defect of era647. Both also suppressed the thermosensitive defect of an ftsZ84 allele (Table 1) because they increase FtsZ protein levels in the cell (36). Thus, high FtsZ levels are needed to suppress the toxicity of the Era647 protein.
TABLE 1.
Multicopy suppression of Era647 overproduction toxicity
| Plasmid clone | Origin | Suppression of era647 | Complementation of ftsZ84 |
|---|---|---|---|
| pBR322 | colE1 | − | − |
| pXM14 (nusB) | colE1 | + | − |
| pXM16 (rof) | colE1 | + | − |
| pXM17 (sdiA) | colE1 | + | + |
| pXM15 (ftsZ) | colE1 | + | + |
| pJPB71 (ftsZ) | pSC101 | + | + |
| pJPB223 (ftsQAZ) | pSC101 | + | + |
| pSEB25 (ftsQZ) | pSC101 | + | + |
| pSEB162 (ftsQA) | pSC101 | − | − |
| pBS58 (ftsQAZ) | pSC101 | + | + |
| pACS1 (rnc era) | colE1 | − | N.D.a |
N.D., not determined.
To find out whether FtsZ is sufficient for suppression of the era647-mediated cell division defect or if some other cell division proteins might also be capable of suppression, we tested a set of five plasmids carrying combinations of the ftsQAZ operon genes for suppression (37). These plasmids harbor the pSC101 origin and have a lower copy number than the pBR322-derived library clones. All four plasmids carrying ftsZ, including pJPB71, which carries only ftsZ, complemented era647 for colony formation at 42° (Table 1). However, plasmid pSEB162 carrying ftsQ and ftsA but not ftsZ did not complement era647 (Table 1), indicating that increasing FtsZ levels is sufficient to suppress the lethal filamentation caused by era647.
To rule out unexpected effects of thermoinduction of pL-era647, we also tested whether extra FtsZ could suppress the toxicity of Era647 overproduced from an arabinose-inducible promoter on a plasmid. As shown in Fig. 4, arabinose induction of pBAD30-era or pBAD30-gfp did not cause significant cell filamentation, but induction of pBAD30-era647 did. Moreover, when pBS58 (ftsQAZ) was introduced into the strain carrying pBAD30-era647, it efficiently suppressed the arabinose-induced cell filamentation.
FIG 4.
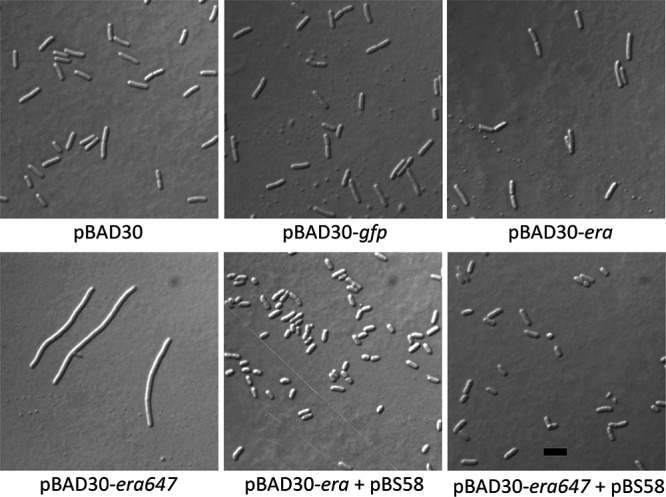
Arabinose induction of era647 on a pBAD plasmid mimics the effects of thermoinduction of pL-era647 and is suppressed by multicopy ftsQAZ. Cells carrying the plasmids indicated were grown in LB plus ampicillin plus spectinomycin and induced with 0.2% l-arabinose for 2 h until they reached late logarithmic phase and then were examined by DIC microscopy. Scale bar, 3 μm.
One potential explanation for why multicopy ftsZ can suppress era647 was that era647 overexpression reduces cellular levels of FtsZ and that ectopically produced FtsZ can restore these levels. However, although FtsZ levels clearly increased in cells harboring the multicopy pBS58 plasmid (data not shown), immunoblotting showed that cellular FtsZ levels did not decrease significantly when either era or era647 was overexpressed (see Fig. S3 in the supplemental material), ruling out this possibility.
Notably, we did not find era among the multicopy suppressors, probably because era647 is dominant to wild-type era. To confirm this, we showed that pACS1 (3), a pBR322-derived clone of the rnc era operon, could not suppress era647 (Table 1). Thus, pL expression of era647 is dominant for inhibition of cell division even in the presence of high levels of wild-type Era.
Era647 inhibits cell division by complete disassembly of Z rings.
In E. coli, cell division is mediated by a collection of proteins that localize to the mid-cell division site, where they assemble into a multiprotein complex called the septal or Z ring (38). FtsZ, a tubulin-like protein with GTP-binding, GTPase, and GTP-dependent polymerization activities (39–42) is the first protein of this collection to localize at the mid-cell division site (43, 44) and is conserved in all bacteria except Chlamydiales and Planctomycetes (45, 46). As mentioned above, homologues of Era also exist in all bacterial species with the exception of Chlamydiales and Planctomycetes (8, 47). To our knowledge, no other cell division protein shares this cluster of orthologous groups (COG).
The COG result specifically linking FtsZ with Era melds well with our genetic results showing that the toxic effects caused by high levels of the Era647 protein can be suppressed by increasing the levels of the FtsZ protein. As a result, we asked whether Era647 overproduction blocked cell division by affecting the assembly of Z rings. We analyzed cells overproducing Era or Era647 by immunofluorescence microscopy, using anti-FtsZ antibodies. As expected, all uninduced cells or cells overproducing wild-type Era were short and most cells (>68%) had sharp Z rings at mid-cell division sites (Fig. 5, first three rows; see Table S2 in the supplemental material). In contrast, all cells overproducing Era647 were filamentous, and their FtsZ staining pattern consisted of irregularly shaped and spaced FtsZ aggregates, with no Z rings visible in 95% of cells (Fig. 5, bottom row; Table S2). These aggregates were almost always located between the multiple segregated nucleoids within these filamentous cells (see Fig. S4 in the supplemental material).
FIG 5.
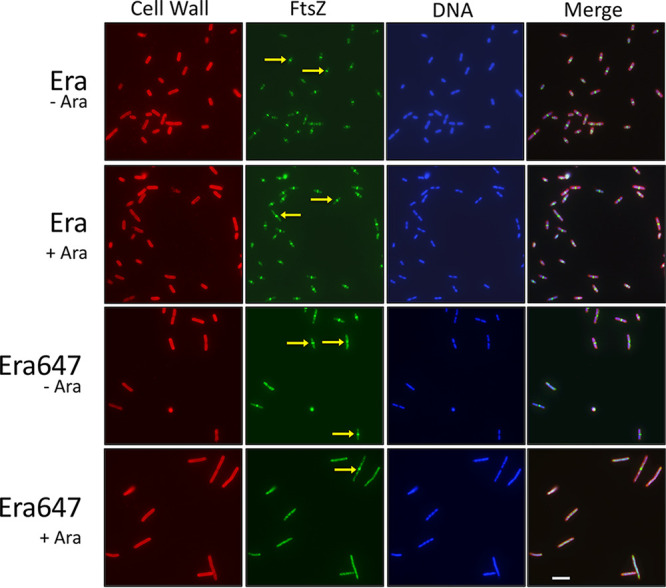
Overproduction of Era647 inhibits Z ring assembly. Cells containing pBAD30 derivatives of Era or Era647 were induced with l-arabinose (Ara) for 1 h in mid- to late logarithmic phase and then fixed and stained with DAPI (blue) and fluorescent lectin (red) and immunostained with anti-FtsZ (green). Arrows highlight examples of sharp Z rings at midcell, which were found in many of the cells expressing wild-type Era. A rare Z ring in the Era647-induced cells at the bottom. Scale bar, 5 μm.
To test whether the Era647 effect on the Z rings might be mitigated under slow growth conditions, we repeated this type of experiment with cells grown in minimal glycerol medium with similar results. Era647 when overproduced in minimal medium generated filamentous cells that contained bright areas of diffuse FtsZ fluorescence, and all cells lacked Z rings (see Fig. S5 in the supplemental material). As in rich medium, excess FtsQAZ from pBS58 rescued cell filamentation in minimal glucose medium (data not shown).
To confirm the results found in fixed cells and to rule out artifacts due to cell fixation or immunostaining, we also examined the effects of excess Era or Era647 in live cells expressing an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible FtsZ-GFP fusion as a merodiploid. For this purpose, we used a higher-copy-number plasmid to overexpress Era647 from the pBAD promoter (pAW5), as well as the wild-type Era control (pAW4). In accord with the immunofluorescence results, uninduced cells were short and ∼90% had sharp Z rings at mid-cell division sites (see Fig. S6, rows A and B, in the supplemental material), whereas all cells induced for Era647 overproduction for several hours became filamentous and exhibited irregularly spaced FtsZ aggregates instead of regularly spaced rings (Fig. S6, row D). Arabinose induction of wild-type Era in cells with FtsZ fused to GFP caused cells to elongate. Elongation in cells carrying FtsZ-GFP has been described and is due to a toxic synergy caused by the partially functional FtsZ-GFP fusion protein inhibiting cell division on its own (48). Importantly, however, these cells overproducing wild-type Era all contained sharply defined Z rings (Fig. S6, row C), which is in clear contrast to the aberrant FtsZ aggregates in Era647 overproducers.
To investigate the process of Z ring disassembly by Era647 in greater detail, we examined the localization of FtsZ-GFP in a time course after arabinose addition of Era647. After 10 to 15 min of induction, most (∼90%) cells still displayed sharp Z rings at the mid-cell division site (Fig. 6). However, by 30 min, the majority of cells lacked midcell Z rings, and by 40 min, only ∼5% of cells contained Z rings, with the remainder of cells displaying only diffuse cytoplasmic fluorescence (Fig. 6). Eventually, these induced cells stopped dividing, and the diffuse fluorescence transformed into multiple fluorescent aggregations observed at later time points similar to those shown in Fig. 5, S4, and S5. As expected, overproduction of wild-type Era from pAW4 had no effect on Z rings (data not shown).
FIG 6.
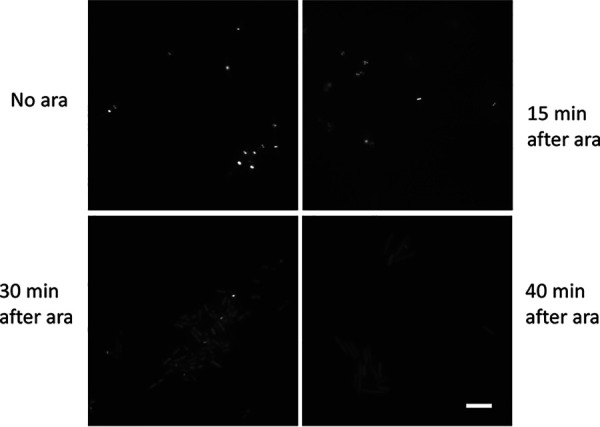
FtsZ-GFP rings rapidly disassemble upon Era647 induction. WM5570 cells carrying pAW5 (pBAD-Era647) and producing moderate levels of FtsZ-GFP (50 μM IPTG) were grown in LB plus chloramphenicol to mid-logarithmic phase, induced with l-arabinose (ara) for the times shown, and rapidly mounted on agarose slabs for imaging by fluorescence microscopy. Scale bar, 5 μm.
These results indicate that Z rings are disassembled completely and relatively rapidly by Era647 expression, and instead of forming aberrant but visible structures such as helices (49) or distributed foci (50, 76, 77), FtsZ delocalizes into a diffuse pattern within the cytoplasm. This is similar to the effects on the ring by the presence of SulA or MciZ proteins, which are FtsZ inhibitors that sequester FtsZ subunits or cap FtsZ polymer ends, respectively (51, 52). It is not clear what causes FtsZ to form the periodic aggregates in the cytoplasm of filamentous cells after longer expression of Era647.
SulA is not responsible for Era647-mediated cell filamentation.
This similarity to the effects of SulA prompted us to test whether Era647 itself might be inducing SulA. Expression of the sulA/sfiA gene is induced during the SOS response to DNA damage, whereupon the SulA protein binds FtsZ directly. This inhibits FtsZ polymerization, Z ring formation, and cell division in a checkpoint control response to DNA damage (53–55). To test whether the Era647 effect might be caused indirectly by overproduction of SulA under these conditions, we introduced a deletion of sulA in strain TUC303, which carries the pL-era647 construct. We found that this strain still exhibited a lethal filamentation phenotype at 42°C (data not shown), indicating no role for SulA in the Era647-mediated disruption of Z rings.
Era and Era647 do not seem to interact directly with FtsZ.
The evidence so far suggests that Era647 in particular might interact directly with FtsZ in order to sequester it away from the Z ring. We tested this possibility by measuring Era-FtsZ interactions in a bacterial two-hybrid assay that produces beta-galactosidase if two proteins fused to separate domains of adenylate cyclase (T18 and T25) interact sufficiently to bring these domains together (56). For this purpose, we fused Era or Era647 to the T18 domain in plasmids pUT18 or pUT18c to fuse T18 to either end of Era or Era647 and introduced all four of these plasmids into a cya BTH101 strain carrying FtsZ fused to the T25 domain in plasmid pKNT25 (57). As a positive control, we introduced pUT18c-FtsA, which is known to interact with FtsZ (58), into the same BTH101 strain carrying pKNT25-FtsZ.
As shown in Fig. S7 in the supplemental material, although the FtsA-FtsZ interaction yielded a strong blue color from colonies grown on plates with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), neither wild-type Era nor Era647 showed a detectable color when paired with FtsZ. This finding suggests that Era/Era647 do not interact with FtsZ, or if they do, it may be transient. We also purified Era and Era647 (see Fig. S8 in the supplemental material) and asked whether they affected FtsZ assembly in vitro and saw no clear effects of Era or Era647 on the ability of FtsZ to polymerize in sedimentation assays (D. P. Haeusser and W. Margolin, unpublished data). In addition, there was no definitive evidence of an interaction in pulldown assays (X. Zhou and D. L. Court, unpublished data). Therefore, despite the effectiveness of Era647 in disassembling Z rings and the ability of extra FtsZ to overcome this effect in vivo, there is currently no evidence to support a direct interaction between FtsZ and Era647 or wild-type Era.
Effects of hypermorphic cell division alleles on the Era647 phenotype.
The ability of increased FtsZ levels to counteract the cell division block by excess Era647 supports a model in which Era647 sequesters FtsZ subunits, preventing them from assembling into FtsZ polymers at the Z ring. However, the lack of evidence for a direct Era-FtsZ interaction prompted us to test whether stimulation of cell division through alternative pathways might be able to resist Era647 toxicity. For this, we exploited two hypermorphic cell division mutants, namely, ftsZ* and ftsL*.
The ftsZ* hypermorph (FtsZL169R) was originally isolated as a suppressor of Kil, a phage lambda-encoded inhibitor of cell division (50). In addition to resisting the effects of Kil, ftsZ* efficiently resists the toxic effects of SulA, MinC, and excess FtsA, all of which block cell division efficiently in cells with wild-type FtsZ. FtsZ normally requires an interaction with both the ZipA and FtsA membrane-associated proteins to tether efficiently to the mid-cell membrane. The ftsZ* mutation bypasses the requirement for the ZipA protein (59). As FtsZ* polymers bundle much more strongly in vitro than those of wild-type FtsZ, the current model is that strong lateral interactions between FtsZ polymers create a more robust Z ring that resists FtsZ inhibitors and, possibly through FtsA, may be able to activate downstream biosynthesis of septal peptidoglycan independently of normal checkpoint controls (60). Cells with ftsZ* in place of ftsZ at the native locus can grow and divide fairly normally, although they often exhibit spiral FtsZ morphologies and abnormal cell shapes (59).
The ftsL* allele, which encodes a point mutation (E88K) in the periplasmic segment of FtsL, is a strong hypermorph that accelerates cell division and, like ftsZ*, bypasses some of the normal regulatory controls for septal peptidoglycan synthesis (61, 62). As a result, cells with ftsL* are significantly shorter than normal and similar to cells overexpressing ftsQAZ.
To test the effects of Era647 with either the FtsZ* or FtsL* proteins present, we introduced pAW5, expressing pBAD-Era647, either into WM4915 carrying the chromosomal ftsZ* allele or into WM6326 carrying the chromosomal ftsL* allele. In the latter strain, the chromosomal ftsZ gene was fused to mNeonGreen (mNG) for visualization (63). After induction with arabinose to overexpress era647 in the presence of chromosomal ftsZ*, cell division remained normal. This result indicated that ftsZ* can effectively resist the cell division inhibition caused by Era647 (Fig. 7). In contrast, after induction with arabinose to overexpress era647 in the presence of chromosomal ftsL*, cells became filamentous, and the pattern of FtsZ-mNG in these filaments was identical to the overexpression of Era647 in wild-type cells by immunostaining or FtsZ-GFP fluorescence (Fig. 7). Therefore, we conclude that the FtsZ* protein prevents the Era647 inhibition of cell division, but the FtsL* hypermorph has no effect.
FIG 7.
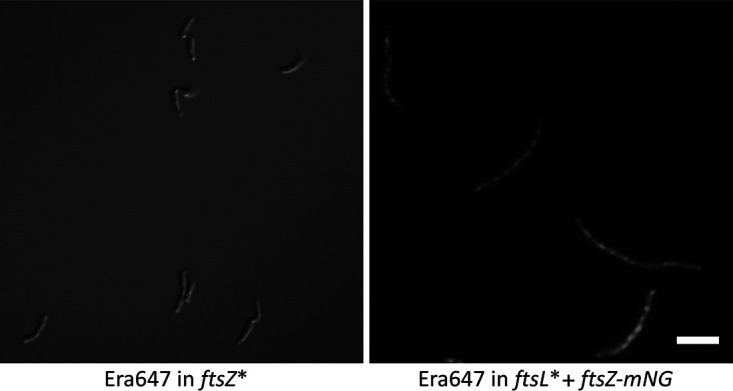
Effects of Era647 on ftsZ* and ftsL* hypermorphic mutants. Strains WM6529 and WM6530, carrying pAW5 (pBAD-Era647) in a chromosomal ftsZ* or ftsL* background, respectively, were grown in LB plus chloramphenicol at 37°C and induced with l-arabinose for 2 h. Cells were imaged live, by either DIC (left) or fluorescence using FtsZ-mNG (right). Scale bar, 5 μm.
Conclusions.
These results demonstrate that while excess Era or Era647 allows cell growth to continue, only excess Era647 blocks cell division and it does so by complete disruption of Z rings. The FtsZ staining patterns, along with the ability to suppress the division block by providing extra FtsZ, suggest that overproduced Era647, either directly or indirectly, prevents FtsZ monomers from forming normal polymers at the septal ring. FtsZ polymers undergo dynamic treadmilling with binding and coordinated assembly of the septal peptidoglycan layer at the mid-cell division site. During this process, FtsZ undergoes constant GTP hydrolysis and requires a large pool of monomers to maintain ring integrity. Our results show that high levels of Era647 not only prevent new Z rings from forming but also efficiently destabilize existing rings, consistent with a model in which Era647 sequesters FtsZ monomers by an indirect mechanism.
The ftsZ* allele, producing the FtsZ* mutant protein at native levels from the native locus, is able to suppress the Era647-mediated cell division block. This result suggests that high levels of FtsZ, while clearly effective in countering the Era647 effects, are not required if FtsZ polymers are able to make strong lateral interactions as FtsZ* is able to do. Moreover, the inability of the strong periplasmic ftsL* hypermorph to suppress the Era647 effect supports the idea that Era647 mainly exerts its effects on the cytoplasmic components of the cell division machinery. We speculate that if nonribosomal Era647 is in its GDP-bound form at the cytoplasmic membrane, then high local levels of Era647 might outcompete FtsZ for GTP exchange. This competition could affect the ability of membrane-associated FtsZ to bind GTP and maintain the stability of FtsZ polymers. How this might be the case for Era647 but not for wild-type Era and whether this is even plausible remain to be investigated.
Interestingly, it was recently discovered that a mutant variant of another highly conserved GTPase, ObgE (ObgE*), exerts a specific dominant negative effect on E. coli cell division when overproduced (64). However, the effects of ObgE* are different from Era647 in that the former induces production of cell chains, indicating a late-stage defect in cell division that probably involves periplasmic components (65), whereas here, we have shown that Era647 inhibits division at the early stage of Z ring formation in the cytoplasm. Despite these differences, such dominant negative variants of these and other essential GTPases should be useful tools in understanding how they function in coupling ribosome assembly with cell cycle control.
MATERIALS AND METHODS
Bacterial strains, phages, and plasmids.
The strains and plasmids used in this study are listed in Table S1 in the supplemental material. Cells were grown in lysogeny broth (LB) or on LB agar (66), unless otherwise indicated. Kanamycin was used at 30 μg/ml, chloramphenicol at 10 μg/ml, tetracycline at 12.5 μg/ml, and ampicillin at 30 μg/ml when the amp cassette is in single copy or at 100 μg/ml for plasmid selection.
Genetic manipulations.
Strain construction involved standard methods using P1 transduction to move markers from one strain to another (67) and recombineering to create point mutations, small substitutions, and gene knockouts (68, 69).
Creating the era647 mutation on a pBAD vector carrying era under arabinose promoter control.
The wild-type era gene was cloned into the expression vector pBAD30, generating plasmid pHKP60. To do this, the era gene from pCE31 (5) was removed on a BstXI to NruI fragment in which the BstXI 3′ overhang was removed in the presence of a T4 DNA polymerase. This fragment was ligated into the SmaI site of pBAD30 to place era under arabinose promoter control (70).
The era647 mutation was created in the cloned era+ gene on plasmid pHKP60 using the QuikChange method as described by Stratagene to generate pHKP67. The nucleotide codon sequence within era was changed from CAGGAGAAAGCCGATCTGCTGCCGCAC, encoding QEKADLLPH, to CAGttagtctatctgcaggggCAC, encoding QLVYLQGH (mutated bases in lowercase). Both the wild-type era gene and the era647 allele were also cloned into pBAD24, in which the bla gene was replaced with a cat cassette (pBAD24Cm), allowing higher-level induction by arabinose and chloramphenicol resistance. These plasmids were named pAW4 (era) and pAW5 (era647), respectively.
Placing the era647 mutation in the chromosomal rnc operon by recombineering.
The era647 mutation was crossed into the rnc operon by recombineering. An era DNA segment of 519 bp encompassing the era647 changes at the center was amplified by standard PCR using the pBADera647 plasmid (pHKP67) as the template. A W3110 derivative was used in which a ΔTn10 insertion (ΔTn10 A17) (3) was present near the rnc operon in the yfhB gene. The strain was transformed with the pSIM5 plasmid carrying the λ red genes to carry out the recombineering (71). After electroporation of the DNA, cells were grown for 4 h in 10 ml of LB at 32°C to allow recombination and complete segregation of any recombinant and wild-type alleles. A total of 100 μl of cells from 10−5 and 10−6 dilutions of this culture were plated onto LB plates and incubated at 32°C. Seven small colonies were identified among ∼150 normal sized colonies. Sequencing the entire gene was used to confirm the presence of the era647 mutation in all seven colonies. The era647 allele in the rnc operon was transferred to other strains with the linked yfhB::ΔTn10 A17 tetracycline resistance marker on the chromosome.
Placing the era alleles in the chromosome under the control of the λpL promoter.
The target gene cassettes era+ and era647 were amplified from pHKP60 and pHKP67 with PCR using a high-fidelity Taq DNA polymerase (Invitrogen, Carlsbad, CA) and inserted in the chromosome under the control of the λpL promoter using recombineering (see Fig. S2 for details). The era genes replaced a cat-sacB cassette by counter selection for sucrose-resistant recombinants placing the ATG start of era precisely at the ATG start of the λcIII gene, as described for previous gene replacements in this λ region (72). The final era constructs were confirmed by sequence analysis. Strains TUC540 and TUC05 are era+ and era647, respectively, under pL control.
Construction of an E. coli genome library and a screen for suppressors of era647.
Chromosomal DNA from MG1655 was partially digested with Sau3AI, DNA fragments of 2 to 6 kb were isolated from a gel and ligated into the BamHI site of pBR322, and a plasmid was prepared. Six 1-μl aliquots from the primary library were used to generate six transformations of TUC05, which were outgrown in LB for 3 hours at 32°C before plating onto 42°C prewarmed LB plates with ampicillin. Colonies from each were pooled and six plasmid minipreps were prepared, which were used to retransform TUC05 on ampicillin at 42°C as before. Twelve colonies (large and small) from the six pools of transformants were purified, plasmid preps were made from each, and the flanking regions of the insert were sequenced using primers in pBR322 on each side of the BamHI site to determine the genomic segment cloned. Different independent plasmids were used to transform TUC05 on LB ampicillin at 32°C, and the resultant colonies were tested at 42°C to verify suppression.
Constructing a sulA deletion.
A sulA<>cat gene replacement was generated in the TUC05 background (pL-era647) strain by recombineering. The recombinant carrying this deletion still exhibited the filamentation phenotype at 42°C. To confirm its phenotype, the sulA<>cat construct was moved to a lon mutant background. The Lon protease degrades SulA, returning SulA to low levels in the cell after DNA damage is repaired. In lon mutants, however, SulA levels remain high, blocking cell division, generating filaments, and causing cell death (73). The lon sulA<>cat mutant strain survives mitomycin treatment.
Microscopy.
The protocol for the examination of 4′,6-diamidino-2-phenylindole (DAPI)-stained cells for microscopy is a modified version of previously published methods (6, 74). Overnight cultures were diluted 1:100 in LB and grown to an optical density at 600 nm (OD600) of about 0.3 at 32°C. Part of the culture was kept at 32°C and the rest shifted to 42°C with shaking for 2 h. Cultures were centrifuged to pellet cells. The pellet was washed twice with 1 ml of 0.84% NaCl. The final pellet was suspended in 100 μl of 0.84% NaCl, 5 μl of the cells were spread onto a Poly-Prep slide (coated with poly-l-lysine, Sigma-Aldrich Inc., St. Louis, MO), and 5 μl 1% paraformaldehyde (in phosphate-buffered saline [PBS]) was added to fix the cells. After 5 min, the slide was washed once with tap water in a beaker and dried at room temperature. A total of 2 μl of DAPI (0.5 μg/ml in dH2O) and one drop of gel mount were added. A clean glass coverslip was pressed down to remove excess liquid. The prepared slide was refrigerated at 4°C for at least 15 min before observing with the microscope. Cells were visualized on a Nikon Eclipse E-1000 microscope equipped with a Nikon Plan fluor 100 objective as described previously (75). Images were acquired using Openlab 4.2 software.
Immunofluorescence microscopy was performed exactly as described previously (50), with DAPI to visualize nucleoids (blue), rhodamine-labeled wheat germ agglutinin to visualize cell wall (red), and goat anti-rabbit secondary antibody conjugated to Alexa Fluor 488 along with rabbit polyclonal anti-FtsZ to visualize FtsZ (green). Pseudocolored and merged images were generated using layers in Adobe Photoshop.
To visualize live cells by differential interference contrast (DIC) or fluorescence microscopy, cells were grown to the appropriate time points and either spotted directly on a glass microscope slide and covered with a coverslip or spotted on a thin layer of solidified 1% LB-agarose and covered with a coverslip. Fluorescence micrographs were acquired with an Olympus BX63 microscope with a Hamamatsu C11440 ORCA-Spark digital complementary metal oxide semiconductor (CMOS) camera. Image data were acquired and analyzed with cellSens software (Olympus).
Bacterial two-hybrid analysis.
To fuse Era or Era647 to the C terminus of the T18 domain of adenylyl cyclase in pUT18c, the respective genes were amplified from plasmids pAW4 or pAW5 by PCR using the forward primer aaatctagagAGCATCGATAAAAGTTACTGCGG (codon 2 and following era sequence in CAPS) and reverse primer aaaggaattcTTAAAGATCGTCAACGTAACCG (era sequence reverse complement, including stop codon in CAPS) and cloned using XbaI and EcoRI. To fuse Era or Era647 to the N terminus of T18, primers aaaagcttgAGCATCGATAAAAGTTACTGCGG (forward) and aaaggaattcccAAGATCGTCAACGTAACCG (reverse, no stop) were used similarly to clone as HindIII-EcoRI fragments into pUT18.
After their sequences were verified, the four resulting plasmids were transformed into the cya strain BTH101 carrying pKNT25-FtsZ, which produces a fusion between the C terminus of FtsZ and the T25 domain of adenylyl cyclase that has been shown previously to interact strongly with T18 fusions to the FtsA protein. Several independent transformants isolated from LB–ampicillin (Amp)-kanamycin (Kan) plates were then patched onto the same media supplemented with X-Gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside) and isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated overnight at 30°C to screen for blue or white colonies, which is indicative of positive or negative two-hybrid interactions, respectively.
Preparation of Era and Era647 proteins.
The Era protein was prepared as described previously (14). To prepare the Era647 protein, the pAW5 plasmid carrying era647 was grown overnight in 5-ml LB culture containing 100 μg/ml ampicillin, and the overnight grown culture was used to inoculate 500 ml of the same culture. The cells were grown with shaking at 200 rpm/min at 32°C until an OD600 value of 0.5 was reached. Then, the cells were induced by raising the temperature to 39°C and grown for another 2 h. The cells were harvested by centrifugation at 10,000 × g for 10 min. The cell pellet was stored at –80°C. The Era647 protein was purified as described for Era purification (14).
Supplementary Material
ACKNOWLEDGMENTS
We thank James Sawitzke, Lynn Thomason, and Carolyn Court for discussion and help with the manuscript and Anna Weaver for generating plasmids used in this work. We thank Joe Lutkenhaus for the gift of the plasmids carrying ftsQAZ genes and Harold Erickson for the gift of the chromosomal ftsZ-mNG strain.
This work was supported by National Institutes of Health grants GM61074 and GM131705 to W.M.; in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research; and by federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400, to D.L.C. and X.J.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the United States Government.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Ahnn J, March PE, Takiff HE, Inouye M. 1986. A GTP-binding protein of Escherichia coli has homology to yeast RAS proteins. Proc Natl Acad Sci U S A 83:8849–8853. doi: 10.1073/pnas.83.23.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inada T, Kawakami K, Chen SM, Takiff HE, Court DL, Nakamura Y. 1989. Temperature-sensitive lethal mutant of era, a G protein in Escherichia coli. J Bacteriol 171:5017–5024. doi: 10.1128/jb.171.9.5017-5024.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takiff HE, Chen SM, Court DL. 1989. Genetic analysis of the rnc operon of Escherichia coli. J Bacteriol 171:2581–2590. doi: 10.1128/jb.171.5.2581-2590.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardwell JC, Régnier P, Chen SM, Nakamura Y, Grunberg-Manago M, Court DL. 1989. Autoregulation of RNase III operon by mRNA processing. EMBO J 8:3401–3407. doi: 10.1002/j.1460-2075.1989.tb08504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SM, Takiff HE, Barber AM, Dubois GC, Bardwell JC, Court DL. 1990. Expression and characterization of RNase III and Era proteins. Products of the rnc operon of Escherichia coli. J Biol Chem 265:2888–2895. [PubMed] [Google Scholar]
- 6.Britton RA, Powell BS, Dasgupta S, Sun Q, Margolin W, Lupski JR, Court DL. 1998. Cell cycle arrest in Era GTPase mutants: a potential growth rate-regulated checkpoint in Escherichia coli. Mol Microbiol 27:739–750. doi: 10.1046/j.1365-2958.1998.00719.x. [DOI] [PubMed] [Google Scholar]
- 7.Britton RA, Chen SM, Wallis D, Koeuth T, Powell BS, Shaffer LG, Largaespada D, Jenkins NA, Copeland NG, Court DL, Lupski JR. 2000. Isolation and preliminary characterization of the human and mouse homologues of the bacterial cell cycle gene era. Genomics 67:78–82. doi: 10.1006/geno.2000.6243. [DOI] [PubMed] [Google Scholar]
- 8.Cummings L, Riley L, Black L, Souvorov A, Resenchuk S, Dondoshansky I, Tatusova T. 2002. Genomic BLAST: custom-defined virtual databases for complete and unfinished genomes. FEMS Microbiol Lett 216:133–138. doi: 10.1111/j.1574-6968.2002.tb11426.x. [DOI] [PubMed] [Google Scholar]
- 9.Dennerlein S, Rozanska A, Wydro M, Chrzanowska-Lightowlers ZMA, Lightowlers RN. 2010. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem J 430:551–558. doi: 10.1042/BJ20100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akiyama T, Gohda J, Shibata S, Nomura Y, Azuma S, Ohmori Y, Sugano S, Arai H, Yamamoto T, Inoue J. 2001. Mammalian homologue of E. coli Ras-like GTPase (ERA) is a possible apoptosis regulator with RNA binding activity. Genes Cells 6:987–1001. doi: 10.1046/j.1365-2443.2001.00480.x. [DOI] [PubMed] [Google Scholar]
- 11.Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D. 2010. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res 38:5554–5568. doi: 10.1093/nar/gkq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tu C, Zhou X, Tarasov SG, Tropea JE, Austin BP, Waugh DS, Court DL, Ji X. 2011. The Era GTPase recognizes the GAUCACCUCC sequence and binds helix 45 near the 3′ end of 16S rRNA. Proc Natl Acad Sci U S A 108:10156–10161. doi: 10.1073/pnas.1017679108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu C, Zhou X, Tropea JE, Austin BP, Waugh DS, Court DL, Ji X. 2009. Structure of ERA in complex with the 3′ end of 16S rRNA: implications for ribosome biogenesis. Proc Natl Acad Sci U S A 106:14843–14848. doi: 10.1073/pnas.0904032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Chen SM, Powell BS, Court DL, Ji X. 1999. Purification, characterization and crystallization of ERA, an essential GTPase from Escherichia coli. FEBS Lett 445:425–430. doi: 10.1016/s0014-5793(99)00178-7. [DOI] [PubMed] [Google Scholar]
- 15.Hang JQ, Meier TI, Zhao G. 2001. Analysis of the interaction of 16S rRNA and cytoplasmic membrane with the C-terminal part of the Streptococcus pneumoniae Era GTPase. Eur J Biochem 268:5570–5577. doi: 10.1046/j.1432-1033.2001.02493.x. [DOI] [PubMed] [Google Scholar]
- 16.Hang JQ, Zhao G. 2003. Characterization of the 16S rRNA- and membrane-binding domains of Streptococcus pneumoniae Era GTPase: structural and functional implications. Eur J Biochem 270:4164–4172. doi: 10.1046/j.1432-1033.2003.03813.x. [DOI] [PubMed] [Google Scholar]
- 17.Meier TI, Peery RB, Jaskunas SR, Zhao G. 1999. 16S rRNA is bound to era of Streptococcus pneumoniae. J Bacteriol 181:5242–5249. doi: 10.1128/JB.181.17.5242-5249.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meier TI, Peery RB, McAllister KA, Zhao G. 2000. Era GTPase of Escherichia coli: binding to 16S rRNA and modulation of GTPase activity by RNA and carbohydrates. Microbiology 146:1071–1083. doi: 10.1099/00221287-146-5-1071. [DOI] [PubMed] [Google Scholar]
- 19.Lu Q, Inouye M. 1998. The gene for 16S rRNA methyltransferase (ksgA) functions as a multicopy suppressor for a cold-sensitive mutant of era, an essential RAS-like GTP-binding protein in Escherichia coli. J Bacteriol 180:5243–5246. doi: 10.1128/JB.180.19.5243-5246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma MR, Barat C, Wilson DN, Booth TM, Kawazoe M, Hori-Takemoto C, Shirouzu M, Yokoyama S, Fucini P, Agrawal RK. 2005. Interaction of Era with the 30S ribosomal subunit implications for 30S subunit assembly. Mol Cell 18:319–329. doi: 10.1016/j.molcel.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 21.Sayed A, Matsuyama S, Inouye M. 1999. Era, an essential Escherichia coli small G-protein, binds to the 30S ribosomal subunit. Biochem Biophys Res Commun 264:51–54. doi: 10.1006/bbrc.1999.1471. [DOI] [PubMed] [Google Scholar]
- 22.Britton RA. 2009. Role of GTPases in bacterial ribosome assembly. Annu Rev Microbiol 63:155–176. doi: 10.1146/annurev.micro.091208.073225. [DOI] [PubMed] [Google Scholar]
- 23.Bunner AE, Nord S, Wikström PM, Williamson JR. 2010. The effect of ribosome assembly cofactors on in vitro 30S subunit reconstitution. J Mol Biol 398:1–7. doi: 10.1016/j.jmb.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosal A, Babu VMP, Walker GC. 2018. Elevated levels of Era GTPase improve growth, 16S rRNA processing, and 70S ribosome assembly of Escherichia coli lacking highly conserved multifunctional YbeY endoribonuclease. J Bacteriol 200:e00278-18. doi: 10.1128/JB.00278-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gollop N, March PE. 1991. A GTP-binding protein (Era) has an essential role in growth rate and cell cycle control in Escherichia coli. J Bacteriol 173:2265–2270. doi: 10.1128/jb.173.7.2265-2270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin YP, Sharer JD, March PE. 1994. GTPase-dependent signaling in bacteria: characterization of a membrane-binding site for era in Escherichia coli. J Bacteriol 176:44–49. doi: 10.1128/jb.176.1.44-49.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gollop N, March PE. 1991. Localization of the membrane binding sites of Era in Escherichia coli. Res Microbiol 142:301–307. doi: 10.1016/0923-2508(91)90045-c. [DOI] [PubMed] [Google Scholar]
- 28.Lerner CG, Gulati PS, Inouye M. 1995. Cold-sensitive conditional mutations in Era, an essential Escherichia coli GTPase, isolated by localized random polymerase chain reaction mutagenesis. FEMS Microbiol Lett 126:291–298. doi: 10.1111/j.1574-6968.1995.tb07432.x. [DOI] [PubMed] [Google Scholar]
- 29.Pillutla RC, Sharer JD, Gulati PS, Wu E, Yamashita Y, Lerner CG, Inouye M, March PE. 1995. Cross-species complementation of the indispensable Escherichia coli era gene highlights amino acid regions essential for activity. J Bacteriol 177:2194–2196. doi: 10.1128/jb.177.8.2194-2196.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Britton RA, Powell BS, Court DL, Lupski JR. 1997. Characterization of mutations affecting the Escherichia coli essential GTPase era that suppress two temperature-sensitive dnaG alleles. J Bacteriol 179:4575–4582. doi: 10.1128/jb.179.14.4575-4582.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morimoto T, Loh PC, Hirai T, Asai K, Kobayashi K, Moriya S, Ogasawara N. 2002. Six GTP-binding proteins of the Era/Obg family are essential for cell growth in Bacillus subtilis. Microbiology 148:3539–3552. doi: 10.1099/00221287-148-11-3539. [DOI] [PubMed] [Google Scholar]
- 32.Friedman DI, Court DL. 1995. Transcription antitermination: the lambda paradigm updated. Mol Microbiol 18:191–200. doi: 10.1111/j.1365-2958.1995.mmi_18020191.x. [DOI] [PubMed] [Google Scholar]
- 33.Luo X, Hsiao H-H, Bubunenko M, Weber G, Court DL, Gottesman ME, Urlaub H, Wahl MC. 2008. Structural and functional analysis of the E. coli NusB-S10 transcription antitermination complex. Mol Cell 32:791–802. doi: 10.1016/j.molcel.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pichoff S, Alibaud L, Guédant A, Castanié MP, Bouché JP. 1998. An Escherichia coli gene (yaeO) suppresses temperature-sensitive mutations in essential genes by modulating Rho-dependent transcription termination. Mol Microbiol 29:859–869. doi: 10.1046/j.1365-2958.1998.00981.x. [DOI] [PubMed] [Google Scholar]
- 35.Adhya S, Gottesman M, Court D. 1977. Independence of gene N and tof functions of bacteriophage lambda. J Mol Biol 112:657–660. doi: 10.1016/s0022-2836(77)80171-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang XD, de Boer PA, Rothfield LI. 1991. A factor that positively regulates cell division by activating transcription of the major cluster of essential cell division genes of Escherichia coli. EMBO J 10:3363–3372. doi: 10.1002/j.1460-2075.1991.tb04900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pichoff S, Lutkenhaus J. 2001. Escherichia coli division inhibitor MinCD blocks septation by preventing Z-ring formation. J Bacteriol 183:6630–6635. doi: 10.1128/JB.183.22.6630-6635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haeusser DP, Margolin W. 2016. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol 14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Boer P, Crossley R, Rothfield L. 1992. The essential bacterial cell-division protein FtsZ is a GTPase. Nature 359:254–256. doi: 10.1038/359254a0. [DOI] [PubMed] [Google Scholar]
- 40.Raychaudhuri D, Park JT. 1992. Escherichia coli cell-division gene ftsZ encodes a novel GTP-binding protein. Nature 359:251–254. doi: 10.1038/359251a0. [DOI] [PubMed] [Google Scholar]
- 41.Mukherjee A, Dai K, Lutkenhaus J. 1993. Escherichia coli cell division protein FtsZ is a guanine nucleotide binding protein. Proc Natl Acad Sci U S A 90:1053–1057. doi: 10.1073/pnas.90.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bramhill D, Thompson CM. 1994. GTP-dependent polymerization of Escherichia coli FtsZ protein to form tubules. Proc Natl Acad Sci U S A 91:5813–5817. doi: 10.1073/pnas.91.13.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bi E, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 44.Addinall SG, Bi E, Lutkenhaus J. 1996. FtsZ ring formation in fts mutants. J Bacteriol 178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Margolin W. 2000. Themes and variations in prokaryotic cell division. FEMS Microbiol Rev 24:531–548. doi: 10.1111/j.1574-6976.2000.tb00554.x. [DOI] [PubMed] [Google Scholar]
- 46.Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH, Petroni G. 2008. Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J Bacteriol 190:3192–3202. doi: 10.1128/JB.01797-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma X, Ehrhardt DW, Margolin W. 1996. Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc Natl Acad Sci U S A 93:12998–13003. doi: 10.1073/pnas.93.23.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Yehuda S, Losick R. 2002. Asymmetric cell division in B. subtilis involves a spiral-like intermediate of the cytokinetic protein FtsZ. Cell 109:257–266. doi: 10.1016/S0092-8674(02)00698-0. [DOI] [PubMed] [Google Scholar]
- 50.Haeusser DP, Hoashi M, Weaver A, Brown N, Pan J, Sawitzke JA, Thomason LC, Court DL, Margolin W. 2014. The Kil peptide of bacteriophage lambda blocks Escherichia coli cytokinesis via ZipA-dependent inhibition of FtsZ assembly. PLoS Genet 10:e1004217. doi: 10.1371/journal.pgen.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, Milam SL, Erickson HP. 2012. SulA inhibits assembly of FtsZ by a simple sequestration mechanism. Biochemistry 51:3100–3109. doi: 10.1021/bi201669d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bisson-Filho AW, Discola KF, Castellen P, Blasios V, Martins A, Sforca ML, Garcia W, Zeri ACM, Erickson HP, Dessen A, Gueiros-Filho FJ. 2015. FtsZ filament capping by MciZ, a developmental regulator of bacterial division. Proc Natl Acad Sci U S A 112:E2130–2138. doi: 10.1073/pnas.1414242112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huisman O, D'Ari R, Gottesman S. 1984. Cell division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proc Natl Acad Sci U S A 81:4490–4494. doi: 10.1073/pnas.81.14.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cordell SC, Robinson EJ, Löwe J. 2003. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc Natl Acad Sci U S A 100:7889–7894. doi: 10.1073/pnas.1330742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dajkovic A, Mukherjee A, Lutkenhaus J. 2008. Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J Bacteriol 190:2513–2526. doi: 10.1128/JB.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shiomi D, Margolin W. 2007. Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Mol Microbiol 66:1396–1415. doi: 10.1111/j.1365-2958.2007.05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haeusser DP, Rowlett VW, Margolin W. 2015. A mutation in Escherichia coli ftsZ bypasses the requirement for the essential division gene zipA and confers resistance to FtsZ assembly inhibitors by stabilizing protofilament bundling. Mol Microbiol 97:988–1005. doi: 10.1111/mmi.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krupka M, Margolin W. 2018. Unite to divide: oligomerization of tubulin and actin homologs regulates initiation of bacterial cell division. F1000Res 7:235. doi: 10.12688/f1000research.13504.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu B, Persons L, Lee L, de Boer PA. 2015. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol Microbiol 95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsang M-J, Bernhardt TG. 2015. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol Microbiol 95:925–944. doi: 10.1111/mmi.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore DA, Whatley ZN, Joshi CP, Osawa M, Erickson HP. 2017. Probing for binding regions of the FtsZ protein surface through site-directed insertions: discovery of fully functional FtsZ-fluorescent proteins. J Bacteriol 199:e00553-16. doi: 10.1128/JB.00553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dewachter L, Verstraeten N, Monteyne D, Kint CI, Versées W, Pérez-Morga D, Michiels J, Fauvart M. 2015. A single-amino-acid substitution in Obg activates a new programmed cell death pathway in Escherichia coli. mBio 6:e01935-15. doi: 10.1128/mBio.01935-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dewachter L, Verstraeten N, Jennes M, Verbeelen T, Biboy J, Monteyne D, Pérez-Morga D, Verstrepen KJ, Vollmer W, Fauvart M, Michiels J. 2017. A mutant isoform of ObgE causes cell death by interfering with cell division. Front Microbiol 8:1193. doi: 10.3389/fmicb.2017.01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bertani G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186:595–600. doi: 10.1128/jb.186.3.595-600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomason LC, Costantino N, Court DL. 2007. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol Chapter 1:Unit 1.17. [DOI] [PubMed] [Google Scholar]
- 68.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. 2007. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol 421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- 69.Sawitzke JA, Costantino N, Li X-T, Thomason LC, Bubunenko M, Court C, Court DL. 2011. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Datta S, Costantino N, Court DL. 2006. A set of recombineering plasmids for gram-negative bacteria. Gene 379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 72.Datta S, Costantino N, Zhou X, Court DL. 2008. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc Natl Acad Sci U S A 105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mizusawa S, Gottesman S. 1983. Protein degradation in Escherichia coli: the lon gene controls the stability of SulA protein. Proc Natl Acad Sci U S A 80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hiraga S, Niki H, Ogura T, Ichinose C, Mori H, Ezaki B, Jaffé A. 1989. Chromosome partitioning in Escherichia coli: novel mutants producing anucleate cells. J Bacteriol 171:1496–1505. doi: 10.1128/jb.171.3.1496-1505.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nielsen HJ, Ottesen JR, Youngren B, Austin SJ, Hansen FG. 2006. The Escherichia coli chromosome is organized with the left and right chromosome arms in separate cell halves. Mol Microbiol 62:331–338. doi: 10.1111/j.1365-2958.2006.05346.x. [DOI] [PubMed] [Google Scholar]
- 76.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in Escherichia coli. EMBO J 21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Araujo-Bazan L, Ruiz-Avila LB, Andreu D, Huecas S, Andreu JM. 2016. Cytological profile of antibacterial FtsZ inhibitors and synthetic peptide MciZ. Front Microbiol 7:1558. doi: 10.3389/fmicb.2016.01558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.