

Abstract
To examine the interactive effects of two proposed risk factors which may contribute to symptom severity of Autism Spectrum Disorder (ASD): prenatal antidepressant exposure and likely gene-disrupting (LGD) mutations. Participants included 2748 individuals with ASD from the Simons Simplex Collection. We examined the effects of prenatal antidepressant exposure, maternal depression, presence of an LGD mutation and their interaction on ASD severity. We found a significant interactive effect between antidepressant exposure and the presence of an LGD mutation on ASD severity in the ADOS and ADI-R verbal communication domains. We consider a “two-hit” model in which one variable lays the foundation for an initial risk which is compounded by a second variable.
Keywords: Autism, ASD, Antidepressants, SSRI, Genetics
Introduction
Autism Spectrum Disorder (ASD) is defined by DSM-5 as a single developmental disorder with a broad phenotype and wide-ranging severity (American Psychiatric Association 2013). With recent genetic sequencing advances, researchers have identified structural abnormalities and likely gene-disrupting (LGD) mutations which appear to contribute to ASD (Fischbach and Lord 2010; O’Roak et al. 2012; Sanders et al. 2015; Iossifov et al. 2014; Krumm et al. 2014; Hanson et al. 2015; Weiss et al. 2008; Girirajan et al. 2013).
To date, genetic events appear to contribute to ASD in approximately 40% of cases, although the phenotype ranges considerably even among individuals with similar events, implicating additional contributory factors (Hanson et al. 2015; Shishido et al. 2014). For example, one of the more common genetic contributions to ASD is a 16p11.2 deletion (Hanson et al. 2015; Weiss et al. 2008; Sanders et al. 2011; Zufferey et al. 2012) yet the phenotype among carriers is highly variable, with high rates of speech sound disorders and language disorders and less than 25% of carriers meeting diagnostic criteria for ASD (Hanson et al. 2015). Secondary genetic hits (Girirajan and Eichler 2010; Girirajan et al. 2010, 2012), genetic background (Chaste and Leboyer 2012), environmental exposures (Landrigan 2010; Shelton et al. 2014; Kalkbrenner et al. 2015), and epigenetic interactions (Mazina et al. 2015b) during early fetal development have been proposed as additional contributory factors. For example, prenatal exposure to maternal infection and medication show promise as likely early fetal development exposures associated with increased rates of ASD (Gardener et al. 2009; Mazina et al. 2015a).
That prenatal exposure to medication might be a factor involved in risk for ASD is important given the current prevalence of maternal use of a variety of medications. A recent study of medication use in pregnancy shows that maternal medication use during the first trimester has been steadily increasing since the 1970s (Mitchell et al. 2011). Of particular importance to this paper, antidepressant use during pregnancy has seen a dramatic change in prevalence (Cooper et al. 2007). The prevalence of maternal use of antidepressants during the first trimester was less than 1% in 1990, whereas by 2008 it was over 7% and rising, with the majority of antidepressants used being selective serotonin reuptake inhibitors (SSRIs) (Mitchell et al. 2011).
While there have been conflicting results (Hviid et al. 2013; Sorensen et al. 2013; Viktorin et al. 2017), some data suggest that prenatal exposure to SSRIs may increase the risk or severity of ASD (Croen et al. 2011; Man et al. 2015; Rai et al. 2013; Gentile 2015; Gidaya et al. 2014; Boukhris et al. 2015). These findings are supported by rodent models showing SSRI use during late pregnancy may have effects on social behavior of offspring, with implications for increased risk or severity of ASD symptomatology in humans (Zimmerberg et al. 2015). Serotonin-related genetic abnormalities have been identified as a potential underlying genetic vulnerability that may be exacerbated by prenatal SSRI exposure, ultimately affecting neurological changes in the fetal brain that are consistent with an ASD phenotype (Croen et al. 2011). However this explanation is speculative, and the above mentioned literature is conflicted regarding causality. Maternal depression plays a complex and potentially confounding role as an independent risk factor for increased ASD symptomatology in offspring, while driving maternal SSRI use and the associated risk factor of fetal SSRI exposure (Croen et al. 2011; Rai et al. 2013). Given that the heterogeneity of phenotype even within certain defined genetic groups (e.g. 16p11.2 CNV) is suggestive of moderating factors, these medication exposure findings raise the question of whether an interactive role exists between prenatal antidepressant exposure and LGD mutations associated with ASD.
In this paper, we sought to examine the interactive effects of prenatal antidepressant exposure, maternal depression, and the presence of LGD mutations on both clinician-observed and parent-reported ASD severity. We evaluate this risk in the broader context of treated versus untreated maternal psychiatric illness and its associated risks to both mother and fetus, a topical investigation given the prevalence of antidepressant use among pregnant women in the United States (Mitchell et al. 2011).
Methods
Study Population
Participants included 2748 individuals from the Simons Simplex Collection (SSC), including, 372 (13.5%) females and 2376 (86.5%) males with ASD. All selected participants utilized in this study have undergone whole exome sequencing (Iossifov et al. 2014), have complete phenotypic assessment data, and have extensive medical history including reported presence of prenatal exposure to antidepressants (Fischbach and Lord 2010).
The SSC represents a collaboration among 12 university-affiliated research clinics to recruit, evaluate and record data from over 2000 simplex families (Fischbach and Lord 2010; O’Roak et al. 2012; Iossifov et al. 2014; Sanders et al. 2011). Each family included one child with a diagnosis of ASD plus unaffected parents and siblings. Probands underwent comprehensive diagnostic evaluation by highly trained clinicians. The Autism Diagnostic Observation Schedule (ADOS) and Autism Diagnostic Interview-Revised (ADI-R) were performed to confirm an ASD diagnosis as defined by DSM-4-TR, thus fulfilling inclusion criteria. A full family medical history and blood sample for DNA sequencing were also taken. Local institutional review boards confirmed that all study participants gave informed consent.
Genetic Events
Genetic status for all participants in the study was culled from the study conducted by Iossifov et al. (2014), in which germline de novo (“new”) missense and likely gene-disrupting (LGD) mutations were identified through whole exome sequencing. Following the definitions outlined in Iossifov et al. 2014, de novo LGD mutation events are nonsense, splice site and frame-shift mutations that compromise protein function and whose targets are enriched for functional categories including FMRP-associated genes, chromatin modifiers, neurotransmitter receptors, ion channels, synaptic proteins, cytoskeletal remodelers and transcriptional regulators (Iossifov et al. 2014). The biological relatedness of these targets to neurological functions potentially relevant to ASD symptomatology ranges from direct to indirect to unknown. For the current analysis, genetic status was a dichotomous variable; individuals were identified as having an LGD mutation defined and listed in Supplementary Table #2 in Iossifov et al. 2014 or not.
Maternal Antidepressant Use
Antidepressant exposure was collected as part of SSC through parent interview and is defined as maternal antidepressant use at any point during pregnancy. Amtriptyline, buproprion, citalopram, escitalopram, fluoxetine, imipramine, paroxetine, sertraline and venlafaxine were noted on the SSC Medical History Form as examples of antidepressant medication. Data on maternal antidepressant use was collected by category: current, trimester one, trimester two, trimester three and “unsure.” Unsure was defined as maternal antidepressant use during pregnancy without specification regarding the time of use. Data about medication name, dosage, duration of treatment were not collected as part of participation in the SSC. We defined prenatal antidepressant exposure as exposure during one or more trimesters of pregnancy using a dichotomous variable.
Maternal History of Depression
Data on history of maternal psychiatric illness was also collected through parent interview using a standardized medical history interview. Endorsement of the Depressive Disorder and Major Depressive Disorder items were used to establish presence of maternal depressive disorder. If neither of the above diagnoses were endorsed, the mother was classified as not having a depressive disorder in our subsequent analysis. The interviewer instruction included the following sample question script: “Does or any of his/ her blood relatives have any [disorder name]? What about [disorder name]? Who?” Data related to the specific time or duration of maternal psychiatric illness—including if maternal psychiatric illness was present during pregnancy—were not collected.
Autism Severity
Clinician observation of ASD severity was measured through the ADOS, the Autism Diagnostic Observation Schedule, a semi-structured 30-min evaluation of play, social interaction and communication, which is used as a diagnostic tool for ASD (Lord et al. 2000). Clinicians administer one of five modules selected based generally on the individual’s expressive language abilities and age. The ADOS calibrated severity score (CSS) is based on total raw scores and standardized across modules to generate a metric of impairment ranging from 1 (unimpaired) to 10 (most impaired), with scores of 1–3 falling in the “no diagnosis” range (Gotham et al. 2009). The CSS is also calculated for social affect (SA) and restricted and repetitive behavior (RRB) subdomains, to estimate ASD severity in these subcategories (Hus et al. 2014).
Parent report of ASD severity was measured through the Autism Diagnostic Interview-Revised (ADI-R), a semi-structured caregiver interview emphasizing three behavioral domains: reciprocal social interaction; language and communication; and restricted and repetitive, stereotyped behaviors and interests (Lord et al. 1994). Scores are totaled for each domain.
Statistical Analysis
Using two 2 × 2 × 2 multifactorial ANOVAs, we examined the main effects of prenatal exposure (i.e. exposure or not), maternal depression (i.e., maternal diagnosis of depression or not), presence of an LGD mutation (i.e. mutation or not) and their interaction on ASD severity, assessed using first clinical report (ADOS overall and domain comparison scores) and second parent report (ADI-R domain total scores) of impairment.
In additional exploratory analyses, we examined in greater detail the study participants who had an LGD mutation and were exposed to antidepressants in utero. Because this restricted the sample size for each factor level, the additional analyses were descriptive and exploratory in nature.
Results
Sample characteristics and rates of LGD mutations, antidepressant exposure, and maternal depression are presented in Table 1. Mean age at testing for all 2748 participants was 108.3 months (approximately 9 years) with a standard deviation of 42.8 months and upper and lower ranges being 4 years 0 months and 17 years 11 months (these time frames would also represent the time between pregnancy and collection of maternal pregnancy information such as antidepressant use). In the study sample, 361 (13.1%) participants had a de novo LGD mutation and 114 (4.1%) participants were exposed to antidepressants in utero. There were nine study participants who had both an LGD mutation and prenatal antidepressant exposure. Details regarding specifically effected genes, maternal use of antidepressants by trimester, and maternal psychiatric diagnosis is presented in Table 2.
Table 1.
Study population broken down by presence of LGD mutation, prenatal antidepressant exposure, mean age and sex
| Mean age in months at ADOS | n Female | n Male | n Total (% total) | |
|---|---|---|---|---|
| LGD mutation (−) | 107.5 (~9.0 years) | 307 | 2080 | 2387 (86.9) |
| LGD mutation (+) | 113.2 (~9.4 years) | 65 | 296 | 361 (13.1) |
| Prenatal antidepressant exposure (−) | 108.9 (~9.0 years) | 359 | 2275 | 2634 (95.9) |
| Prenatal antidepressant exposure (+) | 94.3 (~7.9 years) | 13 | 101 | 114 (4.1) |
| Maternal depression (−) | 107.6 (~9.0 years) | 317 | 1972 | 2289 (83.3) |
| Maternal depression (+) | 111.6 (~9.3 years) | 54 | 392 | 446 (16.2) |
| Maternal Depression (missing) | N/A | N/A | N/A | 13 (0.5) |
| Total (n) | N/A | 372 | 2376 | 2748 |
Table 2.
LGD mutation, antidepressant use by trimester, & maternal psychiatric history for LGD and antidepressant interaction group individuals
| Individual ID | 11453.p1 | 12521.p1 | 12645.p1 | 12673.p1 | 13069.p1 | 13647.p1 | 13711.p1 | 13809.p1 | 14667.p1 |
|---|---|---|---|---|---|---|---|---|---|
| Variant | sub(G->A) | del(4) | sub(C->T) | del(4) | sub(G->A) | del(1), del(2) | ins(T) | del(2) | sub(G->A) |
| Effect gene | PTPN13 | USP15 | ANK2 | TNRC6B | TCF7L2 | IL3, THAP5 | HECTD1 | CDC23 | RUNDC3B |
| Effect type | Splice-site | Frame-shift | Nonsense | Frame-shift | Splice-site | Both frame-shift | Frame-shift | Frame-shift | Nonsense |
| Antidepressants—trimester 1 use | Missing | No | No | Yes | Yes | Yes | Yes | Yes | Yes |
| Antidepressants—trimester 2 use | Missing | Yes | No | No | Missing | No | Yes | Yes | Yes |
| Antidepressants—trimester 3 use | Missing | Yes | Yes | No | Missing | No | Yes | Yes | Yes |
| Antidepressants—trimester not sure | Yes | No | No | No | Missing | No | No | No | No |
| Antidepressants during pregnancy | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Maternal depressive disorder | Yes | Yes | No | No | Missing | Yes | No | Yes | Yes |
| Maternal anxiety disorder | No | Yes | No | No | Missing | No | No | No | No |
| Maternal bipolar disorder | No | No | No | No | Missing | No | No | No | No |
Self-reported maternal psychiatric illness and antidepressant use during pregnancy were not in total correspondence. Of the 114 individuals exposed to antidepressants in utero, 61 had a history of maternal depression, 28 had a history of maternal anxiety disorder and 10 had a history of maternal bipolar disorder. This leaves 15 individuals exposed to antidepressants in utero whose mothers reported no history of depressive, anxiety or bipolar disorder. Of course, antidepressants can be prescribed for reasons other than mood or anxiety (insomnia, pruritis, etc.), but data was not available which specifically illuminated this discrepancy in our study sample.
MANOVA revealed no main effects for prenatal antidepressant exposure, maternal depression, or presence of LGD mutations on clinician-measured (ADOS) or parent-reported (ADI-R) autism severity. However, a significant interactive effect between prenatal antidepressant exposure and the presence of LGD mutation was found on ASD severity in the overall ADOS comparison score: F (1, 2542) = 4.882, p = 0.027 (Fig. 1). A trend was observed for the ADOS social affect subdomain: F (1, 2542)=3.753, p=0.053 but no effect was observed for the restricted and repetitive subdomain: F (1, 2542) = 0.863, p = 0.353. Post-hoc analysis shows that the interaction was driven by differences between participants with both LGD mutations and prenatal antidepressant exposure versus those with LGD mutations only: F (1, 2542) = 4.540, p = 0.033 (Fig. 1).
Fig. 1.
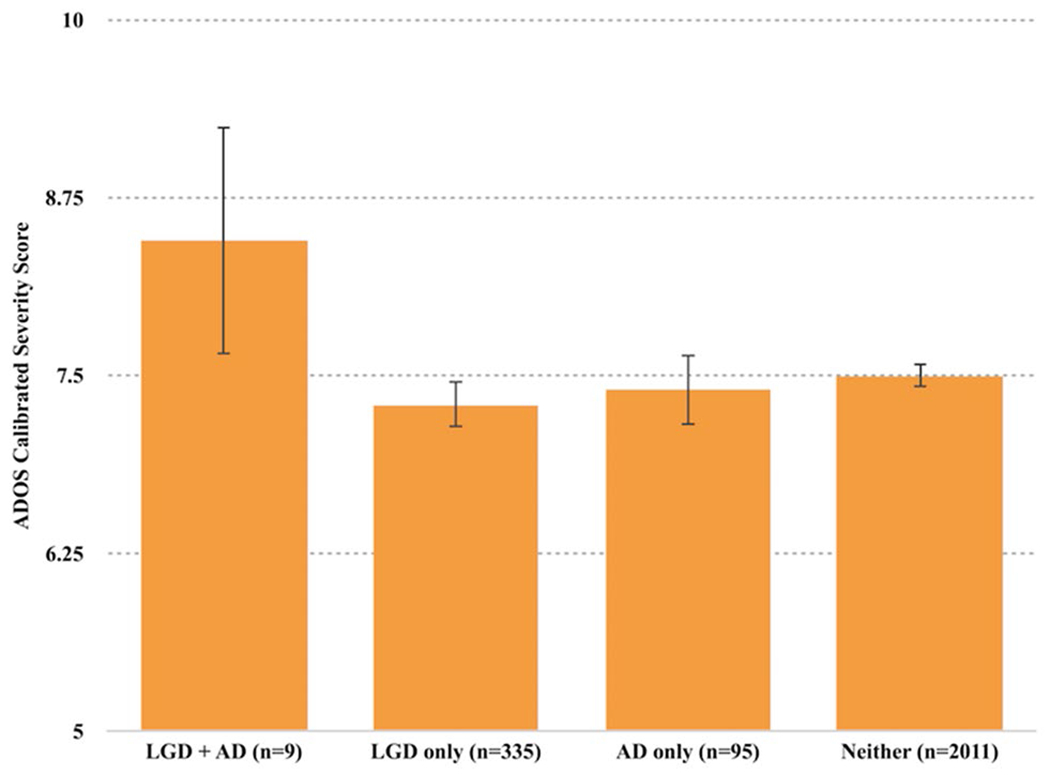
Interaction between antidepressant (AD) exposure and LGD mutation on ASD symptoms (ADOS CSS): significant interactive effect: F (1, 2542) = 4.882, p = 0.027. Error bars 95% CI
A significant interactive effect between prenatal antidepressant exposure and the presence of LGD mutations was found on the verbal communication domain score of the ADI-R as well: F (1, 2397) = 4.554, p = 0.033 (Fig. 2). Post-hoc analysis shows that the interaction was driven by differences between participants with prenatal antidepressant exposure and LGD mutations versus those with prenatal antidepressant exposure and without LGD mutations: F (1, 2397) = 4.174, p = 0.041 (Fig. 2).
Fig. 2.
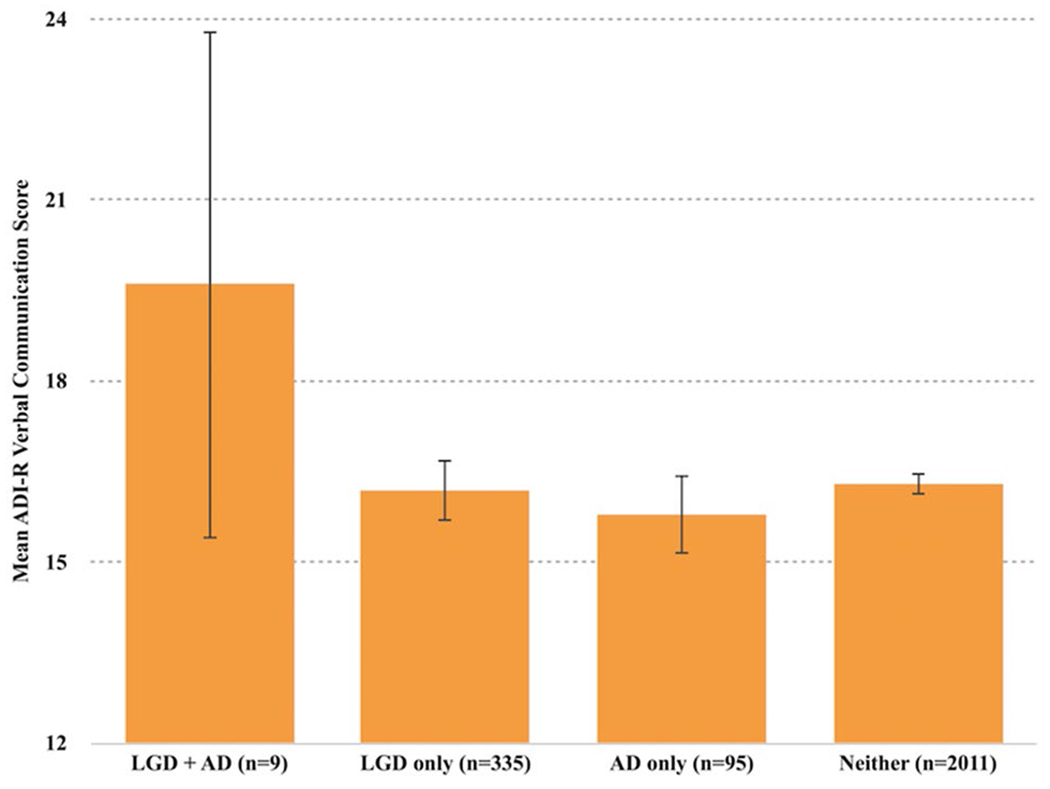
Interaction between antidepressant (AD) exposure and LGD mutation versus ASD severity (ADI-R): significant interactive effect in ADI-R verbal communication domain: F (1, 2397) = 4.554, p = 0.033. Error bars 95% CI
No other significant interactions were observed, including no interactive effect between maternal depression and LGD mutation on clinician-measured (ADOS) or parent-reported (ADI-R) autism severity.
In our exploratory analyses, which we pursued in the hopes a possible genetic mechanism for antidepressant and LGD interaction could be identified, we examined the nine individuals who had both an LGD mutation and prenatal antidepressant exposure. Factors we examined among these individuals included disrupted gene, location, type, variant, duration of antidepressant exposure during pregnancy by trimester and maternal psychiatric history. No obvious patterns emerged from this investigation that suggested a possible genetic mechanism for antidepressant and LGD mutation interaction.
In a second round of exploratory analysis, we examined the only three genes—ANK2, TNRC6B and TCF7L2—which had an LGD mutation in study participants with prenatal antidepressant exposure as well as those without prenatal antidepressant exposure. Three individuals in the entire study population had an ANK2 LGD mutation; however only one of those individuals was also exposed to antidepressants in utero. Two individuals, one exposed to antidepressants in utero and one not, had a TNRC6B LGD mutation. Likewise, two individuals, one exposed to antidepressants in utero and one not, had a TCF7L2 LGD mutation. Visual inspection of the data showed that all three individuals with both an LGD mutation plus prenatal antidepressant exposure showed increased ASD severity in the ADOS calibrated severity score and ADI-R verbal communication domain scores as compared to the individual(s) with the same LGD mutation and without prenatal antidepressant exposure.
A third round of exploratory analysis focused on the sex of the participants, given previous work suggesting the risk for ASD in those exposed to SSRIs in utero was strongest in males (Harrington et al. 2014). Of the 9 participants with both LGD mutations and antidepressant exposure, 8 (89%) are male. Of the 114 children with antidepressant exposure, 101 (also 89%) are male. There is a null finding when using Fisher’s exact test to examine the rate of males in the interaction group.
Discussion
In our analysis of over 2700 well-characterized, exome-sequenced children with ASD, we found a significant interaction between antidepressant exposure and the presence of an LGD mutation on clinician reported and parent reported ASD severity. We did not find an effect for antidepressant exposure on ASD severity, nor an effect for the presence of an LGD mutation or maternal depression on ASD severity. Further, there were no other significant interactions found, including no interaction between maternal depression and LGD mutation on ASD severity.
Given that our study was inspired by previous findings that prenatal antidepressant exposure increased ASD risk (Croen et al. 2011; Man et al. 2015; Rai et al. 2013; Gentile 2015; Gidaya et al. 2014; Boukhris et al. 2015), it is notable that we found no such effect in our own study. However methodological differences exist. Primarily, our study investigated whether prenatal antidepressant exposure increased ASD severity among those with an ASD diagnosis, not risk for the general population. In this sense our findings do not contradict previous studies.
We interpret our findings regarding maternal depression similarly. Our findings indicate that maternal depression is not associated with increased ASD symptomatology as measured by the ADOS and ADI-R domains. Whether maternal depression has an effect on ASD risk in the general population is a different question.
In the absence of main effects on ASD severity for prenatal antidepressant exposure, maternal depression or LGD mutations, we consider a “two-hit” (Girirajan et al. 2010; Leblond et al. 2012) model to explain why a significant interaction between prenatal antidepressant exposure and LGD mutations was found. By the logic of this model, one variable engenders an initial risk which is then compounded by a second variable, the result being that ASD symptomatology is more likely when both variables are present than either one alone. This model is distinct but perhaps related in theory to a dosage effect, such as that reported by Iossifov et al. identifying that the greater number of LGD mutations was associated with more cognitive impairment (2014). In our secondary analysis, we were compelled by the finding that those individuals with both an LGD mutation and prenatal antidepressant exposure were more impaired according to the ADOS calibrated severity score and ADI-verbal communication domain score than those individuals with the same LGD mutation and without prenatal antidepressant exposure. These findings are merely anecdotal given the sample size, but they also support a two-hit model whereby individuals with prenatal antidepressant exposure plus an LGD mutation exhibit increased ASD severity. Further analyses in individuals with the same genetic event are required.
While no obvious patterns emerged from our secondary analysis examining underlying genetic mechanisms guiding antidepressant and LGD mutation interaction, it is conceivable that future work may reveal connections between time and/or duration of prenatal antidepressant exposure and those genes expressed during corresponding stages of embryological development. Additionally, while the disrupted genes in our sample of children exposed to antidepressants appeared not to be overtly related, we can imagine a scenario in which antidepressants preferentially interact with certain gene products linked by pathway and/ or function.
Of note, previous work has found that antidepressant use in pregnancy seemed to be associated more with ASD particularly without intellectual disability (Rai et al. 2013). Some of the LGD mutations found in our participants are strongly associated with ASD and intellectual disability (Stessman et al. 2017) and some are not. This variability in our genes suggests the relationship is either beyond ASD alone or there is a different process associated with the interaction with LGD mutations.
There are a few limitations to this study that must be considered when interpreting the findings. First, the existing work associating antidepressant exposure and ASD is generally confined to SSRIs, whereas our data is concerning the broad category of antidepressant use. This should be considered an inevitable limitation of our study, given that maternal medication data in SSC was classified by class (e.g. “antidepressant”) rather than name (e.g. “sertraline”). That stated, the vast majority of antidepressants influence serotonergic pathways and an assumption could be made that most of the antidepressant use was via SSRIs. In support of this assumption, we emphasize that SSRIs were overwhelmingly prescribed for depression during the time period when all SSC participants were in utero, 1989–2007 (Mitchell et al. 2011). Three SSRIs are among the most typically used medications in pregnancy—fluoxetine, sertraline, and escitalopram—with no SNRIs, mood stabilizers, or other medications used as antidepressants commonly prescribed (Mitchell et al. 2011). Second, we also relied on parent report for data on maternal antidepressant use during pregnancy. While this data is likely not without flaws, a recent study on infection during pregnancy and ASD found that report of infection episodes by mothers matched corresponding hospital records reasonably well (Atladottir et al. 2012), substantiating the credibility of parent report. Third, while the Simons Simplex Collection is a very large study, the subset of participants we were most interested in—those with exposure to both LGD and prenatal antidepressant exposure—was small. This small sample also limited any ability to meaningfully examine various issues, such as timing of medication by trimester. This points to a need for larger or combined genetic studies with medical history collected as well.
There are some limitations of genetic analysis to take into account as well. We capitalized on the genomic analysis reported in Iossifov et al. (2014), through which the investigators applied whole exome sequencing to identify de novo protein truncating mutations associated with ASD. It is important to recognize that the sample under study in this project includes individuals with these identified protein truncating mutations, but does not mean that all other children in the sample are without genetic events that may or may not be contributing to ASD. That is, structural changes, inherited truncating variants to genes not associated with ASD at this time, rare variants that are equally as deleterious as protein truncating mutations, as well as variants and disruptive events occurring outside the exonic regions may be present in the individuals without LGD mutations. Despite the considerable noise in the data, the identification of the interactive effect suggests a distinct contribution of these high confidence ASD risk genes previously identified in this sample. Also, while not necessarily a limitation of the study, it is worth noting that while studies have shown the general population rate for some CNVs associated with ASD (Stefansson et al. 2014), population based rates are not available for the LGD mutations identified through exome sequencing in the SSC.
In summary, we found a significant interactive effect between antidepressant exposure and the presence of an LGD mutation on ASD severity in the ADOS and ADI-R verbal communication domains. We consider a “two-hit” model in which one variable lays the foundation for an initial risk which is compounded by a second variable. However, given the small sample size and other limitations, further research is needed to better understand the possible interactions of maternal depression, antidepressant exposure, and LGD mutations in regards to the etiology of autism.
Acknowledgments
We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman). We appreciate obtaining access to phenotypic data on SFARI Base.
Funding This work was supported by a grant from the Simons Foundation (SFARI #89368) and from the National Institute of Mental Health (#01MH100047 to R.B.). This study was funded by the Medical Student Research Training Program (MSRTP), University of Washington School of Medicine, Seattle, WA. The funding source did not participate in any part of the performance of the study.
Footnotes
The original version of this article was revised: The spelling of the co-author Sarah Schoenbrun was misspelled. This has been corrected in this version.
Approved researchers can obtain the SSC population dataset described in this study by applying at https://base.sfari.org.
Conflict of interest All authors report no conflict of interest.
Ethical Approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent Informed consent was obtained from all individual participants included in the study.
References
- American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders (5th edn.). Arlington, VA: American Psychiatric Publishing. [Google Scholar]
- Atladóttir HÓ, Henriksen TB, Schendel DE, et al. (2012). Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics, 130(6), e1447–e1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhris T, Sheehy O, Mottron L, et al. (2015). Antidepressant use during pregnancy and the risk of autism spectrum disorder in children. JAMA Pediatrics, 170(2), 117–124. [DOI] [PubMed] [Google Scholar]
- Chaste P, & Leboyer M (2012). Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues in Clinical Neuroscience, 14(3), 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper WO, Willy ME, Pont SJ, & Ray WA (2007). Increasing use of antidepressants in pregnancy. American Journal of Obstetrics and Gynecology, 196(6), 544.e1–5. [DOI] [PubMed] [Google Scholar]
- Croen LA, Grether JK, Yoshida CK, et al. (2011). Antidepressant use during pregnancy and childhood autism spectrum disorders. Archives of General Psychiatry, 68(11), 1104–1112. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, & Lord C (2010). The Simons Simplex Collection: A resource for identification of autism genetic risk factors. Neuron, 68(2), 192–195. [DOI] [PubMed] [Google Scholar]
- Gardener H, Spiegelman D, & Buka SL (2009). Prenatal risk factors for autism: Comprehensive meta-analysis. The British Journal of Psychiatry, 195(1), 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile S (2015). Prenatal antidepressant exposure and the risk of autism spectrum disorders in children. Are we looking at the fall of Gods? Journal of Affective Disorders, 182, 132–137. [DOI] [PubMed] [Google Scholar]
- Gidaya NB, Lee BK, Burstyn I, et al. (2014). In utero exposure to selective serotonin reuptake inhibitors and risk for autism spectrum disorder. Journal of Autism and Developmental Disorders, 44(10), 2558–2567. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Dennis MY, Baker C, et al. (2013). Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. American Journal of Human Genetics, 92(2), 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, & Eichler EE (2010). Phenotypic variability and genetic susceptibility to genomic disorders. Human Molecular Genetics, 19(R2), R176–R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Coe BP, et al. (2012). Phenotypic heterogeneity of genomic disorders and rare copy-number variants. The New England Journal of Medicine, 367(14), 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, et al. (2010). A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nature Genetics, 42, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotham K, Pickles A, & Lord C (2009). Standardizing ADOS scores for a measure of severity in autism spectrum disorders. Journal of Autism and Developmental Disorders, 39(5), 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Bernier R, Porche K, et al. (2015). Simons Variation in Individuals Project Consortium. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biological Psychiatry, 77(9), 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington RA, Lee LC, Crum RM, et al. (2014). Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics 133(5), 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hus V, Gotham K, & Lord C (2014). Standardizing ADOS domain scores: Separating severity of social affect and restricted and repetitive behaviors. Journal of Autism and Developmental Disorders, 44(10), 2400–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hviid A, Melbye M, & Pasternak B (2013). Use of selective serotonin reuptake inhibitors during pregnancy and risk of autism. The New England Journal of Medicine, 369(25), 2406–2415. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515(7526):216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkbrenner AE, Windham GC, Serre ML, et al. (2015). Particulate matter exposure, prenatal and postnatal windows of susceptibility, and autism spectrum disorders. Epidemiology 26(1), 30–42. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, et al. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends in Neurosciences, 37(2), 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ (2010). What causes autism? Exploring the environmental contribution. Current Opinion in Pediatrics 22(2), 219–225. [DOI] [PubMed] [Google Scholar]
- Leblond CS, Heinrich J, Delorme R, et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genetics, 8(2), e1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, et al. (2000). The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. Journal of Autism and Developmental Disorders, 30(3), 205–223. [PubMed] [Google Scholar]
- Lord C, Rutter M, & Le Couteur A (1994). Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders, 24(5), 659–685. [DOI] [PubMed] [Google Scholar]
- Man KK, Tong HH, Wong LY, et al. (2015). Exposure to selective serotonin reuptake inhibitors during pregnancy and risk of autism spectrum disorder in children: A systematic review and meta-analysis of observational studies. Neuroscience and Biobehavioral Reviews, 49, 82–89. [DOI] [PubMed] [Google Scholar]
- Mazina V, et al. (2015a). Interactive effects of copy number variation and maternal infection on autism impairment. Journal of Developmental and Behavioral Pediatrics: JDBP, 36(2), 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina V, Gerdts J, Trinh S, et al. (2015b). Epigenetics of autism-related impairment: Copy number variation and maternal infection. Journal of Developmental and Behavioral Pediatrics: JDBP, 36(2), 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AA, Gilboa SM, Werler MM, et al. (2011). Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. American Journal of Obstetrics and Gynecology, 205(51), e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485(7397), 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai D, Lee BK, Dalman C, et al. (2013). Parental depression, maternal antidepressant use during pregnancy, and risk of autism spectrum disorders: Population based case-control study. BMJ 346, f2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70(5), 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 Risk Loci. Neuron, 87(6), 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JF, Geraghty EM, Tancredi DJ, et al. (2014). Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The CHARGE study. Environmental Health Perspectives, 122(10), 1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido E, Aleksic B, & Ozaki N (2014). Copy-number variation in the pathogenesis of autism spectrum disorder. Psychiatry and Clinical Neurosciences, 68(2), 85–95. [DOI] [PubMed] [Google Scholar]
- Sorensen MJ, Gronborg TK, Christensen J, et al. (2013). Antidepressant exposure in pregnancy and risk of autism spectrum disorders. Clinical Epidemiology 15(5), 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, et al. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505(7483):361. [DOI] [PubMed] [Google Scholar]
- Stessman HA, Xiong B, Coe BP, et al. (2017). Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nature Genetics, 49(4), 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viktorin A, Uher R, Reichenberg A, et al. (2017). Autism risk following antidepressant medication during pregnancy. Psychological Medicine, 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. The New England Journal of Medicine, 358(7), 667–675. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, & Germeyan SC (2015). Effects of neonatal fluoxetine exposure on behavior across development in rats selectively bred for an infantile affective trait. Developmental Psychobiology, 57(2), 141–152. [DOI] [PubMed] [Google Scholar]
- Zufferey F, Sherr EH, Beckmann ND, et al. (2012). A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. Journal of Medical Genetics, 49(10), 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]