

Abstract
Purpose:
For refractory/relapsed Hodgkin lymphoma (HL) patients (roughly 20% of total cases), few effective therapeutic options exist. Currently, Brentuximab Vedotin (BV), a drug-conjugated anti-CD30 antibody, is one of the most effective approved therapy agents for these patients. However, many patients do not achieve complete remission and ultimately develop BV-resistant disease, –necessitating a more detailed understanding of the molecular circuitry that drives BV sensitivity and the mechanism of BV resistance.
Experimental Design:
Here, we established a ubiquitin regulator-focused CRISPR library screening platform in HL and carried out a drug sensitization screen against BV to identify genes regulating BV treatment sensitivity.
Results:
Our CRISPR library screens revealed the ubiquitin-editing enzymes A20 and RBX1 as key molecule effectors that regulate BV sensitivity in HL line L428. A20 negatively regulates NF-κB activity which is required to prevent BV cytotoxicity. In line with these results, the RNA-seq analysis of the BV-resistant single cell clones demonstrated a consistent upregulation of NF-κB signature genes, as well as the ABC transporter gene ABCB1. Mechanically, NF-κB regulates BV treatment sensitivity through mediating ABCB1 expression. Targeting NF-κB activity synergized well with BV in killing HL cell lines, augmented BV sensitivity and overcame BV resistance in vitro and in HL xenograft mouse models.
Conclusions:
Thus, our identification of this previously unrecognized mechanism provides novel knowledge of possible BV responsiveness and resistance mechanism in HL, as well as leads to promising hypothesis for the development of therapeutic strategies to overcome BV resistance in this disease.
Introduction
Hodgkin lymphoma (HL) is one of the most common cancers in young adulthood (1), representing about 1 in 6 of all cancers affecting young people between 15 and 24 years old. Although there has been great progress in treating HL over the last few decades, the survival rate for patients diagnosed at an advanced stage or with relapsed/refractory disease remains low, especially for elderly patients who do not tolerate intensive treatment (2). For these patients, few effective therapeutic options exist. In the last 30 years, only three drugs have been approved for relapsed HL: the PD-1 blockage nivolumab and pembrolizumab, as well as brentuximab vedotin (BV), a drug-conjugated anti-CD30 antibody.
The malignant component of classical HL (cHL) tumors, the Hodgkin and Reed/Sternberg (HRS) cells, have high surface expression of several TNF receptor family members, including CD30, CD40, RANK, TACI and BCMA (3–6). This unique characteristic property of this disease has been translated into the development and approval of the first effective targeted therapy for relapsed/refractory HL, BV, an anti-CD30 antibody-drug conjugate that specifically delivers a cytotoxic agent, monomethyl auristatin E (MMAE), to cells expressing surface CD30. BV was approved by the FDA first in 2011 for relapsed or refractory HL after an autologous stem cell transplantation (ASCT) or following 2 prior lines of multiagent chemotherapy; then was approved as part of the initial, front-line treatment of advanced stage HL in 2018. Although BV elicits a high response rate (75%) in HL, patients who do not achieve a complete response (CR) will eventually develop resistance to BV, and progressive disease despite active treatment (7,8). Therefore, the optimal clinical development of BV will likely depend upon its pairing with other agents to increase response rates and durability, as well as to circumvent potential resistance mechanisms. Downregulation of surface CD30 expression was found only in a small portion of BV resistant HL cases (9), hence the question of BV resistance mechanisms remains open.
Protein ubiquitination is an essential and ubiquitous mechanism used by eukaryotic cells to regulate responses to multiple cellular stimuli and stresses (10). In general, protein ubiquitination proceeds by a three-step cascade mechanism (11), which initiates with an ATP-dependent ubiquitin activation by an E1 (ubiquitin-activating enzyme), followed by the transfer of an activated ubiquitin to a cysteine residue within the E2 (ubiquitin-conjugating enzyme), and ends with the conjugation of ubiquitin to a target protein through the activity of a ubiquitin-protein ligase (E3). Like protein phosphorylation, ubiquitination is a reversible process mediated by specific proteases named deubiquitinating enzyme (DUB). The human genome encodes two ubiquitin E1 enzymes, 38 E2 enzymes, more than 600 ubiquitin E3 ligases, and roughly 100 DUBs. Recently, many studies have revealed the critical functions of the protein ubiquitination system in multiple steps of human lymphoid malignancies, particularly in HL (12). Therefore, it is likely that the protein ubiquitination pathway might impact HL responsiveness to BV and contribute to BV resistance; however, this possibility has not been examined. Therefore, it will be important to gain a complete understanding of how the ubiquitin modifying machinery regulates BV treatment sensitivity of HL in order to identify and exploit critical therapeutic vulnerabilities.
The newly established RNA-guided CRISPR–associated nuclease Cas9 provides a next-generation approach for genome-scale functional screening (13,14). We have successfully established this screening platform, and have utilized this powerful approach to identify novel therapeutic targets in a very recent study (15). Therefore, we decided to use the CRISPR library screening technology to address the gaps in knowledge and to gain a complete understanding of how the ubiquitin modifying machinery regulates HL responsiveness to BV and BV resistance.
Materials and Methods
(See Supplemental Experimental Procedures for details)
Cell line Authentication (See Supplemental Experimental Procedures for details).
All the cell lines used in this study, including all the parental and the engineered cell lines (Cas9 single cell clones and BV-resistant single cell clones) have been authenticated and verified by the STR analysis (DNA fingerprinting) method described in (16).
Ubiquitin regulator-focused sgRNA library.
A 10-sgRNA-per-gene CRISPR–Cas9 deletion library was designed to target ~1300 genes in the human genome focused on ubiquitin regulators, including all E3 ligases, deubiquitinating enzymes (DUB), and immune cell signaling components as controls. The library contained 100 negative control sgRNAs: non-targeting control sgRNA with no binding sites in the genome.
BV sensitization CRISPR library screen (see Supplemental Experimental Procedures for details).
In brief, 40 million cells were infected with the pooled lentiviral ubiquitin regulators-focused sgRNA library. Doxycycline was then added to induce sgRNAs expression. Library transduced and induced cells were treated with an IC50 dosage of BV for 6 days or left untreated. Genomic DNA was extracted and sgRNA sequences were amplified by two rounds of PCR. The resulting libraries were sequenced with single end read with dual-index 75 bp.
RNA sequencing.
For RNA sequencing analysis, the total RNA was extracted from the BV-resistant single cell clones and parental controls using the RNeasy Kit (Qiagen). The bulk RNA-seq libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB). The quality-ensured RNA-seq libraries were also pooled and sequenced on the Illumina HiSeq 2500 platform with 100 or 150 bp paired-end mode (sequenced by Novogene).
For RNA-seq data processing, raw sequence reads were aligned to the human genome (hg38) using the Tophat algorithm (17); the Cufflinks algorithm (18) was implemented to assemble transcripts and estimate their abundance. Cuffdiff (19) was used to statistically assess expression changes in quantified genes in different conditions.
Data access.
The high-throughput RNA sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA) under accession number: SUB6466040
Tumor Model and Therapy Study (see Supplemental Experimental Procedures for details).
For xenograft tumor model, the human Hodgkin lymphoma L428 or KMH2 cells were subcutaneously (s.c.) injected into the flanks of female NSG mice. All animal experiments were approved by the National Cancer Institute Animal Care and Use Committee (NCI ACUC) and Fox Chase Cancer Center Animal Care and Use Committee (FCCC ACUC), and were performed in accordance with ACUC guidelines.
Statistical analysis.
All experiments have been repeated and results reproduced. Where possible, error bars or P values are shown to indicate statistical significance. In some figures, error bars are not visible due to their short heights relative to the size of the symbols. P < 0.05 was considered statistically significant.
Results
Using drug sensitization CRISPR library screen to identify genes regulating BV sensitivity
To gain a complete understanding of how the ubiquitin modifying machinery regulates HL responsiveness to BV, we produced a unique ubiquitin regulator-focused sgRNA library for use in genetic screens. This library contains 10 sgRNAs per target that are directed at 5′ constitutive exons of ~800 human genes, including all E3 ligases, deubiquitinating enzymes (DUB), and controls. A drug sensitization CRIPSR screen was performed in the HL line L428, as depicted in Fig. 1A. Briefly, Cas9 inducible L428 cells was transduced with a pool of lentivirus from this library, selected and induced, and then kept untreated or treated with a submaximal lethal dose of BV such that roughly 50% of the cells were killed after 6 days. The sgRNA abundance in both the treated and untreated populations was determined by sequencing, and the relative counts of sgRNAs in each population was calculated and plotted (Supplemental Fig. 1A). This allowed us to identify sgRNAs that increased or decreased cell death in the presence of BV but to ignore sgRNAs that were toxic in a manner that was BV-independent. While the majority of sgRNAs counted roughly evenly in BV+ vs BV− populations, we have identified a set of sgRNAs that significantly promoted resistance to BV cytotoxicity or augmented BV sensitivity (Supplemental Fig. 1A).
Figure 1: BV sensitization CRISPR library screen in HL line.
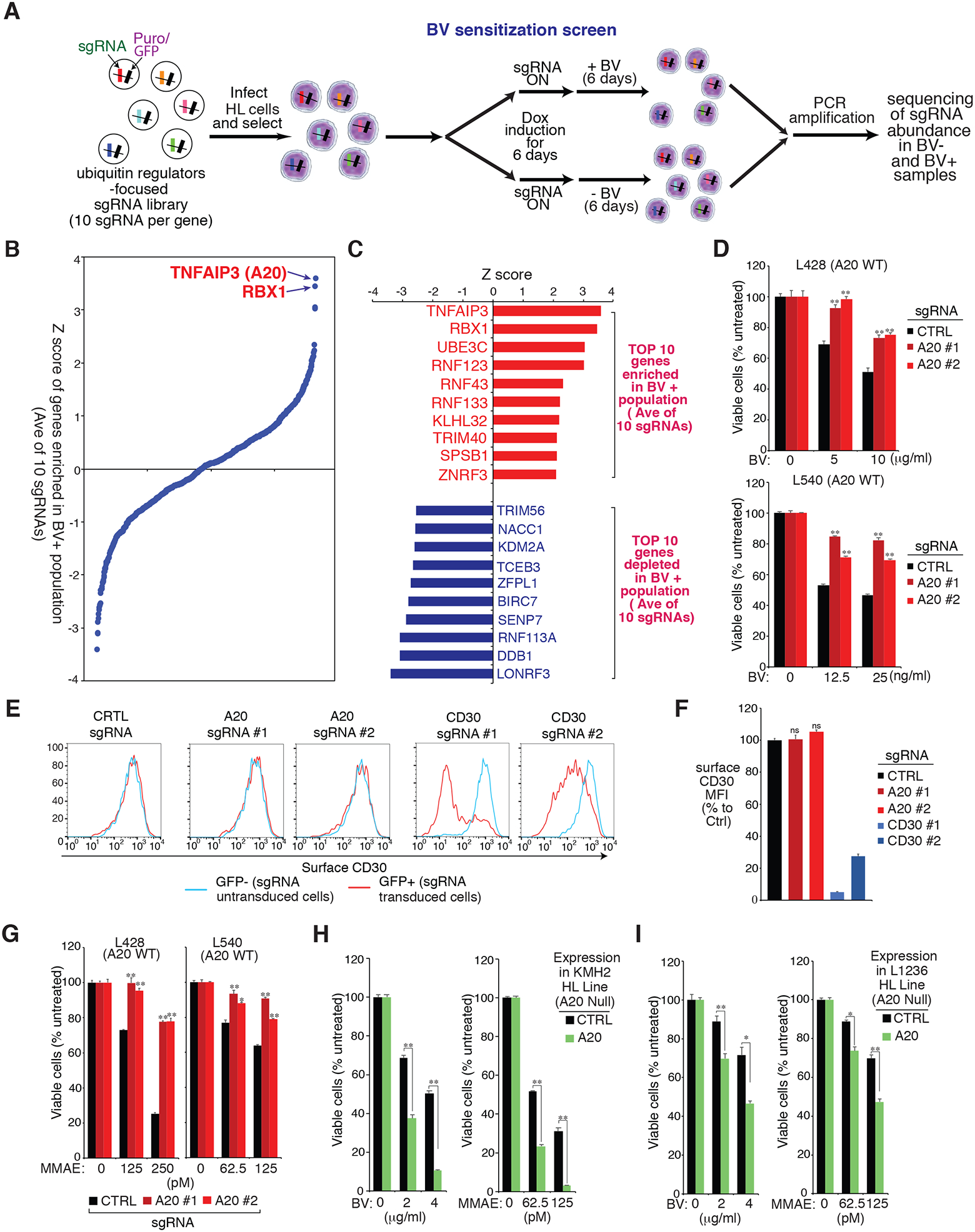
A. Outline of the workflow of the BV sensitization CRISPR library screen in HL line. B. Shown are the ranking of all the genes (average of 10 sgRNAs of each gene) enriched in the treated (BV+) population of the L428 line. Y axis indicates the distribution of standardized enrichment scores (Z-scores) of each gene enrichment. C. List of top 10 genes enriched (upper) or depleted (lower) in the treated (BV+) of L428 line. Y axis indicates the distribution of standardized enrichment scores (Z-scores) of each gene enrichment. D. HL cell lines L428 and L540 were transduced with A20 or Ctrl sgRNAs, selected and expression induced, then treated with BV at the indicated concentrations for 4 days. Viability was measured by the MTS assay and normalized to PBS-treated cells. E. HL cell line L428 was transduced with A20, CD30 or Ctrl sgRNAs along with GFP, and sgRNAs expressions were induced for 4 days. Surface CD30 expression in uninfected (GFP−) cells and sgRNA infected (GFP+) cells was measured by flow cytometry. One of the representative experiments is shown. F. As in the E. the summary of three independent experiments are shown. The relative CD30 MFI was normalized to the uninfected (GFP−) cells. G. The HL cell lines L428 and L540 were transduced with A20 or Ctrl sgRNAs, selected and expression induced, then treated with MMAE at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to DMSO-treated cells. H and I. A20 deficient HL cell lines KMH2 (H) and L1236 (I) were transduced with A20 cDNA or an empty control, selected and expression induced, then treated with BV or MMAE at the indicated concentrations for 4 days. Viability was measured by an MTS assay and normalized to PBS-treated or DMSO-treated cells. For all panels, error bars denote SEM of triplicates. P were calculated comparing Ctrl and A20 sgRNA transduced groups, or Ctrl and A20 expression groups; * indicates P < 0.05; ** indicates P < 0.01; n.s indicates no statistical difference.
We then plotted the screen results for all the ubiquitin regulator genes in the library, ranked by enrichment in the treated (BV+) populations (average of 10 sgRNAs), and analyzed all the molecular effectors responsible for BV sensitivity in the L428 line (Fig. 1B and 1C). Genes whose loss augmented BV sensitivity in HL were several Ring Finger E3 enzymes including LONRF3, RNF113A and BIRC7, as well as Cullin 4 (CUL4) ubiquitin E3 ligase complex adaptor protein DDB1. However, since we are seeking to understand the common mechanism of BV resistance, we are more focused on the genes whose loss promoted resistance to BV cytotoxicity in HL cell lines. Notably, TNFAIP3 (A20), an ubiquitin-editing enzyme, is the top gene in our list whose loss promoted resistance to BV, followed by RBX1 (RING box protein 1) (Fig. 1B and 1C). Remarkably, inactivation of A20 by nonsense/deletions or missense mutations is the most recurrent genetic alteration, occurring in 30–40% of HL cases (20,21), especially in EBV-negative HL (around 70–80%). RBX1 is an essential component of SCF (Skp1/Cullins/F-box) E3 ubiquitin ligases complex, which target diverse proteins for proteasome-mediated degradation. Therefore, our drug sensitization CRISPR library screen identified A20 and RBX1 as essential genes regulating BV treatment sensitivity, which provides insight into the basis for BV resistance.
A20 regulates BV sensitivity of HL in a CD30 independent manner
Our unbiased BV sensitization CRISPR library screen (Fig. 1A–C) revealed the ubiquitin-editing enzyme A20 as a novel regulator of BV sensitivity in HL cell lines. To verify the CRISPR library results, we designed sgRNAs targeting A20 and the positive control CD30, and tested their ability to inhibit BV treatment sensitivity in L428 cells (Fig. 1D upper, Supplemental Fig. 1B, and Supplemental Fig. 1C). As expected, CD30 depletion largely decreased BV sensitivity compared to that in control cells. Importantly, the tumor cells depleted of A20 were resistant to BV treatment in a similar degree as with CD30 depletion (Fig. 1D upper), verifying the CRISPR library results. Similarly, depletion of A20 in another A20 WT HL line L540 also decreased BV sensitivity (Fig. 1D lower and Supplemental Fig. 1C right).
Since BV specifically targets cells expressing surface CD30, we then examined if A20 deletion impacts surface CD30 expression in HL cell line. While the control CD30 sgRNAs largely reduced surface CD30 expression, two individual A20 sgRNAs both had no effect (Fig. 1E and 1F), indicating that A20 regulates BV sensitivity of HL likely not through CD30. Indeed, the A20 depleted HL L428 and L540 cells were much more resistant to MMAE, the cytotoxic agent conjugated to anti-CD30 antibody in BV (Fig. 1G). MMAE can readily enter cells via passive diffusion (22), therefore A20 is likely to mediate certain cellular pathways required for controlling MMAE cytotoxicity in HL. In line with the depletion experiments, reconstitution of WT A20 into the A20-deficient HL line KMH2 and L1236 remarkably increased the sensitivity to both BV and MMAE treatment (Fig. 1H, Fig. 1I, and Supplemental Fig. 1D). Thus, we conclude that A20 promotes BV sensitivity in HL cell lines, likely through its ability to mediate cellular pathways required for directing MMAE cytotoxicity.
A20 regulates BV sensitivity of HL through NF-κB
A20 is an NF-κB target gene, but its induction antagonizes NF-κB activation through the so called “dual ubiquitin-modifying/editing capacity” feature (23). In HL, reconstitution of WT A20 into A20-deficient HL cell lines revealed a significant decrease in NF-κB activity(21). Consistent with previous observations, depletion of A20 in L428 cells promoted NF-κB, as indicated by NF-κB target genes CD83 and IL-6 expression, as well as by p-p65 intracellular level (Fig. 2A). Therefore, loss of A20 revises BV sensitivity in HL cell lines that is likely due to its capability to regulate NF-κB activity.
Figure 2: A20 regulates BV sensitivity through NF-κB.
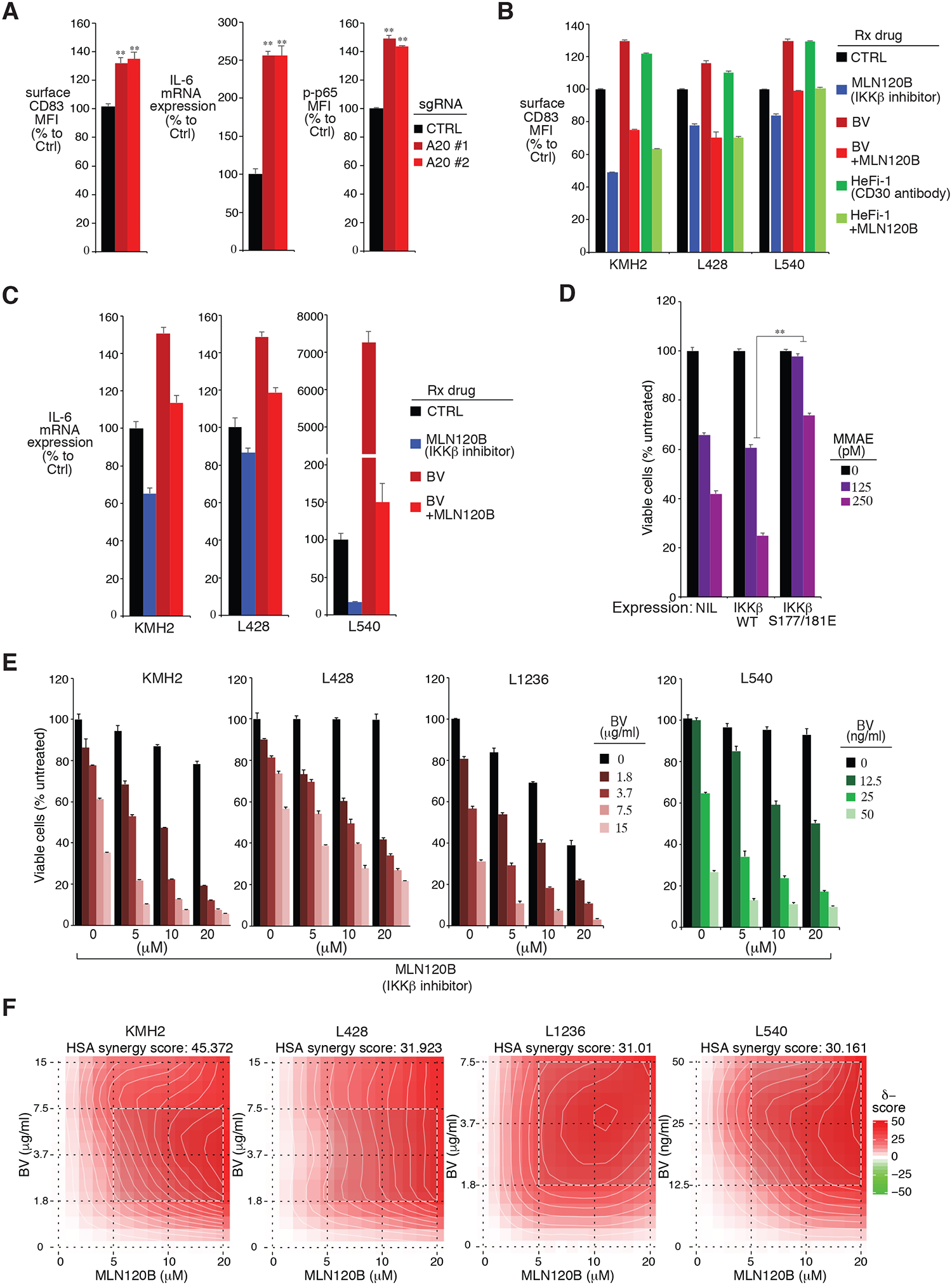
A. The HL cell line L428 was transduced with A20 or Ctrl sgRNAs, selected and expression induced. Surface CD83 and intracellular p-p65 expression were measured by flow cytometry. The relative CD83 and p-p65 MFI were normalized to the Ctrl sgRNA transduced cells. IL-6 gene expression was measured by real-time PCR and normalized to the Ctrl sgRNA transduced cells. Error bars denote SEM of triplicates. P were calculated comparing Ctrl and A20 sgRNA transduced groups; ** indicates P < 0.01. B. HL cell lines KMH2, L428 and L540 were treated with the indicated drugs at the following concentrations: KMH2 and L428 (MLN120B (80μM), BV (50μg/ml), HeFi-1 (50μg/ml), or the combination); L540 (MLN120B (20μM), BV (10μg/ml), HeFi-1 (10μg/ml), or the combination); for 24 hours. Surface CD83 expression was measured by flow cytometry. The relative CD83 MFI was normalized to the untreated cells. Error bars denote SEM of triplicates. C. same as B. IL-6 gene expression was measured by real-time PCR and normalized to the untreated cells. Error bars denote SEM of triplicates. D. The HL cell line L428 was transduced with IKKβ WT, IKKβ S177/181E or empty control, selected and expression induced, then treated with MMAE at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to DMSO-treated cells. Error bars denote SEM of triplicates. P were calculated comparing IKKβ WT and IKKβ S177/181E groups. ** indicates P < 0.01. E. Viability of HL cell lines KMH2, L428, L1236 and L540 after treatment (4 days) with the indicated concentrations of BV, MLN120B, or both. Data are normalized to DMSO-treated cells. Error bars denote SEM of triplicates. F. Formal calculation of synergism between BV and MLN120B. The raw cell viability data from E was entered into the SynergyFinder web application described in (30). The resulting contour plots; the HSA synergy score are shown. Positive HSA synergy score values indicate synergy.
Similar to other TNF superfamily members, CD30 signals lead to strong NF-κB activation, mediated by interactions with TNFR-associated factor (TRAF) 2 and 5 (24–28). To assess if BV treatment induces NF-κB activity in HL cells, we examined surface CD83 expression in a panel of HL cell lines. BV stimulation increased surface CD83 expression in all HL lines tested, so did HeFi-1, a pure murine IgG1 recognizing the ligand-binding site on human CD30 (29) (Fig. 2B). Importantly, a highly specific IKKβ inhibitor MLN120B completely blocked BV or HeFi-1 induced CD83 expression, indicating that this is a specific NF-κB response. Similar results could be achieved when we examined the IL-6 expression in the same settings (Fig. 2C). Thus, NF-κB upregulation is commonly acquired upon BV treatment, which might impact HL cells responsiveness to BV and determine the BV sensitivity. Consistent with this notion, ectopic expression of a constitutively active IKKβ mutant into L428 cells largely mitigated the cytotoxic effect of MMAE, but the IKKβ WT failed to do so (Fig. 2D and Supplemental Fig. 2A).
The results in Fig. 3B–D suggest that blocking NF-κB activity represents an effective way to promote BV sensitivity in HL cell lines. To test this concept, we treated a panel of HL cells lines in vitro with BV in combination with the IKKβ inhibitor MLN120B (Fig. 2E). In all four HL lines we tested, tumor cells were killed more efficiently with the combination of BV and MLN120B than with either drug alone. Importantly, these combinational effects appear to be synergistic, which was assessed using the mathematical algorithm described by Ianevski et al (30) (Fig. 2F). Hence, combination of BV with agents targeting NF-κB activity synergistically killed HL cell lines in vitro, supporting the in vivo evaluation of this treatment regimen.
Figure 3: Analysis of BV-resistant single cell clones of HL line.
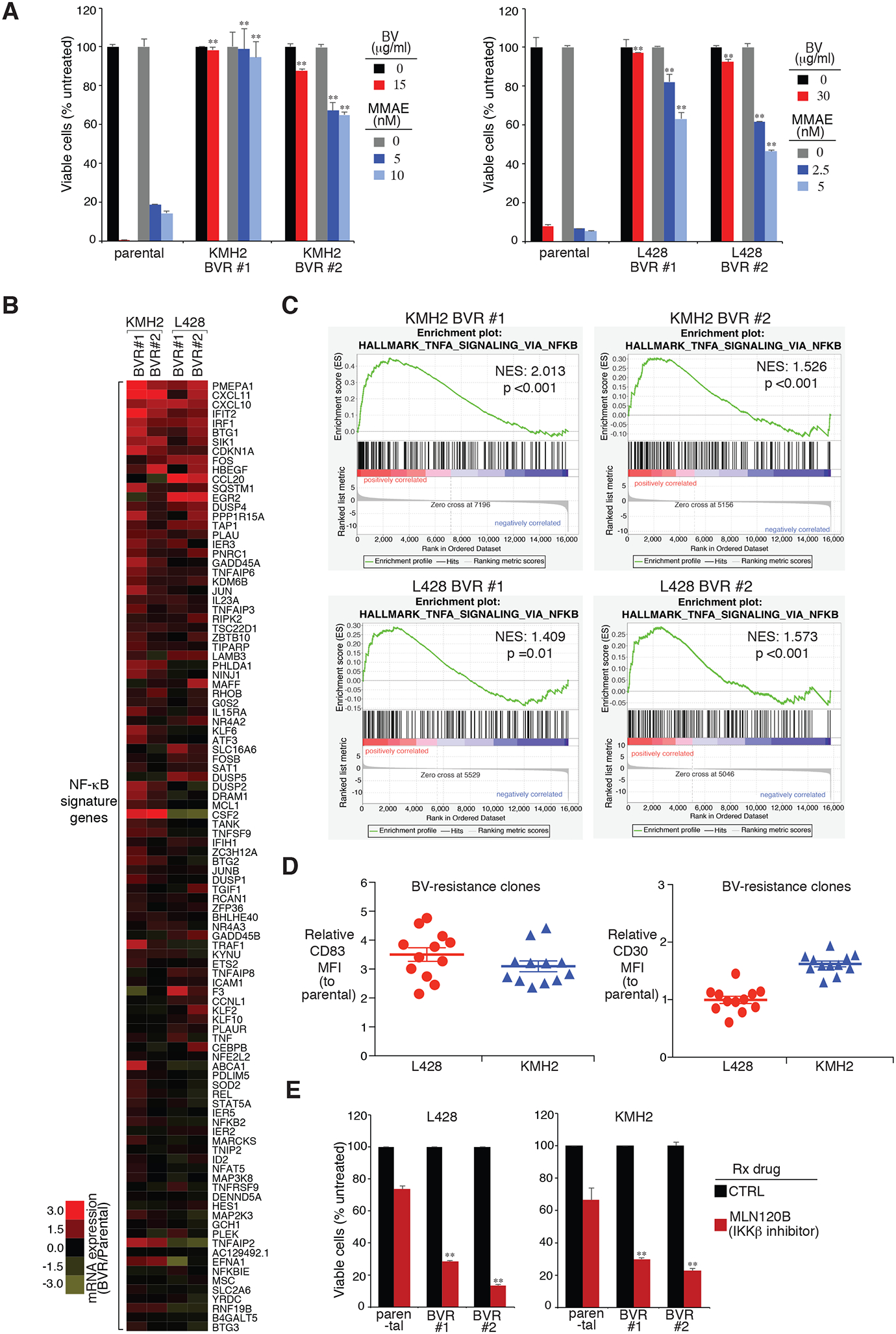
A. Two individual BV-resistant single cell clones of KMH2 and L428 HL lines, as well as their parental controls, were treated with BV or MMAE at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to PBS-treated or DMSO-treated cells. Error bars denote SEM of triplicates. P were calculated comparing BV-resistant clones and its parental controls under the same treatment. ** indicates P < 0.01. B. Heatmap of mRNA induction of NF-κB signature genes in the indicated BV-resistant single cell clones of KMH2 and L428 HL lines, relative to their parental controls. C. GSEA of NF-κB signatures. The analysis was based on BV-resistant clones vs. parental controls in KMH2 and L428 lines. Normalized enrichment score (NES) and normalized (NOM) P value are shown. D. Surface CD83 and CD30 expression were measured in BV-resistant single cell clones of KMH2 and L428 HL lines, as well as their parental controls, by flow cytometry. The relative CD83 MFI was normalized to the parental controls. Error bars denote SEM. E. Two individual BV-resistant single cell clones of KMH2 and L428 HL lines, as well as their parental controls, were treated with MLN120B (80μM) for 3 days. Cell numbers were counted by flow cytometry and normalized to DMSO-treated cells. Error bars denote SEM of triplicates. P were calculated comparing BV-resistant clones and its parental controls under the same treatment. ** indicates P < 0.01.
Upregulation of NF-κB signature in BV-resistant HL clones
To gain insight into the molecular mechanisms of BV resistance in HL cells, we developed BV-resistant HL models in both KMH2 and L428 HL lines. Briefly, HL cells were incubated at the IC90 concentration of BV over 7 weeks to generate BV-resistant pools. Then the pools were single-cell cloned with BV-containing media. For each line, we generated 12 individual BV-resistant single-cell clones. These BV-resistant clones were very resistant to BV treatment compared to the parental lines, as expected (Fig. 3A). Of note, these BV-resistant clones were resistant to MMAE treatment as well (Fig. 3A), indicating that there are cellular pathway alterations inside these clones which inhibit their response to MMAE cytotoxicity, e.g., NF-κB activation.
To determine the pathway alternations of these BV-resistant clones, we profiled gene expression changes by RNA-seq of two individual BV-resistant clones vs parental control, in both KMH2 and L428 models, allowing us to define a set of genes that were consistently upregulated or downregulated. We then compared the gene expression changes with a database of gene expression signatures that reflect regulatory processes in normal and malignant blood cells (31). Among upregulated genes, a signature of NF-κB activated genes was significantly enriched (Fig. 3B and 3C). NF-κB signaling genes were upregulated in all of the BV-resistant KMH2 and L428 clones we examined (Fig. 3B), and demonstrated significant enrichemnt in Gene Set Enrichment Analysis (GSEA) (Fig. 3C). In line with the gene expression profiling, all of these BV-resistant clones exhibited almost normal CD30 expression, but markedly increased induction of the NF-κB response gene CD83, measured by surface flow cytometry (Fig. 3D). This suggests that resistance to BV, at least in our cell line models, is due to upregulation of NF-κB activity, rather than downregulation of surface CD30. Importantly, these BV-resistant clones are much more sensitive to the IKKβ inhibitor MLN120B treatment (Fig. 3E), suggesting that targeting this pathway would provide a way to overcome BV resistance.
ABCB1 upregulation decreases BV sensitivity and promotes BV resistance of HL cell lines
To investigate the molecular mechanism of how NF-κB activity decreases BV treatment sensitivity of HL, we further analyzed our gene expression profiling results of the BV-resistant clones (Fig. 3). Remarkly, in the KEGG pathway emrichment analysis, the ATP-binding cassette (ABC) transporters pathway was the one showing significant enrichment (p value <0.05) in all of the four BV-resistant clones we examined (Supplemental Fig. 2B). Indeed, a set of ABC transporter genes were strongly upregualted in all of the BV-resistant clones (Fig. 4A). Among these genes, ABCB1 (MDR1), encoding a membrane-associated protein in the MDR/TAP subfamily, was the most upregulated. ABCB1 is responsible for transporting various molecules across cellular membranes therefore decreasing drug accumulation inside cells. In recent studies, ABCB1 has been implied to play a role in BV resistance of HL (9,32), and inhibition of ABCB1 was able to overcome resistance to BV in a phase I clinical trial (32). However, the molecular evidence and its regulatory mechanism are missing.
Figure 4: ABCB1 expression is essential for BV sensitivity.
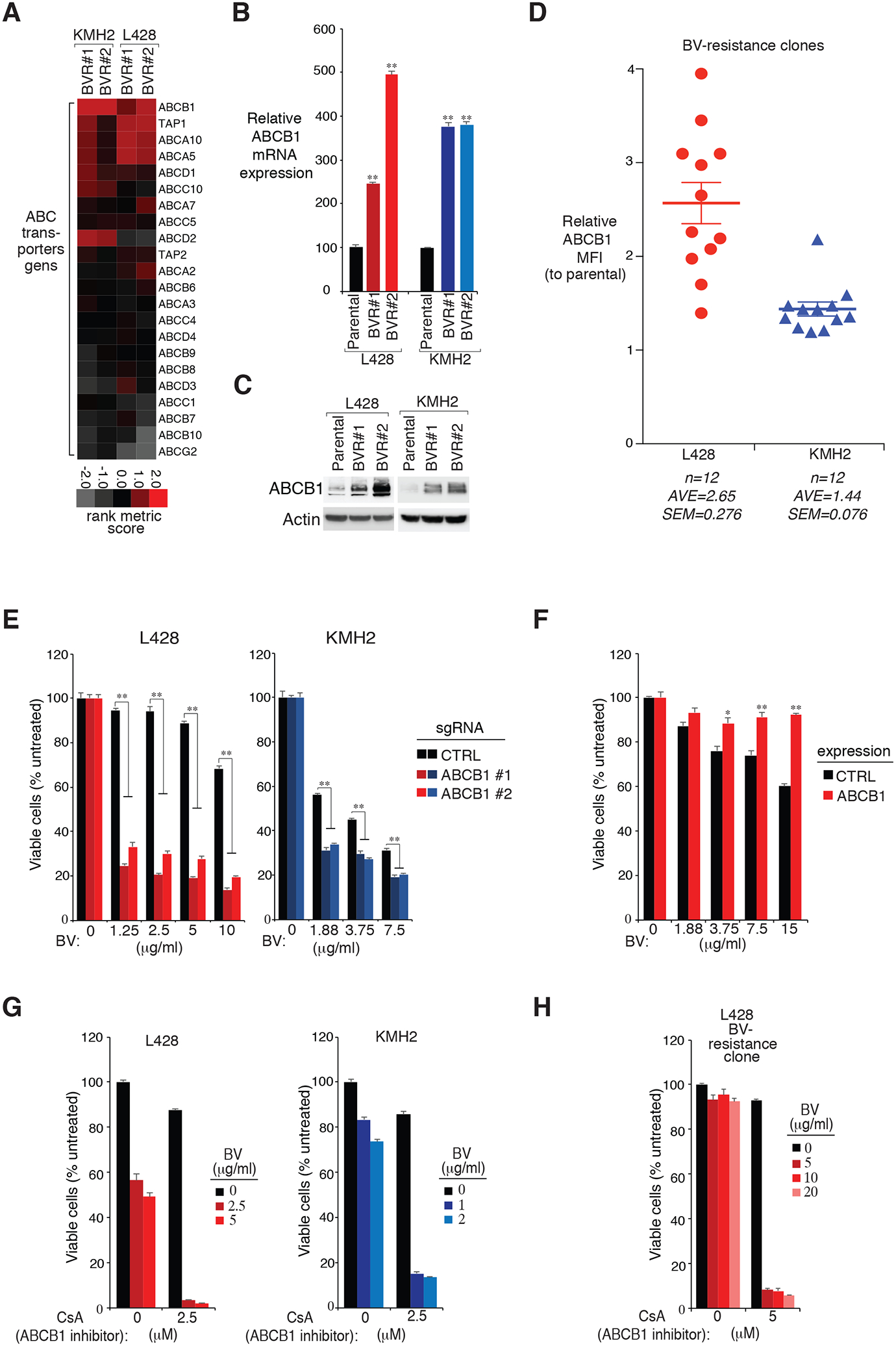
A. Heatmap of rank metric scores of ABC transporter genes in the indicated BV-resistant single cell clones of KMH2 and L428 HL lines, relative to their parental controls. B. and C. ABCB1 expression in two individual BV-resistant single cell clones of KMH2 and L428 HL lines, as well as their parental controls, measured by real-time PCR (B) and immunoblot (C). P were calculated comparing BV-resistant clones and its parental controls. ** indicates P < 0.01. D. Surface ABCB1 expression was measured in all BV-resistant single cell clones of KMH2 and L428 HL lines, as well as their parental controls, by flow cytometry. The relative CD83 MFI was normalized to the parental controls. Error bars denote SEM. E. HL cell lines L428 and KMH2 were transduced with ABCB1 or Ctrl sgRNAs, selected and expression induced, then treated with BV at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to PBS-treated cells. Error bars denote SEM of triplicates. ** indicates P < 0.01. F. HL cell line L428 was transduced with ABCB1 cDNA or empty control, selected and expression induced, then treated with BV at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to PBS-treated cells. Error bars denote SEM of triplicates. * indicates P < 0.05; ** indicates P < 0.01. G. Viability of HL cell lines KMH2, and L428 after treatment (4 days) with the indicated concentrations of BV, ABCB1 inhibitor CsA, or both. Data are normalized to PBS-treated cells. Error bars denote SEM of triplicates. H. Viability of a L428 BV-resistance line after treatment (4 days) with the indicated concentrations of BV, ABCB1 inhibitor CsA, or both. Data are normalized to DMSO-treated cells. Error bars denote SEM of triplicates.
Confirming the RNA-seq results, ABCB1 expression was largely upregulated in both of the KMH2 and L428 BV-resistant clones, revealed by real-time PCR (Fig. 4B) and by immunoblot analysis (Fig. 4C). Similarly, all of the BV-resistant clones exhibited markedly increased induction of surface ABCB1 expression, measured by flow cytometry (Fig. 4D). To determine if ABCB1 expression decreases BV sensitivity in HL, we designed sgRNAs targeting ABCB1 and tested their ability to inhibit BV sensitivity in KMH2 and L428 cells (Fig. 4E and Supplemental Fig. 3A). In both lines, ABCB1 depletion largely increased BV sensitivity compared to the control cells. Consistently, ectopic expression of ABCB1 into L428 cells decreased the cytotoxic effect of BV (Fig. 4F and Supplemental Fig. 3B). Furthermore, cyclosporin A (CsA), a potent ABCB1 inhibitor, strongly promoted BV sensitivity in all four HL lines we tested: KMH2, L428, L1236 and L540 (Fig. 4G and Supplemental Fig. 3C). Lastly, pretreating of BV-resistant clones with ABCB1 inhibitor CsA, completely revised its resistance ability upon BV treatment (Fig. 4H). Thus, ABCB1 is the key molecular whose upregulation is essential for the inhibition of BV sensitivity and increasing BV resistance in HL cell lines.
NF-κB and A20 mediate ABCB1 expression
Given that NF-κB/A20 and ABCB1 are all required to regulate BV sensitivity in HL cells, we sought to understand the epistatic relationships between these key factors. In L428 cells, treatment with the IKKβ inhibitor MLN120B strongly reduced ABCB1 surface expression (Fig. 5A), indicating that NF-κB activity supports ABCB1 expression in HL cell lines. BV treatment stimulated ABCB1 expression, which could also be inhibited by the IKKβ inhibitor MLN120B (Fig. 5A), in line with our previous results (Fig. 2B and 2C). This inhibition appears to be mostly at the transcriptional level, as MLN120B treatment reduced ABCB1 mRNA expression in all of the HL lines we tested (Fig. 5B). Furthermore, ectopic expression of a constitutively active IKKβ mutant into HL cells promoted ABCB1 expression, measured by FACS analysis (Fig. 5C) and immunoblotting (Supplemental Fig. 4A). Hence, NF-κB activation stimulates ABCB1 expression in HL cell lines. We then examined if A20 regulates ABCB1 expression as well. In A20 WT HL line L428, A20 depletion by sgRNAs significantly augmented ABCB1 mRNA transcription (Fig. 5D), surface expression (Fig. 5E), and protein level (Supplemental Fig. 4B).
Figure 5: NF-κB, A20 and RBX1 regulate ABCB1 expression in HL lines.
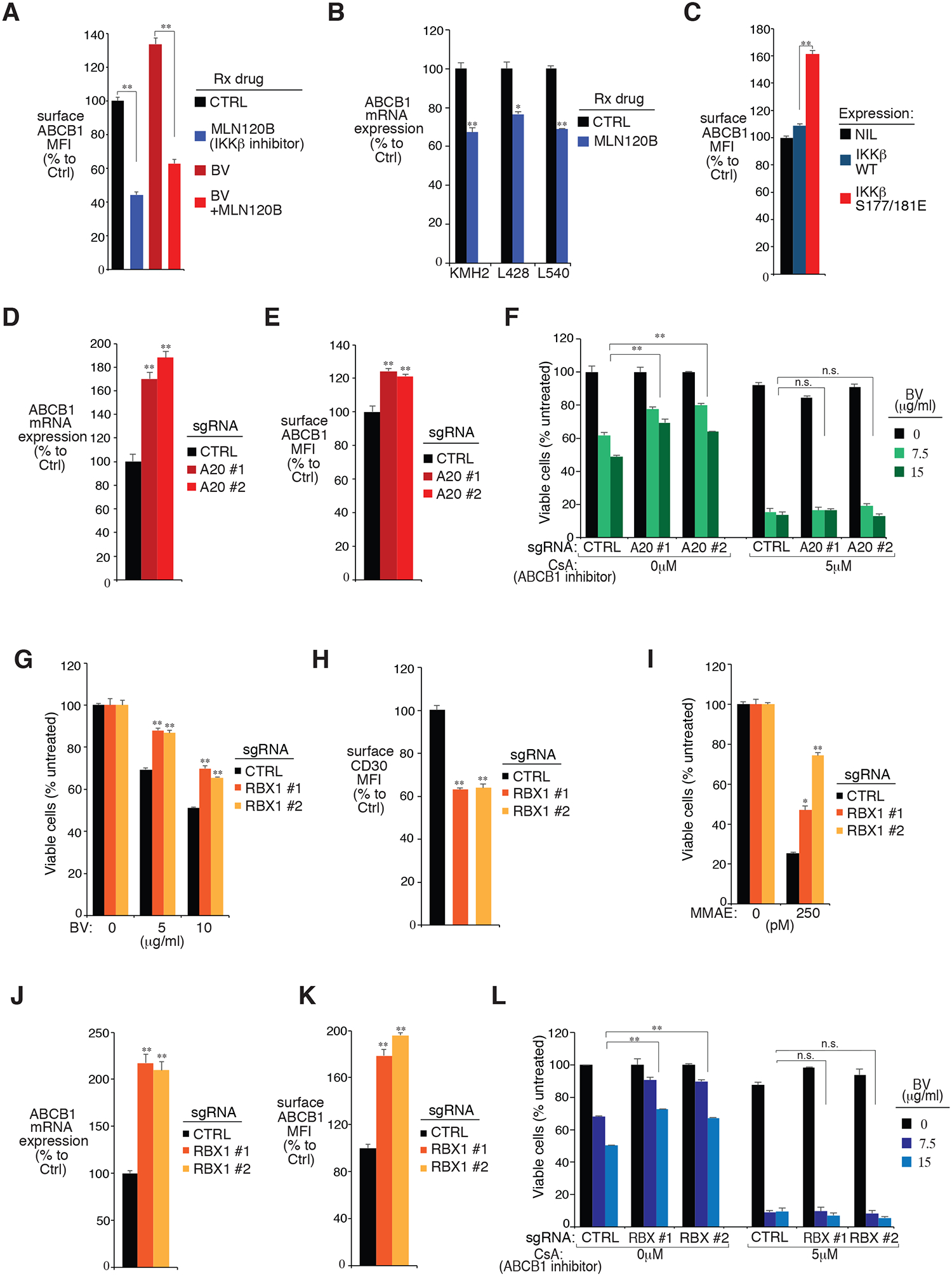
A. The L428 line was treated with indicated drugs at the following concentrations: MLN120B (20μM), BV (10μg/ml), or the combination for 24 hours. Surface ABCB1 expressions were measured by flow cytometry. The relative ABCB1 MFI was normalized to the untreated cells. B. KMH2, L428 and L540 lines were treated with MLN120B (20μM) for 24 hours. ABCB1 mRNA expressions were measured by real-time PCR and normalized to the untreated cells. C. HL cell line L428 was transduced with cDNA of IKKβ WT, IKKβ S177/181E or empty control, selected and expression induced. Surface ABCB1 expressions were measured by flow cytometry and normalized to the empty control. D and E, The L428 line was transduced with A20 or Ctrl sgRNAs, selected and expression induced. Relative ABCB1 expressions were measured by real-time PCR (D) and flow cytometry (E). F. The L428 line was transduced with A20 or Ctrl sgRNAs, selected and expression induced, then treated with the indicated concentrations of BV, ABCB1 inhibitor CsA, or both for 4 days. Viability was measured by MTS assay and normalized to PBS-treated cells. G. The HL cell line L428 was transduced with RBX1 or Ctrl sgRNAs, selected and expression induced, then treated with BV at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to PBS-treated cells. H. The L428 line was transduced with RBX1 or Ctrl sgRNAs along with GFP. Surface CD30 expression in uninfected (GFP−) cells and sgRNA infected (GFP+) cells was measured by flow cytometry. The summary of three independent experiments is shown. The relative CD30 MFI was normalized to the uninfected (GFP−) cells. I. The L428 line was transduced with RBX1 or Ctrl sgRNAs, selected and expression induced, then treated with MMAE at the indicated concentrations for 4 days. Viability was measured by MTS assay and normalized to DMSO-treated cells. J and K. The L428 line was transduced with RBX1 or Ctrl sgRNAs, selected and expression induced. Relative ABCB1 expressions were measured by real-time PCR (J) and flow cytometry (K). L. The L428 line was transduced with RBX1 or Ctrl sgRNAs, selected and expression induced, then treated with the indicated concentrations of BV, ABCB1 inhibitor CsA, or both for 4 days. Viability was measured by MTS assay and normalized to PBS-treated cells. For all the panels, Error bars denote SEM of triplicates. * indicates P < 0.05; ** indicates P < 0.01; n.s indicates no statistical difference.
To further investigate if A20 and NF-κB mediate BV sensitivity through ABCB1, we examined the effect of the ABCB1 inhibitor CsA on BV sensitivity of A20 depleted or constitutively active IKKβ mutant expressing cells. In L428 cells, A20 depletion rescued tumor cells from BV treatment, but not in tumor cells pre-treated with the ABCB1 inhibitor CsA (Fig. 5F). Similarly, ectopic expression of the constitutively active IKKβ mutant largely decreased MMAE sensitivity, as expected, but the CsA pre-treatment completely diminished the differences (Supplemental Fig. 4C). Taken together, A20 and NF-κB activity mediate the key drug-transporter protein ABCB1 expression in HL cells, which is the likely mechanism explaining how they regulate HL responsiveness to BV and eventually BV resistance.
RBX1 regulates BV sensitivity through both CD30 and ABCB1
In addition to A20, RBX1, an essential component of SCF (Skp1/Cullins/F-box) E3 ubiquitin ligases complex, was also identified as a top hit in our drug sensitization CRISPR library screen whose loss promoted resistance to BV cytotoxicity (Fig. 1B and 1C). This screen result was validated by an MTS assay using two individual sgRNAs targeting RBX1 in all 4 HL cell lines tested (Fig. 5G, Supplemental Fig. 5A, and Supplemental Fig. 5B). Interestingly, in contrast to A20 depletion, RBX1 depletion reduced surface CD30 expression in HL cells (Fig. 5H and Supplemental Fig. 5C). Indeed, RBX1 sgRNAs had no effect on NF-κB activity of HL cells, as indicated by NF-κB target gene CD83 expression, as well as by the p-p65 intracellular level (Supplemental Fig. 5D). Therefore, loss of RBX1 decreases BV sensitivity in HL cells, not through NF-κB but likely at least partially through CD30.
However, similar to A20, the RBX1 depleted HL cells were also resistant to MMAE treatment compared to control cells (Fig. 5I, and Supplemental Fig. 5E), indicating that RBX1 might also regulate drug-transporter expression, in addition to its ability to regulate CD30 surface expression. Indeed, RBX1 depletion by sgRNAs significantly augmented ABCB1 mRNA transcription (Fig. 5J), surface accumulation (Fig. 5K), and total protein expression (Supplemental Fig. 5F). Furthermore, pre-treating HL cells with ABCB1 inhibitor CsA completely blocked the ability of RBX1 sgRNAs to rescue tumor cells from BV treatment (Fig. 5L). Hence, loss of RBX1 decreases BV sensitivity also by upregulating ABCB1 expression, although by a mechanism that is not NF-κB dependent. To further assess the role of CRLs/SCF E3 ligase complex in ABCB1 regulation, we treated HL cell lines KMH2 and L428 with the NEDD8 activating enzyme (NAE) inhibitor Pevonedistat, which inactivates CRLs/SCF E3 ligase activity. In both cell lines, treatment of Pevonedistat increased MDR1 protein levels in a dose-dependent manner, indicating the role of CRLs/SCF E3 ligase in MDR1 regulation (Supplemental Fig. 6A). In line with these results, pre-treating HL cells with Pevonedistat largely decreases BV sensitivity in KMH2 and L428 lines (Supplemental Fig. 6B).
Targeting NF-κB improves BV sensitivity and overcomes BV resistance in HL xenograft mouse models
Our in vitro cell line studies revealed that NF-κB activity is the major determinant of BV sensitivity in HL, and that the IKKβ inhibitor MLN120B synergized well with BV in killing HL cells (Fig. 2E and 2F). Therefore, we established two HL xenograft mouse models using the L428 and KMH2 cell lines and sought to evaluate the efficacy of this combinational treatment in mouse xenograft models. In a L428 xenograft mouse model, treatment with BV every 4 days (0.4 mg/kg) slowed tumor growth and yielded a greater prolongation of survival (mice died between days 55 and 61, median survival: 58 d, p < 0.0001), as expected (Fig. 6A and 6B). Furthermore, the combination of BV and MLN120B treatment completely arrested growth of established tumors as was confirmed by survival of all mice for the total 61-d period of observation (combination vs. BV, p =0.0002) (Fig. 6A and 6B). Of note, the pure human CD30 antibody HeFi-1 had little effect on the growth of the xenografts and the survival of engrafted mice, as a single agent or in combination with MLN120B (Fig. 6A and 6B), indicating that the combinational effect of MLN120B plus BV is not due to the nonspecific antibody-dependent cellular cytotoxicity (ADCC). At the doses used the BV plus MLN120B combination was well tolerated by mice, with no change in body weight observed (Fig. 6C). Meanwhile, the serum levels of IL-6 paralleled our cell line observations as a surrogate marker of tumor size and NF-κB activation in all the engrafted mice. Compared with the vehicle treatment group, there were reductions of IL-6 in the BV individual group (P < 0.0001 compared with the vehicle group) at 5.5 weeks after tumor inoculation, but there were greater reductions in IL-6 with the BV plus MLN120B combination group compared with BV only treatment group (P < 0.0001) (Fig. 6D). Hence, targeting NF-κB augments BV sensitivity and minimizes BV resistance in HL.
Figure 6: Targeting NF-κB in HL xenograft mice models.
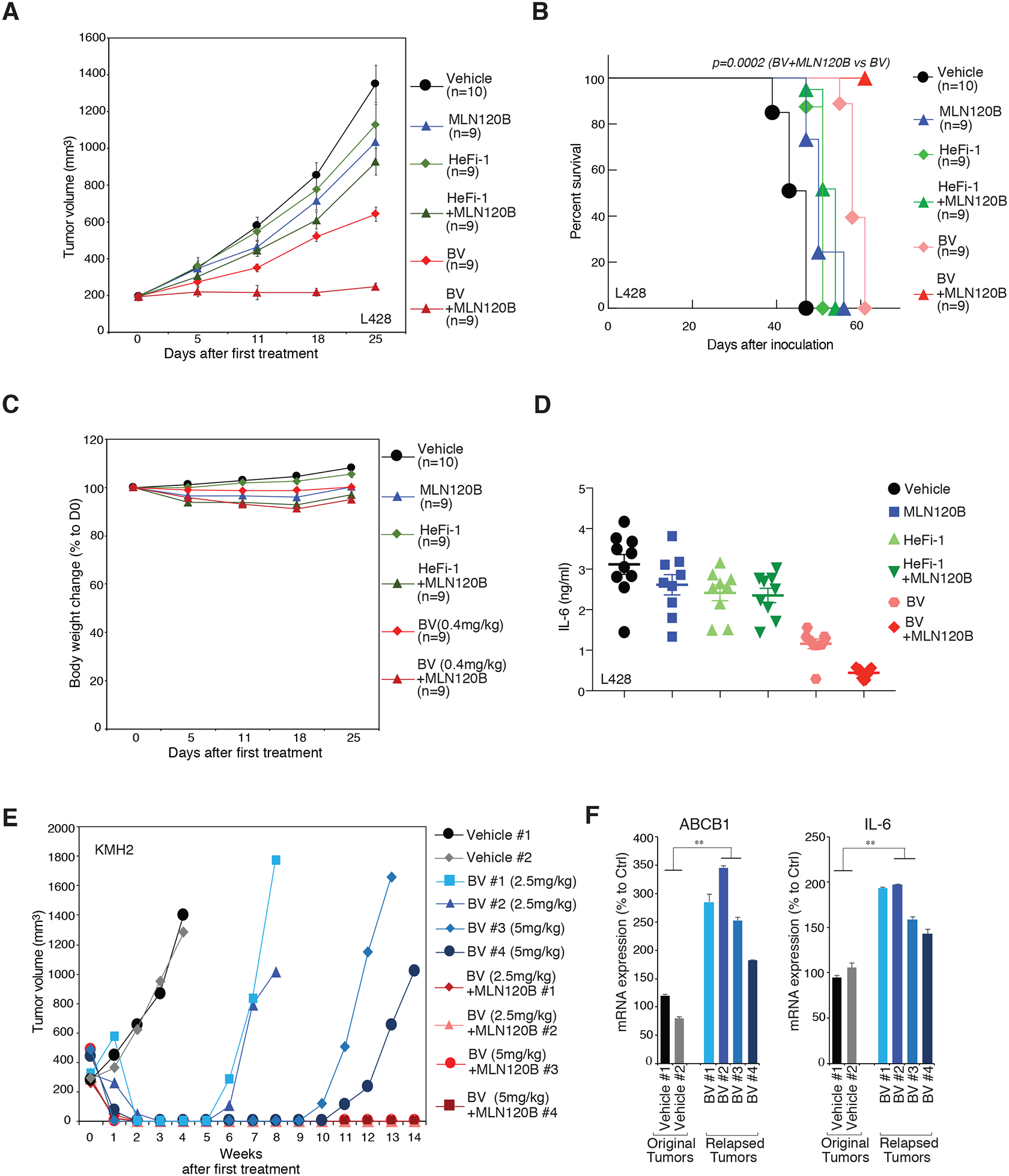
A. NSG mice bearing L428 xenografts were treated with MLN120B (50mg/kg daily, n=9), HeFi-1 (0.1mg/mouse weekly, n=9), BV (0.4mg/kg every 4 days, n=9), the combination of MLN120B and HeFi-1 (same dosage dosing schedules as in single treatment groups, n=9), the combination of MLN120B and BV (same dosage dosing schedules as in single treatment groups, n=9), as well as vehicle controls (n=10). Tumor growth was measured as a function of tumor volume. Error bars denote SEM. B. Kaplan–Meier analysis of the survival of the tumor-bearing mice in the therapeutic studies of A. C. Mouse body weight changes, normalized to D0, in the experiments of A. Error bars denote SEM. D. Serum levels of human IL-6 5.5 weeks after inoculation of tumor cells, in the in the experiments of A. E. NSG mice bearing KMH2 xenografts were treated with BV (2.5mg/kg or 5mg/kg every 4 days), the combination of MLN120B (50mg/kg daily) and BV (2.5mg/kg or 5mg/kg every 4 days), as well as vehicle controls. Tumor growth was measured as a function of tumor volume. F. The relapsed tumor after BV treatment in E., and the original tumor (vehicle treated) were harvested. The mRNA expression of ABCB1 and IL-6 were measured by real-time PCR. Error bars denote SEM of triplicates. P were calculated comparing relapsed group and original group. ** indicates P < 0.01.
Finally, we examined if targeting NF-κB could overcome tumor relapse upon BV treatment in the HL xenograft mouse model. In the KMH2 xenograft mouse model, we treated the tumor with high dosages of BV (2.5 or 5 mg/kg, every 4 days) alone or with the combination with MLN120B. Both the BV only group and the combination group appeared tumor free 1–2 wks after starting therapy (Fig. 6E). However, tumors recurred in the mice of the BV-treated group 5–10 wks after the tumors initially became undetectable. Remarkably, treatments with BV combined with MLN120B yielded a complete response and the mice remained tumor free as was maintained for the 14 weeks of observation (Fig. 6E). Importantly, in the relapsed tumors of BV only group, ABCB1 and IL-6 mRNA levels were substantially higher than in the original (vehicle treated) tumors (Fig. 6F), indicating consistent upregulations of the drug-transporter gene and of NF-κB activity during relapse, in keeping with our previous results with BV-resistant single cell clones (Fig. 3 and Fig. 4).
Discussion
Although BV elicits a high response rate in HL, many patients do not achieve complete remissions and ultimately develop BV-resistant disease. Therefore, new treatments for HL should ideally exploit emerging insights into BV-resistance mechanisms, which will create opportunities for synthetic interactions with drugs that increase BV response rates and durability. In this study, we utilized a combination of two unbiased high-throughput analysis: drug sensitization CRISPR library screen and RNA-seq analysis to comprehensively investigate the BV resistance mechanisms. Both of these analyses lead us to identify the central role of NF-κB activity in regulating BV treatment sensitivity of HL cell lines. The ubiquitin regulator-focused CRISPR library screen identified A20, a negative regulator of NF-κB pathway, augmented BV sensitivity in HL lines. Meanwhile, our RNA-seq analysis revealed a significant upregulation of the NF-κB signature in BV-resistant HL cells. A20 and NF-κB regulate BV sensitivity apparently through mediating the expression of the ABC transporter gene ABCB1. Finally, targeting NF-κB augmented BV sensitivity and overcame BV resistance in HL xenograft mouse models. Taken together, our findings provide a broad understanding of how BV sensitivity is regulated by the protein ubiquitination systems in HL cell lines and offer a rationale for therapeutic approaches to overcome BV resistance in this disease.
Aberrant activation of the NF-κB pathway is indeed the most striking oncogenic mechanism in HL (33), and our study revealed that it is also involved in BV resistance. Genetically, various recurrent somatic genetic lesions have been identified in the NF-κB pathway of HL. For instance, amplification of the REL locus is present in more than 50% of cHL cases (34–36). Frameshift or nonsense mutations in NFKBIA, which encodes IκBα, produce inactive, truncated IκBα isoforms and occur in ~5–20% of Hodgkin lymphoma cases (37–40). Additionally, loss-of-function mutations occur in NFKBIE, encoding IκBε, albeit at a lower frequency (41). Among these genetic events, inactivation of the ubiquitin editing protein A20 by biallelic deletions or frameshift/nonsense mutations is the most recurrent genetic alteration, occurring in 30–40% of HL cases (20,21), and preferentially in EBV-negative HL (around 70–80%). Epigenetically, A20 can also be downregulated by promoter methylation (42). As only 75% of HL cases respond to BV treatment and there are merely 32% CRs, it is very likely that genetic lesions in the NF-κB pathway, especially A20, are associated with BV responsiveness and resistance in the clinic. Therefore, A20 mutation and expression status can be used as biomarkers to predict treatment outcomes.
It is also worth mentioning that our study is mainly based on HL cell lines and xenograft models in NSG mice which lacks immune microenvironment. While HL cell lines recapitulate the physiological feature of HRS cells, they are often unable to fully recreate the genetic complexity of human HL. In patient, HRS cells only represent a minority of the cells within tumors, with the bulk of the tumor composed of various inflammatory cell types including T cells, macrophages, neutrophils, and B cells. The presence of such a characteristic inflammatory microenvironment is a fundamental component of the tumor mass and an essential pathogenetic factor in HL, which would be an important factor to determine BV responsiveness and promote resistance. Unfortunately, currently there are no murine models presenting human HL. Therefore, the clinical association of A20 mutation/expression status to BV responsiveness and resistance will need to be further examined in primary HL cases in future studies. Similarly, the association of NF-κB activity with BV responsiveness will need to be further examined as well, especially in BV relapsed cases.
Increased expression of the plasma membrane drug efflux pumps, the ABC transporters, is generally involved in anticancer drug resistance. Our study identified the ABC transporter gene, ABCB1, as essential for BV resistance of HL cell lines, in line with previous studies (9,32). Importantly, ABCB1 expression is regulated by NF-κB activity in HL cells, which provides a molecular mechanism for NF-κB in BV resistance. In addition to NF-κB, p38 MAPK (43,44) and Wnt/β-catenin pathway (45) have also been implicated in the transcriptional regulation of ABCB1, indicating that other cellular pathways in addition to NF-κB could also contribute to BV resistance in HL. Indeed, our ubiquitin regulator-focused CRISPR library screen also identified RBX1, the key component of SCF (Skp1/Cullins/F-box) E3 ubiquitin ligases complex, as an essential factor that mediates ABCB1 expression in an NF-κB independent manner. Thus, RBX1 could regulate p38 MAPK or Wnt/β-catenin pathways in HL. In addition, ABCB1 could be targeted for proteasomal degradation through SCF and F-box protein FBXO15 (SCFFBX15) (46). FBXO15 was not scored as a strong positive hit in our library screen, indicating that there are other F-box proteins that could compensate for the FBXO15 loss. In this case, RBX1 could regulate the ABCB1 level both transcriptionally and post-transcriptionally. Future work should explicate the intricate mechanisms that RBX1 regulate ABCB1 in HL cells, and assess the potential F-box proteins required to regulate ABCB1 stabilization.
In summary, the present study demonstrates that targeting NF-κB provides an attractive strategy to improve BV sensitivity and overcome BV resistance in HL. These findings deliver support for a therapeutic trial for select patients carrying recurrent somatic genetic lesions in NF-κB pathways with a combination regimen that involves BV with NF-κB inhibitors. Although currently there are no effective therapies that directly target IKK in clinic, the development of next generation of IKKβ inhibitors with high selectivity, nanomolar potency, and flexibility to targeted delivery would be desirable to overcome drug resistance, as well as to kill tumor cells relied on NF-κB to survival. Since ABC transporter genes are also involved in other anticancer drug resistance, it is reasonable to predict that targeting NF-κB might have a broad benefit in this context as well.
Supplementary Material
Statement of translational relevance.
Although there has been great progress in treating Hodgkin lymphoma (HL), the survival rate for patients diagnosed at an advanced stage or with relapsed/refractory disease remains low. The current understanding of the biology of the disease has been translated into the development and FDA approval of the first effective targeted therapy for relapsed/refractory HL, Brentuximab Vedotin (BV), a drug-conjugated anti-CD30 antibody. However, many patients do not achieve complete remission and develop BV-resistant disease, which usually leads to poor outcomes. So far, the mechanisms that underlie BV resistance still remains poorly understood. Giving the fact that the protein ubiquitination system plays a critical role in HL pathogenesis, we decided to use the CRISPR library screening technologies to gain a complete understanding of how the ubiquitin modifying machinery regulates BV effectiveness in HL, which will provide actionable interventions for new therapies of patients suffering with HL, particularly in BV resistant cases.
Acknowledgements
This research was supported by NIH K22 CA197014 (Y. Yang) and American Cancer Society (ASC) IRG-92-027-21 (Y. Yang), and the Medical Research Grant from the W. W. SMITH Charitable Trust (Y. Yang). This research was also funded through the NIH/NCI Cancer Center Support Grant P30 CA006927. The research was also supported by the Intramural Research Program of the National Cancer Institute NIH (T.A. Waldmann).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Brauninger A, Schmitz R, Bechtel D, Renne C, Hansmann ML, Kuppers R. Molecular biology of Hodgkin’s and Reed/Sternberg cells in Hodgkin’s lymphoma. Int J Cancer 2006;118(8):1853–61 doi 10.1002/ijc.21716. [DOI] [PubMed] [Google Scholar]
- 2.Quddus F, Armitage JO. Salvage therapy for Hodgkin’s lymphoma. Cancer J 2009;15(2):161–3 doi 10.1097/PPO.0b013e3181a1438a. [DOI] [PubMed] [Google Scholar]
- 3.Stein H, Gerdes J, Schwab U, Lemke H, Mason DY, Ziegler A, et al. Identification of Hodgkin and Sternberg-reed cells as a unique cell type derived from a newly-detected small-cell population. Int J Cancer 1982;30(4):445–59. [DOI] [PubMed] [Google Scholar]
- 4.Gruss HJ, Hirschstein D, Wright B, Ulrich D, Caligiuri MA, Barcos M, et al. Expression and function of CD40 on Hodgkin and Reed-Sternberg cells and the possible relevance for Hodgkin’s disease. Blood 1994;84(7):2305–14. [PubMed] [Google Scholar]
- 5.Fiumara P, Snell V, Li Y, Mukhopadhyay A, Younes M, Gillenwater AM, et al. Functional expression of receptor activator of nuclear factor kappaB in Hodgkin disease cell lines. Blood 2001;98(9):2784–90. [DOI] [PubMed] [Google Scholar]
- 6.Chiu A, Xu W, He B, Dillon SR, Gross JA, Sievers E, et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007;109(2):729–39 doi blood-2006-04-015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30(18):2183–9 doi 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheah CY, Chihara D, Horowitz S, Sevin A, Oki Y, Zhou S, et al. Patients with classical Hodgkin lymphoma experiencing disease progression after treatment with brentuximab vedotin have poor outcomes. Ann Oncol 2016;27(7):1317–23 doi 10.1093/annonc/mdw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R, Hou J, Newman E, Kim Y, Donohue C, Liu X, et al. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol Cancer Ther 2015;14(6):1376–84 doi 10.1158/1535-7163.MCT-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature 2009;458(7237):430–7 doi 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 11.Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry 1998;67:425–79 doi 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Staudt LM. Protein ubiquitination in lymphoid malignancies. Immunol Rev 2015;263(1):240–56 doi 10.1111/imr.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343(6166):84–7 doi 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014;343(6166):80–4 doi 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang JP, Song Z, Wang HB, Lang L, Yang YZ, Xiao W, et al. A novel model of controlling PD-L1 expression in ALK(+) anaplastic large cell lymphoma revealed by CRISPR screening. Blood 2019;134(2):171–85 doi 10.1182/blood.2019001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dirks WG, Drexler HG. Authentication of scientific human cell lines: easy-to-use DNA fingerprinting. Methods Mol Biol 2005;290:35–50. [DOI] [PubMed] [Google Scholar]
- 17.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25(9):1105–11 doi 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28(5):511–5 doi nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2013;31(1):46–53 doi 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009;459(7247):712–6 doi nature07969. [DOI] [PubMed] [Google Scholar]
- 21.Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 2009;206(5):981–9 doi jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer 2017;117(12):1736–42 doi 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. Deubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004;430(7000):694–9. [DOI] [PubMed] [Google Scholar]
- 24.Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, et al. Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NFkappaB activation. J Biol Chem 1997;272(4):2042–5. [DOI] [PubMed] [Google Scholar]
- 25.Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol 1997;17(3):1535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen JL, Thompson CB. CD30 contains two binding sites with different specificities for members of the tumor necrosis factor receptor-associated factor family of signal transducing proteins. J Biol Chem 1996;271(22):12852–8. [DOI] [PubMed] [Google Scholar]
- 27.Horie R, Aizawa S, Nagai M, Ito K, Higashihara M, Ishida T, et al. A novel domain in the CD30 cytoplasmic tail mediates NFkappaB activation. Int Immunol 1998;10(2):203–10. [DOI] [PubMed] [Google Scholar]
- 28.Lee SY, Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. CD30/TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci U S A 1996;93(18):9699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M, Yao Z, Zhang Z, Garmestani K, Goldman CK, Ravetch JV, et al. Effective therapy for a murine model of human anaplastic large-cell lymphoma with the anti-CD30 monoclonal antibody, HeFi-1, does not require activating Fc receptors. Blood 2006;108(2):705–10 doi 10.1182/blood-2005-11-4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017;33(15):2413–5 doi 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaffer AL, Wright G, Yang L, Powell J, Ngo V, Lamy L, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev 2006;210:67–85. [DOI] [PubMed] [Google Scholar]
- 32.Chen R, Herrera AF, Hou J, Chen L, Wu J, Guo Y, et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-19-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weniger MA, Kuppers R. NF-kappaB deregulation in Hodgkin lymphoma. Semin Cancer Biol 2016;39:32–9 doi 10.1016/j.semcancer.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 34.Barth TF, Martin-Subero JI, Joos S, Menz CK, Hasel C, Mechtersheimer G, et al. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003;101(9):3681–6 doi 10.1182/blood-2002-08-2577. [DOI] [PubMed] [Google Scholar]
- 35.Martin-Subero JI, Gesk S, Harder L, Sonoki T, Tucker PW, Schlegelberger B, et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002;99(4):1474–7. [DOI] [PubMed] [Google Scholar]
- 36.Joos S, Menz CK, Wrobel G, Siebert R, Gesk S, Ohl S, et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002;99(4):1381–7. [DOI] [PubMed] [Google Scholar]
- 37.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene 1999;18(20):3063–70. [DOI] [PubMed] [Google Scholar]
- 38.Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in reed-sternberg cells. Blood 1999;94(9):3129–34. [PubMed] [Google Scholar]
- 39.Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dorken B, Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene 1999;18(4):943–53. [DOI] [PubMed] [Google Scholar]
- 40.Lake A, Shield LA, Cordano P, Chui DT, Osborne J, Crae S, et al. Mutations of NFKBIA, encoding IkappaB alpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int J Cancer 2009;125(6):1334–42 doi 10.1002/ijc.24502. [DOI] [PubMed] [Google Scholar]
- 41.Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Dohner K, et al. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol 2003;201(3):413–20. [DOI] [PubMed] [Google Scholar]
- 42.Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood 2009;114(12):2467–75 doi blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 43.Guo X, Ma N, Wang J, Song J, Bu X, Cheng Y, et al. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008;8:375 doi 10.1186/1471-2407-8-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barancik M, Bohacova V, Kvackajova J, Hudecova S, Krizanova O, Breier A. SB203580, a specific inhibitor of p38-MAPK pathway, is a new reversal agent of P-glycoprotein-mediated multidrug resistance. Eur J Pharm Sci 2001;14(1):29–36. [DOI] [PubMed] [Google Scholar]
- 45.Correa S, Binato R, Du Rocher B, Castelo-Branco MT, Pizzatti L, Abdelhay E. Wnt/beta-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012;12:303 doi 10.1186/1471-2407-12-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katayama K, Noguchi K, Sugimoto Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin--proteasome pathway in cancer cells. Cancer Sci 2013;104(6):694–702 doi 10.1111/cas.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.