

Abstract
Ubiquitination is a crucial component of many immune processes. While ubiquitin-mediated degradation is essential to T cell activation via T cell receptor signaling, the specific E3 ligases and substrates involved are not well-understood. Here, we describe a strategy integrating RNA, protein, and posttranslational modification datasets to identify targets of ubiquitin-mediated degradation. When integrated, these assays can provide broad insight into how this posttranslational modification regulates protein function and influences T cell biology.
Keywords: Ubiquitin, T cell, Lymphocyte, Diglycine remnant profiling, Signaling
1. Introduction
Ubiquitin-mediated degradation is required for many aspects of T cell function. Mutations in E3 ubiquitin ligases or their activators lead to dramatic changes in T cell differentiation [1–3]. Furthermore, T cell activation is blocked when the proteasome is inhibited by MG132 [4]. However, our understanding of proteins that undergo ubiquitin-mediated degradation has been limited by lack of technologies that broadly query these events.
Our laboratory recently developed a strategy for identifying and validating targets of ubiquitin-mediated degradation using combined transcriptomic and proteomic analyses. By comparing whole-cell proteomic changes to whole-cell transcriptomic changes, we can generate a list of proteins that are downregulated at the protein but not mRNA level, indicating they are likely to undergo protein degradation. Additionally, Steven Carr’s group has developed a technique to identify ubiquitinated proteins following trypsin digest, which reveals a diglycine (K-ε-GG) remnant; K-ε-GG-containing proteins can then be enriched using anti-diglycine antibody, and identified using mass spectrometry [5, 6]. Cross-referencing these ubiquitination candidate proteins with degradation candidates generates a list of potential targets of ubiquitin-mediated degradation. These targets can be validated experimentally, for example by measuring protein half-life using cycloheximide treatment followed by Western blotting. Furthermore, performing a pulldown with a tandem ubiquitin-binding entity (TUBE) and blotting for the protein of interest can reveal whether the protein is ubiquitinated.
While we have used this strategy to screen for substrates degraded in stimulated CD4 T cells as compared to cells in a resting state [4], it can be adapted to screen for targets of specific E3 ligases by comparing wild-type cells to cells deficient for the ligase of interest. These studies enable us to understand the biological function of E3 ubiquitin ligases by revealing their substrates.
2. Materials
2.1. T Cell Preparation
Dissection tools (forceps and scissors).
60 mm Dishes.
70 μm Filters.
1 ml Syringes.
50 ml Conical tubes.
10 cm Tissue culture plates.
L3T4 CD4 T cell enrichment kit (Miltenyi, 130-117-043).
Phosphate-buffered saline (PBS).
Fetal calf serum (our lab uses premium FCS from Atlanta Biologicals; see Note 1).
MACS buffer: phosphate-buffered saline, 0.5% FCS, 2 mM ETDA.
LS columns (Miltenyi 130-042-401).
15 ml Conical tubes.
MACS magnet.
Trypan blue.
Hemocytometer.
Light microscope.
Red blood cell (RBC) lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 1 mM EDTA, pH 7.3).
T cell culture media: RPMI 1640 (HyClone, SH30027.01), 10% FCS, 100 U/ml penicillin-streptomycin (Gibco, 15140122), 1× Glutamax (ThermoFisher, 35050061), 1× nonessential amino acids (Gibco, 11140–050), 2 mM HEPES (Gibco, 15630–080), 1 mM sodium pyruvate (Corning, 20115013), 0.008 μl/ml 2-mercaptoethanol (Sigma, M6250).
Recombinant human IL-2 (PeproTech 200–02).
Anti-CD28 (Clone 37.51; BioLegend).
Anti-CD3 (Clone 145–2C11; BioLegend).
Dynabeads (ThermoFisher, 11456D).
2.2. K-ε-GG Pulldown
Note that the materials required for the K-ε-GG pulldown are described in Udeshi et al. [5]. Our protocol requires the following additional reagents:
K-ε-GG lysis buffer: 8 M urea, 75 mM NaCl, 50 mM Tris–HCl pH 8.0, 1 mM EDTA, 2 μg/ml aprotinin (Sigma, A6103), 10 μg/ml leupeptin (Roche, 11017101001), 1 mM PMSF (Sigma, 78830), 10 mM NaF, 25 μM PR619, 5 mM iodoacetamide (Sigma, A3221).
Pierce micro BCA assay kit (ThermoFisher, 23225).
Diglycine antibody (Cell Signaling, 5562).
RapiGest (Waters, 186001860).
Vacuum manifold (Waters, 186001831).
96 well Oasis HLB μElution plate (Waters, 186001828BA).
2.3. Whole-Cell Proteome Preparation
Details on performing an in-gel digest can be found in Shevchenko et al. [7]. Preparing samples requires the following reagents:
Laemmli sample buffer (4×).
DTT.
10% NuPage™ acrylamide gel (ThermoFisher, NP0301PK2).
NuPage™ running buffer (ThermoFisher, NP0001).
Western blotting apparatus.
Power supply.
Excision template.
Unstained protein ladder.
Coomassie stain.
2.4. RNA-Seq Preparation
Qiagen RNeasy Plus Mini kit (Cat: 74134).
RNaseZap™ wipes (ThermoFisher, AM9786).
Filter pipette tips.
Agilent bioanalyzer.
2.5. Data Analysis
STAR aligner [10] (https://github.com/alexdobin/STAR).
2.6. Validation
(See Subheading 2.1 for materials needed for T cell extraction and culture.)
Lysis buffer for TUBE pulldown: RIPA (ThermoFisher, 89900) supplemented with 50 mM Tris–HCl, 0.15 M NaCl, 1 mM EDTA, 1% NP40, 10% glycerol, 1× Complete EDTA-free protease inhibitor dissolved in 300 μl Milli-Q water for a 20× stock (Roche, 11873580001), 0.4 mM, PR619, 200 mM 1,10-phenyanthroline (o-PA).
Lysis buffer for cycloheximide chase: RIPA (ThermoFisher, 89900) supplemented with 1× HALT phosphatase inhibitor (ThermoFisher, 78420) and 1× Complete EDTA-free protease inhibitor (See item 1).
Cycloheximide (Sigma, C4859).
MG132 (10 mM for a 1:1000 stock solution).
Chloroquine (Sigma, C6628—dissolve 0.41 g in 10 ml PBS for a 1:1600 stock).
UbiTest Agarose-TUBE Elution Kit (LifeSensors, UM411).
Uncoupled agarose (LifeSensors, UM400).
Deubiquitinating enzymes (DUBs): USP2 core pan-DUB (LifeSensors, DB501) and/or Otub1 K48-specific DUB (LifeSensors, DB201).
Tris-buffered saline with 0.1% tween (TBS-T).
SDS sample buffer.
4–12% NuPage™ gels.
NuPage™ running buffer (ThermoFisher, NP0001).
NuPage™ Transfer buffer (ThermoFisher NP0006).
Western blotting apparatus.
Power supply.
Odyssey blocking buffer (Li-Cor, 927–50000).
Antibodies for protein(s) of interest.
3. Methods
3.1. Preparing Samples for Mass Spectrometry and RNA Sequencing
Dishes should be prepared prior to starting dissection, using sterile technique in a biosafety cabinet, and kept on ice. See Fig. 1 for a workflow of sample preparation.
Harvest spleen and peripheral lymph nodes (LNs) from mice on C57BL6J background euthanized by CO2 asphyxiation following IACUC-approved standards. Mice should be older than 8 weeks of age, depending on experimental design. For ubiquitin remnant profiling, multiple mice are required. Place harvested spleen and LNs into 70 μM cell strainer which has been placed in a plastic 60 mm petri dish with 5 ml of RPMI supplemented with 1% fetal calf serum (FCS) (see Notes 1–6).
Coat tissue-culture-coated plates with anti-CD3 and anti-CD28 at 5 ng/ml in sterile PBS. Use enough solution to cover the bottom of the plates. Coat plates at least 2 h at 37 °C in a tissue culture incubator. This incubation can take place during cell preparation.
In sterile biosafety hood, mash the tissue through the strainer with the flat end of a plunger from a 1 ml syringe (opened sterilely in hood). All following steps should be performed under sterile conditions, with pre-sterilized serological pipets, tips, etc.
Pipet the cell solution through the filter into a 50 ml conical tube. Wash the filter twice with 5 ml of RPMI supplemented with 1% FCS.
Pellet cells at 1300 rpm for 5 min and 4 °C in a tabletop centrifuge.
Decant supernatant. LNs may be resuspended directly into CD4+ T cell isolation media (i.e., MACS buffer). Lyse red blood cells in spleen samples by resuspending the cells in 1–2 ml of red blood cell (RBC) lysis buffer. Quench lysis with MACS buffer after 30 s, and pellet cells immediately. Resuspend the splenocytes using LN cell solution for the same set of animals, filter through a 70 μm, filter, and pellet cells again.
Proceed to CD4+ T cell enrichment using CD4+ T cell microbeads (L3T4) following Miltenyi manufacturer instructions for enrichment with LS columns or automated selection (max capacity of total cells: 2 × 109; max capacity of labeled cells: 1 × 108). The spleen and LNs from about three unin flamed wild-type mice can be used in one column. MACS buffer should be prepared as indicated in the enrichment protocol, including degassing (see Note 7).
(Recommended) Stain an aliquot of cells with a lineage cocktail (CD11b, CD11c, FCERG1, Ter119, CD8b, GR1, Ly6G, Ly6C), CD3 (17A2), CD4, CD8a, and viability dye to assess purity by flow cytometry.
Resuspend cells in complete T cell media containing 50 U/ml of IL-2. Remove a small aliquot (about 10 μl) to count cells. Adjust volume of the cell resuspension to 1–1.5 × 106 cells/ml.
Aspirate the PBS from the coated plate, rinse plate once with sterile PBS, and add the cell mixture. Cell mixture should be added immediately to avoid drying the plate. Incubate at 37 °C 5% or 10% CO2 (according to the laboratory’s standard culturing conditions) for 72 h, or as called for in experimental workflow.
After 72 h, examine cells visually for signs of blasting and proliferation using a light microscope. Gently harvest cells from plates by gentle pipetting through a serological pipette. Wash plate with cold HBSS or PBS to collect residual cells. Examine the plate by microscope to ensure removal of cells.
Pellet cells, and resuspend in fresh culture media with IL-2 (50 U/ml). Count cells and dilute to 1–1.5 × 106/ml before plating on 10 or 15 cm tissue-culture-treated plates. If polarizing conditions were used, verify T cell differentiation by performing a brief restimulation of a cell aliquot with PMA/ionomycin in the presence of Brefeldin A for intracellular cytokine staining.
Split cells 1:1 with media containing IL-2 on the two consecutive days (see Note 8).
On the third day, harvest cells, count, and resuspend at 4 × 106/ml for short restimulation or other experimental modifications. Restimulation should be performed using Dynabeads, at a minimum of 1:3 beads/cell ratio in complete T cell media (see Notes 9 and 10).
On an aliquot of rested and restimulated samples, stain for CD3, CD4, and CD69 to obtain purity and activation status.
Pellet cells and beads in microcentrifuge tubes (3–5 min ~8000 rpm spin in microcentrifuge). Aspirate the supernatant, and freeze pellets at −80 °C for lysis. Be sure to freeze in appropriate aliquots for experimental purposes. 1 × 106 is sufficient for RNA analysis, and K-ε-GG analysis requires ~250–300 million cells for sufficient protein.
Fig. 1.
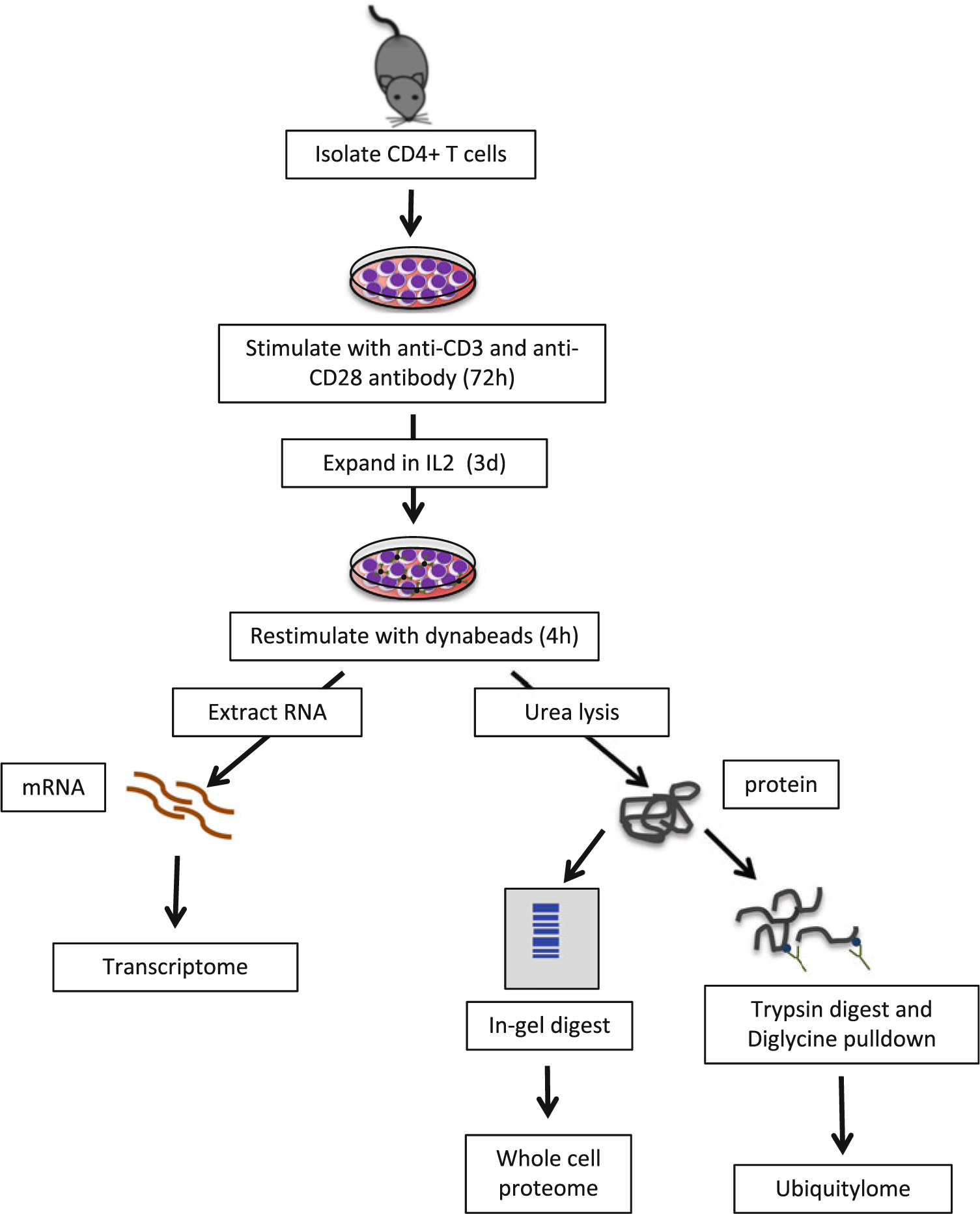
Workflow for sample preparation
3.2. K-ε-GG Pulldown
This K-ε-GG analysis method closely follows the method described by Steven Carr’s group [6] with modifications described here. Prior to beginning enrichment, crosslink antibody to beads and verify by gel electrophoresis.
Perform cell lysis using a slightly adapted lysis buffer (see Subheading 2.2, item 1).
Perform a BCA analysis on lysate to determine protein concentration. Use the manufacturer’s protocol.
Save about 30 μg of protein from the lysate for proteomic analysis (see Note 11).
Proceed to the immunoprecipitation, following the published protocol with the modifications described below. Note that 31ug of crosslinked antibody was used for our experiments, but this ratio may need to be determined empirically for different amounts of input protein.
After the immunoprecipitation, wash the bead/antibody complex as described; however for the third wash, supplement the IAP with 0.05% RapiGest. Continue with three PBS washes.
Perform elution as described.
Condition a 96 well Oasis HLB μElution plate using 100% acetonitrile (200 μl per well) under a Waters vacuum manifold.
Equilibrate the plate using 2× 200 μl washed of water with 0.1% trifluoroacetic acid (TFA).
Apply the eluted peptides to the plate.
Wash 3× 200 μl of water with 0.1% TFA.
Elute the peptides with 3× 80 μl 80% acetonitrile with 0.1% TFA.
3.3. Whole-Cell Proteome Preparation
Details on performing an in-gel digest can be found in Shevchenko et al. [7]. We use the following method to prepare the samples:
Mix proteomic sample (about 30 μg lysate) 1:1 with 4 Laemmli buffer containing DTT.
Fractionate using a 10% acrylamide gel (without a stacking gel). Mark gel cassette at the tenth fraction using a discarded excision template (~2 cm past well).
Run sample with unstained protein ladder at 120 V until dye front has passed the tenth fraction mark to get approximately 10–12 fractions. Additional fractions will yield a more complex proteome.
Stain gel in high-sensitivity Coomassie [11].
3.4. RNA-Seq Preparation
Protect RNA samples from degradation. Wipe down all sur-faces with the RNaseZap wipes to remove any RNases, and do not allow samples to leave the clean area. Use filter pipette tips for all RNA isolation steps, and always use gloves when handling any of the reagents.
Perform RNA isolation using the Qiagen RNeasy Plus Mini kit, following the kit protocol exactly.
RNA quality can be assessed using an Agilent bioanalyzer. RIN greater than eight is optimal. Quantity should be at least 200 ng per sample at a concentration of at least 10 ng/μl in RNase-free water.
RNA-seq can be carried out with Illumina technology using paired-end reads,100 base pair length, at a minimum of 30,000,000 read depth.
3.5. RNA-Seq Data Processing
There are a variety of alignment algorithms that can be implemented to align the RNA-seq reads to the reference genome [12]. This protocol describes the implementation of the STAR alignment program [10] and DESeq2 [8] to calculate differential expressions:
Obtain the reference genome to which the RNA-seq data will be aligned. The genome can be downloaded to a local directory from a genome browser such as Ensembl (https://www.ensembl.org/). The reference genome should match the species from which the RNA was harvested. The genomic sequence data is stored in fasta (*.fa) files and comprises one file for each chromosome in the genome. The corresponding *.gtf annotation file must also be downloaded with the sequence information (see Note 12).
Generate genome indexes of the reference genome using the STAR program. The STAR program is run using the “genomeGenerate” switch for the “--runMode” option. The users must also specify the path to the read files and annotation file using the options “--genomeFastaFiles” and “--sjdbGTFfile,” respectively.
Map the RNA-seq reads to the reference genome using the STAR program. The STAR program is run with the paths to the genome index directory and the read files specified using the options “--genomeDir” and “--readFilesIn,” respectively. Additionally, it is advantageous to use the option “--quant-Mode GeneCounts,” which will provide an output with the counts of aligned reads per gene. This data will be used to compute differential expression and associated statistics.
- Input the read count data into the R statistical software package, and format the data to generate a count matrix where the columns are samples, the rows are the reference genes, and the data comprises the un-normalized read counts generated by STAR. For example, samples identified by the identifications “ctrl_rep_1,” “ctrl_rep_2,” “knockout_rep_1,” and “knockout_rep_2” would be in the format:
# ctrl_rep_1 ctrl_rep_2 knockout_rep_1 knockout_rep_2 # ENSgene0001 count count count count # ENSgene0002 count count count count # ENSgene0003 count count count count # ENSgene0004 count count count count Filter the count matrix to remove genes that contain counts below a specified threshold.
- Generate a table relating the sample identification to the sample description (“condition”). For example, samples identified by identifications “control replicate 1,” “control replicate 2,” “knockout replicate 1,” and “knockout replicate 2” for conditions “control” and “knockout” would be in the format:
# Condition # control replicate 1 control # control replicate 2 control # knockout replicate 1 knockout # knockout replicate 2 knockout Input the data into DESeq2 using the command “DESeqDataSetFromMatrix.”
Calculate differential expression and associated statistics using the command “DESeq.”
Generate the results of DESeq2 differential expression using the command “results.” The results output the base mean, log2 fold change, log2 standard error, the Wald statistic, the Wald p-value, and Benjamini-Hochberg-adjusted p-value. The log2 fold changes and adjusted p-values are used to identify differentially expressed transcripts.
A volcano plot is a common and understandable way of representing differential expression data. A volcano plot graphs each identified transcript log2 fold change (x-axis) and corresponding log transformed adjusted p-value (y-axis) to visualize the magnitude of change and associated significance of expressed genes. Volcano plots can be generated using the software package ggplot2.
3.6. Whole-Cell Proteome Analysis
This protocol describes a mass spectrometry experiment consisting of two conditions with three biological replicates for each condition. Protein abundance is measured by label-free quantification. The raw data must be searched against the appropriate proteome with a program such as MaxQuant [13] or Open Mass Spectrometry Search Algorithm (OMSAA) [14]:
The output of the mass spectrometry search algorithm (e.g., the proteinGroups.txt file generated by MaxQuant) should be read into the preferred data manipulation/data analysis software (R, Excel, etc.). The output is composed of mass spectrometry data for which each identified protein is one row and the data for each protein is given in the respective columns. The relevant data for the differential protein expression analysis is the quantification parameter (iBAQ, LFQ, Intensity, etc.).
Remove the predicted contaminant proteins and reverse proteins from the dataset.
Convert all cases for which a protein was not quantified for the abundance measurement (a “blank” or a zero entry) to “NA” values.
Transform the protein abundance data into log base 2 form. NA values should remain NA.
Calculate the median or mean of the log2 transformed abundances for each sample in the experiment. NA values should not be included in this calculation as to not affect the average of the abundances of the identified proteins within the sample (see Note 13).
Normalize by adjusting the median (or mean) of the sample to “0.” Subtract the sample median (or mean) from each log2 transformed abundance value within the respective sample. This normalization accounts for technical variation, such as amount of protein input, in the mass spectrometry data across each replicate. The normalization by sample median (mean) allows for protein abundance comparison across samples.
Remove proteins with less than two out of three replicates identified for both conditions (see Note 14).
Calculate a z-score of the abundance for each protein by subtracting the protein abundance from the sample mean abundance and dividing by the sample standard deviation. The z-score characterizes the relative abundance of each protein within a sample. Proteins with z-scores of approximately zero exhibit average abundance levels. Proteins with high or low (negative) z-score exhibit relatively high or low abundance, respectively, within the sample.
Average protein abundance across replicates within each condition. Protein abundance fold changes measure the differential abundance of a respective protein in each condition. Log2 fold changes are calculated by subtracting protein log2 transformed abundance values of a “control” sample from the protein log2 transformed abundance of a “test” sample. Fold changes calculated in the log2 can be interpreted as increased abundance in the “test” condition for positive log2 fold changes and increased abundance in the control condition for negative log2 fold changes. Log2 fold changes near zero indicate approximately equal abundance in each condition.
Perform a t-test (two-tailed) using log2 transformed abundance values for each identified replicate comparing the “control” and “test” conditions. The null hypothesis assumes no change in abundance between the conditions, and a p-value <0.05 is generally considered statistically significant (see Note 15).
Protein differential expression is assessed by considering fold change and corresponding statistical significance.
A volcano plot is a common and understandable way of representing differential expression data. A volcano plot graphs each identified protein log2 fold change (x-axis) and corresponding log transformed p-value (y-axis) to visualize the magnitude of change and associated significance of expressed protein.
3.7. K-ε-GG Enrichment Analysis
The K-ε-GG analysis is very similar to the whole-cell proteome analysis. The major difference is that the K-ε-GG data abundance is generated on the peptide level rather than the protein level for whole-cell proteomics. However, the normalization, filtering, fold change, and statistics are computed the same way as described for the whole-cell proteome, except these calculations are done on the peptide level (i.e., using peptide abundance):
Peptide-based K-ε-GG modification abundance fold changes are converted to a protein-based fold change by calculating a weighted average of the peptide fold changes for each peptide identified within the respective protein. The peptide-based fold changes are weighted by modified peptide abundance, as measured by intensity level, such that the most abundant peptides constitute the highest impact to the protein fold change.
K-ε-GG modification fold changes are weighted by total protein changes, as determined from the corresponding whole-cell proteome, if a corresponding whole-cell proteome is available. The weighting is performed to determine whether the K-ε-GG fold change is driven by increases or decreases in total protein abundance. If there are increases or decreases in total protein abundance, the possibility of enriching for a K-ε-GG peptide from that protein is similarly increased or decreased. Differential changes in K-ε-GG abundance, after normalization for protein abundance, are used to classify increases or decreases in modification for purposes of identifying ubiquitination changes between the tested conditions.
Hypothesis testing is implemented to assess the significance of the changes observed in protein-based fold changes across conditions. The one sample t-test (two-tailed) is performed using the protein-based K-ε-GG fold change. The null hypothesis assumes no change in abundance between the conditions, and a p-value <0.05 is generally considered statistically significant (see Note 15).
A volcano plot is a common and understandable way of representing differential expression data. A volcano plot graphs each identified protein log2 fold change (x-axis) and corresponding log transformed p-value (y-axis) to visualize the magnitude of change and associated significance of expressed protein.
3.8. Validating Substrate Ubiquitination
We have used the UbiTest Agarose-TUBE elution kit from Life-Sensors to detect ubiquitination. The enriched ubiquitinated proteins can be treated with deubiquitinating enzymes (DUBs) that cleave all ubiquitin chains, allowing for a single band on a gel to identify a protein. The enriched proteins can also be treated with cleavage-specific DUBs to identify the type of ubiquitin chains. This protocol includes an option for a K48-specific DUB. For the enrichment, we recommend the manufacturer’s protocol with the following modifications:
Isolate and stimulate cells as described in Subheading 3.1, corresponding to the methods used for RNA-seq and proteomics data collection. Harvest cells in cold PBS, and wash three times. Pellet cells, and lyse immediately, or store at −80 °C.
Lyse cells in a Pierce RIPA (ThermoFisher)-based lysis buffer (see Subheading 2.6, item 1). Lyse cells in approximately 500 μl lysis buffer per 50 × 106 cells.
Lyse for 30 min on ice; vortex every 10 min.
Following incubation, centrifuge lysate for 20 min at maximum speed. Transfer supernatant to a clean tube (this is your lysate).
Perform BCA assay to test protein concentration.
Use 3–4 mg total protein as input for the pan-TUBE enrichment (approximately 100–150 × 106 cells)—keep in mind this is for three enrichments.
Equilibrate 150 μl slurry for the uncoupled agarose according to the manual. It is crucial to keep spin speed to 1000 × g.
Dilute input lysate 1:10 in PBS (or MilliQ). Add 0.1 mM PR619 and 50 μM o-PA and protease inhibitors at 1× concentrations.
Equilibrate 150 μl slurry for the TUBE-agarose according to the manual. It is crucial to keep spin speed to 1000 × g.
Add TUBE-agarose to the diluted lysate, and incubate at 4 °C, with rotation, for approximately 12 h.
Following incubation, spin agarose 1000 × g for 5 min. Collect supernatant for the unbound fraction if desired.
Wash agarose 3× with 1–2 ml TBST (0.1% tween). Following each wash collect beads by centrifuging at 1000 × g for 5 min, discard supernatant.
Resuspend agarose in 200–400 μl of supplied wash buffer.
Use desktop vortexer set to lowest speed to shake for 15 min at room temperature.
Centrifuge at 1000 × g for 5 min; discard supernatant.
Resuspend agarose in 150 μl of supplied elution buffer, and add 0.1 mM PR619 and 50 μM o-PA.
Use desktop vortexer set to lowest speed to shake for 25 min at room temperature.
Centrifuge at 4500 × g for 5 min and collect supernatant—this is your enrichment.
Neutralize supernatant by adding supplied 10× neutralization buffer at appropriate volume.
- If using DUBs, split sample into three equal parts:
- Leave one sample untreated.
- Add USP2core pan-DUB to second sample.
- Add Otub1 K48-specific DUB to third sample.
Incubate samples at 30 °C for 1.5 h.
Stop reaction by adding SDS sample buffer and boiling at 100 °C for 3–5 min. Your samples are now ready to load (see next section for Western blotting recommendations).
3.9. Validating Substrate Degradation
Since the proteomics studies require cells that have been expanded in vitro, using expanded, restimulated cells for initial validation studies is recommended. However, if you are interested in measuring degradation at earlier time points, it is also possible to treat with protein synthesis and degradation inhibitors within the first 24 h after activation:
Coat a plate with anti-CD3 anti-CD28 antibody as described in Subheading 3.1, step 3. We use 500 μl of this mixture per well of a 24 well plate or 200 μl per well of a 48 well plate. Incubate the plate at 37 °C for at least 2 h (this can be during the cell isolation).
- Harvest lymph nodes and spleens from mice as described in Subheading 3.1 (steps 1–6), and proceed to magnetic separation in step 7:
- When focusing on time points after the first 48 h of initial activation, we recommend using the Miltenyi CD4+ microbeads (see Note 7).
- If testing for degradation within the first 48 h of activation, we recommend using the Miltenyi naı¨ve CD4+ negative selection kit, in order to synchronize activation of the isolated cells.
Resuspend the cells in complete media with 50 U/ml IL-2 at a concentration of 1–1.5 million cells per ml.
Remove the coated plate, and aspirate off PBS.
Add the cell mixture to the plate (1–2 ml for a 24 well plate; 500 μl to 1 ml for a 48 well plate).
Spin the plate at 300 × g for 1 min to bring the cells to the bottom of the plate.
Place in the incubator for 72 h (if expanding/restimulating the cells) or for the desired time point.
- Inhibiting protein synthesis or degradation:
- For each experiment, at least four conditions are needed:
- An untreated sample collected prior to any drug treatments.
- An untreated sample allowed to remain in culture for the duration of the experiment (any protein changes between samples i and ii are reflective of the “normal” behavior of this protein).
- A cycloheximide-treated sample (to measure protein changes when new protein synthesis is blocked. Use cycloheximide stock at 1:10,000).
- An MG132- and chloroquine-treated sample (to measure protein changes when protein degradation is inhibited). Use 10 mM MG132 at 1:1000 (for 10 μM final concentration) and chloroquine at 1:1600 (dissolved as described in Subheading 2.6, item 5).
- Since chloroquine inhibits lysosomal degradation and MG132 inhibits proteasomal degradation, you can opt to use these drugs in two separate conditions to differentiate between these mechanisms.
- It is predicted that targets of ubiquitin-mediated degradation will decrease in abundance (relative to untreated) when treated with inhibitors of protein synthesis (e.g., cycloheximide) but will remain stable or increase when treated with inhibitors of degradation (e.g., MG132 and chloroquine) (see Note 16).
- Do not treat samples with drugs for longer than 6 h as cells will start to lose viability at this time.
- After desired treatment time (between 2 and 6 h), collect samples, and pellet for Western blot. They can be stored at −80 °C for several months.
Lyse samples in RIPA buffer supplemented with HALT phosphatase inhibitors (1:100) and 1× Roche Complete ETDA-free tablet (Described in Subheading 2.6, item 2).
Lyse on ice for 20 min; vortex occasionally.
Spin samples down at maximum speed for 10 min, and transfer supernatant to a new tube (this is your lysate).
Perform BCA assay to measure protein content.
Add 4× or 6× Laemmli sample buffer with 1:5 DTT added to each sample. Boil at 95 °C for 5 min. Your samples are now ready to load.
We typically use NuPage 4–12% precast gels, but other gel types may be appropriate depending on the size of your protein of interest. Run gel for 2 h at 120 V using NuPage running buffer diluted to 1×.
Transfer: We typically use PVDF membranes activated in methanol and transfer in 1× NuPage transfer buffer with 10% methanol on ice. Run transfer for 75 min at 45 V (may need to adjust for your protein).
Block in Odyssey blocking buffer diluted 1:1 with PBS for at least 30 min.
Blot with primary antibody overnight in Odyssey blocking buffer diluted 1:1 with PBS at 4 °C.
Wash 3× for 10 min each with PBS+ 0.1% tween.
Incubate with secondary antibody for 1 h at room temperature.
Repeat wash step.
Expose membrane using a Li-Cor imager or traditional film methods.
4. Notes
It is recommended to test reagents before expanding a large number of cells. Over the course of the stimulation and expansion, CD3+CD4+ T cells should retain their phenotype with minimal presence of contaminating populations (less than 5% desired), and cells should expand approximately tenfold in number.
Quantification of proteomic data can be through labeling or label-free methods. Common labeling strategies include stable isotope labeling with amino acids (SILAC) or alternative labeling such as TMT [15]. Note that no ratiometric quantification is possible if desired protein/peptide is absent from one sample. Additionally, for SILAC labeling, it is recommended to perform a label switch experiment as modified carbon isotopes can impact cellular phenotypes.
Consider the reproducibility of experimental manipulations to determine which stage is appropriate for sample pooling.
For harvesting lymph nodes, we typically use inguinal, cervical, axial, brachial, popliteal, and lumbar. Using lymph nodes from obese mice can result in significant cell death during preparation due to excess adipose. Younger mice are recommended. Depending on experimental conditions, co-housing of different genotypes (greater than 2 weeks) or use of littermates is highly preferred. Comparing mice from different colonies/ facilities should be avoided.
Lymph nodes and spleens from 2 to 3 mice can be processed together as one sample, but spleens should be kept separate until after RBC lysis.
Fetal calf serum (FCS) contains cytokines that may interfere with T cell differentiation. If the experiment involves polarizing conditions, cells should be harvested in serum-free media or plain Hank’s balanced salt solution (HBSS).
While positive selection using directly conjugated microbeads has been reliable, cost effective, fast and provides high level of purity after culture, there are several alternatives for enriching for CD4+ T cells. We have used negative selection using Miltenyi CD4+ T cell isolation kit, positive selection using CD4-PE and anti-PE microbeads, and positive selection using anti-CD4 hybridoma and anti-FC magnetic beads. Sorting is not recommended for large-scale cultures due to cell loss during preparation and the large starting numbers of cells required for the expanded cultures.
When culturing T cells, keep the cell concentration between one and two million cells per ml of media. Lower cell concentrations will result in loss of proliferation, and higher cell concentrations will lead to overgrowth.
Restimulation should be done with bead-bound anti-CD3/CD28 as opposed to plate-bound antibody to increase ease of harvesting cells after stimulation and reduce cell death; agitation in the presence of cross-linked soluble anti-CD3 and anti-CD28 may also be sufficient. A suboptimal bead/cell ratio of 1:3 in restimulation reduces costs and achieves approximately 70% stimulation, but note the manufacturer recommended restimulation condition is 1:1 bead/cell.
Protein modifications other than ubiquitination (such as neddylation) can leave behind diglycine remnants. Consider adding a Nedd8 inhibitor for the final 1–2 h of the restimulation if neddylation is expected to be a complicating factor in the experiment.
If using label-based quantification for proteomics, mix the comparator sample and experimental sample together after lysing based on protein quantification. Start by mixing a small fraction of each sample, and validate the mixture using LC/MS before scaling up.
The alignment of RNA-seq data to the reference genome must be performed on a high-performance computing cluster implementing a scheduling protocol, such as Sun Grid Engine, that allows for submission of jobs to compute nodes.
For proteomic data, lack of detection of a particular protein should not be interpreted as an abundance of zero. Typically, these values are assigned “NA” to indicated lack of data.
Reproducibility can be assessed in a variety of ways including by identification of unique peptides, number of peptides identified, or repeated abundance quantification across biological replicates. This protocol assumes reproducibility is assessed by quantification across biological replicates.
Depending on the power of the dataset and the number of proteins identified, it may be appropriate to perform a multiple testing correction [16].
MG132, chloroquine, and cycloheximide are nonspecific and may interfere with protein expression indirectly.
References
- 1.Fang D et al. (2002) Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol 3:281–287 [DOI] [PubMed] [Google Scholar]
- 2.Layman AAK et al. (2017) Ndfip1 restricts mTORC1 signalling and glycolysis in regulatory T cells to prevent autoinflammatory disease. Nat Commun. 10.1038/ncomms15677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Layman AAK, Oliver PM (2016) Ubiquitin ligases and deubiquitinating enzymes in CD4+ T cell effector fate choice and function. J Immunol 196:3975–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dybas JM et al. (2019) Integrative proteomics reveals that CD4+ T cell activation promotes predominantly non-degradative ubiquitylation. Nat Immunol 20(6):747–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Udeshi ND et al. (2013) Refined preparation and use of anti-diglycine remnant (K-e-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics 132:825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mertins P et al. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods 10:634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2007) In-gel digestion for mass spec-trometric characterization of proteins and proteomes. Nat Protoc. 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- 8.Love MI, Huber W, Anders S (2011) Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biol 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: ISBN 978-3-319-24277-4 [Google Scholar]
- 10.Dobin A et al. (2013) Sequence analysis STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyballa N, Metzger S (2009) Fast and sensitive colloidal Coomassie G-250 staining for proteins in polyacrylamide gels. Part 1: Two-dimensional (2-D) gel electrophoresis using cup-loading Part 2: Colloidal Coomassie staining with CBB G-250. J Vis Exp. 10.3791/1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baruzzo G et al. (2017) Simulation-based comprehensive benchmarking of RNA-seq aligners HHS Public Access. Nat Methods 14:135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319 [DOI] [PubMed] [Google Scholar]
- 14.Geer LY et al. (2004) Open mass spectrometry search algorithm, J Proteome Res. 10.1021/PR0499491 [DOI] [PubMed] [Google Scholar]
- 15.Tan H et al. (2017) Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46:488–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pascovici D, Handler DCL, Wu JX, Haynes PA (2016) Multiple testing corrections in quantitative proteomics: a useful but blunt tool. Proteomics 16:2448–2453. 10.1002/pmic.201600044 [DOI] [PubMed] [Google Scholar]