

Abstract
Background
Lacrimo‐auriculo‐dento‐digital (LADD) syndrome is a rare autosomal dominant disorder caused by mutations in one of the three genes: fibroblast growth factor receptor 2 (FGFR2), FGFR3, or FGF10. Affected patients have hypoplasia/aplasia of lacrimal ducts/glands, hypoplasia/aplasia of salivary glands, dental anomalies, ear anomalies, hearing loss, and digital anomalies.
Case Presentation
Proband was an 11‐year‐old male with xerostomia, xerophthalmia, and a referring diagnosis of Sjogren syndrome. He presented with microdontia, hypodontia, low‐set/cupped ear auricles, and hearing loss in the left ear.
Methods
Whole exome sequencing (WES) was performed on proband. Variations and segregation within the family were verified using Sanger sequencing.
Results
Molecular studies revealed a novel heterozygous missense mutation in exon 11 of FGFR2: c.1547C>T (p.Ala516Val), compatible with LADD syndrome.
Conclusion
To the best of our knowledge, this is the first report of a family with LADD syndrome in Korea. The combination of xerostomia and xerophthalmia, seen in patients with LADD syndrome, may be misdiagnosed as Sjogren syndrome. WES may be a useful clinical tool in ascertaining the affected gene in patients with suspected genetic disorders. Here, a literature review and summary of 23 case reports/series of LADD syndrome are presented, which may help to identify patients with this condition.
Keywords: lacrimo‐auriculo‐dento‐digital (LADD) syndrome, salivary gland agenesis, Sjogren syndrome, xerostomia
Lacrimo‐auriculo‐dento‐digital (LADD) syndrome is a rare autosomal dominant genetic disease. Patients affected have malformations of the lacrimal ducts or glands, aplasia or hypoplasia of major salivary glands, dental anomalies, low‐set cup‐shaped ears, hearing loss, and digital anomalies. Here we report a novel mutation in a Korean family with LADD syndrome and a review of previous literature.

1. INTRODUCTION
Lacrimo‐auriculo‐dento‐digital (LADD; OMIM#129730) syndrome is a rare autosomal dominantly inherited genetic disorder first described in 1973. (Hollister, Klein, De Jager, Lachman, & Rimoin, 1973, 1974; Levy, 1967). The term “LADD” was first introduced by Temtamy (1974). LADD syndrome is characterized by varying degrees of hypoplasia/aplasia of lacrimal ducts/glands, hypoplasia/aplasia of salivary glands, dental anomalies such as hypoplastic enamel, hypodontia, oligodontia, microdontia, inner and outer ear malformations, possible hearing loss, and digital anomalies (Wiedemann & Drescher, 1986). Affected individuals complain of xerophthalmia with irritation or infection, xerostomia, dental caries, and dysphagia (Kreutz & Hoyme, 1988; Lacombe, Serville, Marchand, & Battin, 1993; Wiedemann & Drescher, 1986). Digital malformations include aplasia, hypoplasia, syndactyly, clinodactyly, or duplications (Hollister, Klein, De Jager, Lachman, & Rimoin, 1973; Levy, 1967).
This genetic disorder is caused by mutations in any of the three known genes, fibroblast growth factor receptor 2 (FGFR 2) (OMIM#134920), fibroblast growth factor receptor 3 (FGFR3) (OMIM#134934), and fibroblast growth factor 10 (FGF10) (OMIM#602115), which is the ligand of FGFR2 (Rohmann et al., 2006; Shams et al., 2007). Mutations in FGF10 also result in aplasia of the lacrimal and salivary glands (ALSG; OMIM#180920), an allelic disorder (Scheckenbach, Balz, Wagenmann, & Hoffmann, 2008). The diagnosis of LADD syndrome depends on clinical, radiologic, and genetic findings. The major features of LADD syndrome (xerostomia, xerophthalmia) may also be seen in Sjogren syndrome, which may lead to a misdiagnosis.
2. CASE PRESENTATION
2.1. Case description
Proband was an 11‐year‐old Korean male with a history of xerostomia. He initially visited a local dentist for dental caries, enamel hypoplasia, and hypodontia, and was referred to a pediatric rheumatologist under specific concerns for Sjogren syndrome. He had dry skin and xerophthalmia, allergic conjunctivitis, and rarely shed tears. The patient had bilateral cryptotia and ear hypoplasia. He did not complain of any other neurologic, genitourinary, or gastrointestinal symptoms. His father was similarly affected but did not seek medical attention.
At the time of assessment, the patient weighed 33 kg (10 percentile) and was 141 cm tall (25 percentile). He had low‐set, small, cup‐shaped ears. An ophthalmic examination showed bilateral corneal punctate epithelial erosions, a bilateral tear break up time of 3 s, Schirmer test 3 and 4 mm for the right and left eye, respectively, and SICCA scores 4 and 5 for his right and left eye, respectively. There was no swelling of the parotid glands or submandibular glands. An intraoral examination showed a bald tongue and absence of the Stensen's duct orifice in both buccal mucosae. A dental examination showed multiple caries, enamel hypoplasia, microdontia, and hypodontia (mandibular second premolars). Digital findings included clinodactyly of the second and third toes of both feet (Figure 1). There was no facial dysmorphism, nail dysplasia, or other malformations such as cleft lip or palate.
Figure 1.
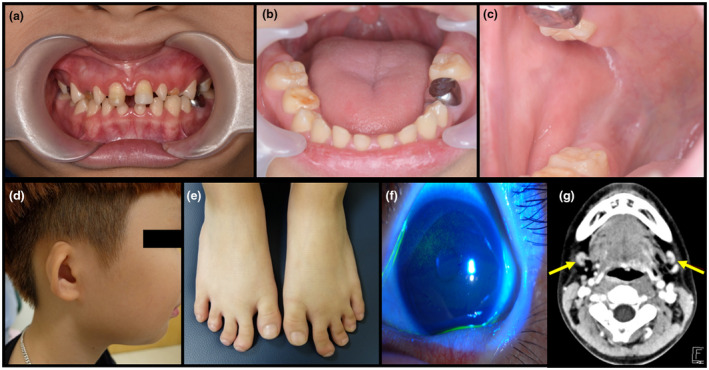
Clinical photographs and radiologic study of the proband. Proband displays severe xerostomia, multiple caries, enamel hypoplasia, microdontia, missing teeth, and a bald tongue (a and b). The orifice of the Stensen's duct is unidentifiable (c). He has low‐set, cup‐shaped ears (d). He has clinodactyly in the second and third toes of both feet (e). Slit lamp examination reveals punctate epithelial erosions on the cornea due to xerophthalmia (f). Computed tomography of the neck shows no visible parotid glands, with small enhancing nodular lesions in both sides of the neck, level IB (g)
Laboratory routine investigations were all normal, including Anti‐SSA/SSB antibodies. Ultrasonographic studies showed no identifiable submandibular or parotid gland structures, with small lymph nodes in its place. A contrast neck and orbit computed tomography scan showed no visible parotid glands, with small enhancing nodular lesions, possibly reactive lymph nodes, or hypoplastic submandibular glands (Figure 1‐g). Audiometry revealed left sensorineural hearing loss.
2.2. Ethical compliance
This study was independently reviewed and approved by the institutional review board of Seoul National University Dental Hospital and Seoul National University Hospital (H‐1904‐164‐1031). Appropriate informed consent was obtained from all participating individuals and blood samples were collected according to the principles of the Declaration of Helsinki.
2.3. Genetic analysis
Genomic DNA was isolated from peripheral blood samples of the proband and both of his parents. As a candidate gene approach, exons and intron‐exon boundaries of FGF10 were screened using Sanger sequencing, as previously described (Seymen et al., 2017). With no pathologic variant in FGF10, WES was performed on the proband. Paired‐end sequencing reads of 101 bp were obtained with the illumine HiSeq 2500 (Theragen Etex Bio Institute) after exome capturing with the Agilent SureSelect XT Human All Exon V5 Target Enrichment System. Sequencing reads were aligned to human reference genome hg19 and a series of bioinformatics analyses were performed as previously described (Koruyucu et al., 2018). Sanger sequencing was used to verify the genetic mutation in the proband and his father. In silico computational prediction analysis of variant was done to predict the pathogenic potential of the variant: Varsome (varsome.com) (Kopanos et al., 2019).
WES revealed a heterozygous novel missense mutation in exon 11 of FGFR2 (FGFR2: NM_022970.3): c.1547C>T (p.Ala516Val), compatible with the diagnosis of LADD syndrome. The novel variant and segregation within the family were verified using Sanger sequencing. His father had the same heterozygous missense mutation (Figure 2‐b). The proband and his father were clinically and genetically diagnosed with LADD syndrome. This pathologic variant was submitted to the ClinVar database (submission ID: SUB6906532).
Figure 2.
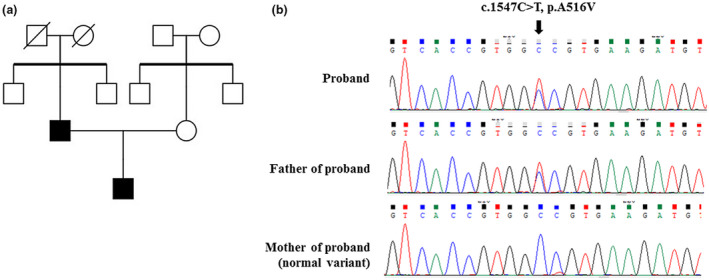
Family pedigree of the proband and genetic analysis of the proband and his parents. The family pedigree reflects an autosomal dominant inheritance pattern from father to son (a). Sequence chromatogram reveals a heterozygous variant c.1547C>T in FGFR2 leading to the A516V amino acid change in father and son (b).
3. DISCUSSION
To the best of our knowledge, this is the first report of a family with LADD syndrome in Korea. Both the patient and his father presented with xerostomia, cryptotia, and sensorineural hearing loss. Proband also had radiologically proven agenesis of parotid glands, no visible Stensen's duct orifices, dental malformations, xerophthalmia, and clinodactyly.
LADD syndrome, also known as Levy‐Hollister syndrome, is a rare autosomal dominant genetic disorder, with varying degrees of anomaly penetration. Previously reported cases had indicated varied phenotypes, including facial dysmorphism, genitourinary and renal anomalies, gastrointestinal symptoms, skeletal involvement other than digital or limb involvement, and airway involvement. Twenty‐three previous case reports and case series are summarized in Table 1 (Azar, Arnold, Scott, & Robin, 2000; Guven, Rosti, Tuna, Kayserili, & Aktoren, 2008; Hajianpour et al., 2017; Haktanir, Degirmenci, Acar, Albayrak, & Yucel, 2005; Hollister et al., 1973; Inan et al., 2006; Kreutz & Hoyme, 1988; Lacombe et al., 1993; Lehotay, Kunkel, & Wehrbein, 2004; Levy, 1967; Mathrawala & Hegde, 2011; McKenna, Burke, & Mellan, 2009; Meuschel‐Wehner, Klingebiel, & Werbs, 2002; Milunsky, Zhao, Maher, Colby, & Everman, 2006; Moses, 2013; Ostuni et al., 1995; Pathivada, Krishna, & Rallan, 2016; Ramirez & Lammer, 2004; Rohmann et al., 2006; Roodhooft, Brussaard, Elst, & Van Acker, 1990; Seymen et al., 2017; Shiang & Holmes, 1977; Talebi, Mardasi, Asl, Bavarsad, & Tizno, 2017; Thompson, Pembrey, & Graham, 1985). LADD syndrome may be genetically diagnosed, as it is known to be caused by mutations in FGFR2, FGFR3, and FGF10 (Rohmann et al., 2006; Shams et al., 2007). In the current case, the proband and his father showed a pathologic mutation in FGFR2.
Table 1.
Review of literature on LADD syndrome
| Reference | Age | Family history | Involved organs | Gene | Pathologic variant | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| F#(ind) | Lacrimal | Auricular/Hearing | Salivary | Dental | Digital/Limb | Other | ||||
| Levy (1967) | F1(1) | |||||||||
| 12 years | − | + | +/NR | + | + | + | ||||
| Hollister et al. (1973) | F1(6) | |||||||||
| 15 months | + | + | +/+ | NR | + | + | Congenital hip dislocation | |||
| 4 years | + | + | +/+ | NR | + | + | Left renal agenesis | |||
| 6 years | + | + | +/+ | NR | + | + | ||||
| 11 years | + | − | +/+ | NR | + | + | ||||
| 17 years | + | + | +/+ | NR | + | + | ||||
| 43 years | + | + | +/+ | NR | + | + | ||||
| Shiang and Holmes (1977) | F1(2) | |||||||||
| 21 y | + | + | −/− | + | + | + | Hypertension, nephrosclerosis | |||
| NR | + | + | −/+ | − | + | + | Diabetes mellitus | |||
| Thompson et al. (1985) | F1(2) | |||||||||
| 6 years | + | + | +/+ | + | + | + | Hiatus hernia, asthma | |||
| 32 years | + | + | +/+ | + | + | − | Stillbirth of second child | |||
| Kreutz and Hoyme (1988) | F1(3) | |||||||||
| 9 years | + | + | +/+ | NR | + | + | ||||
| NR | + | − | +/− | NR | + | + | ||||
| 18 hr | + | − | +/NR | NR | NR | + | Hyaline membranous disease, death | |||
| Roodhooft et al. (1990) | 11 years | − | + | +/− | − | + | + | Caliectasis and lithiasis of kidneys, Henoch‐Schonlein purpura with nephrotic syndrome | ||
| Lacombe et al. (1993) | F1(2) | |||||||||
| 22 months | + | + | +/+ | NR | + | + | Delayed psychomotor development, facial dysmorphism | |||
| 25 years | + | − | −/− | NR | + | + | Split foot deformity, split hand deformity | |||
| Ostuni et al. (1995) | F1(2) | |||||||||
| 24 years | + | + | +/NR | + | + | + | Facial dysmorphism, ptosis, bifid uvula | |||
| 4 years | + | + | NR/NR | NR | NR | + | ||||
| Azar et al. (2000) | 5 years | − | + | +/+ | NR | + | + | Bilateral hydronephrosis, epiglottic hypoplasia, micrognathia, undescended testicle, delayed psychomotor development | ||
| Meuschel‐Wehner et al. (2002) | 13 years | − | + | −/+ | + | + | + | Scoliosis, inner ear dysplasia | ||
| Ramirez and Lammer (2004) | F1(8) | |||||||||
| 6 years | + | − | +/− | + | + | + | Cleft lip and palate | |||
| 9 years | + | − | +/+ | NR | + | + | Pneumothorax, pulmonary hypertension, PDA, bilateral hydronephrosis and vesicoureteral reflux | |||
| NR | + | + | +/NR | NR | + | + | Vesicoureteral reflux, hydronephrosis, small scarred kidney, bicornuate uterus | |||
| NR | + | − | NR/NR | NR | + | + | ||||
| NR | + | NR | +/NR | NR | + | + | ||||
| NR | + | − | +/NR | NR | + | + | ||||
| NR | + | − | +/NR | NR | + | + | Arthritis (hands) | |||
| NR | + | NR | −/NR | NR | + | + | Congenitally blind (unknown cause) | |||
| Lehotay et al. (2004) | 13 years | − | NR | +/+ | + | + | + | Midfacial hypoplasia, delayed speech development | ||
| Haktanir et al. (2005) | F1(3) | |||||||||
| 14 years | + | + | +/+ | + | + | + | Ptosis, nail dysplasia, epilepsy, hypocalcemia, basal ganglia calcification | |||
| NR | + | NR | NR/+ | + | NR | + | ||||
| NR | + | + | NR/NR | NR | NR | NR | ||||
| Rohmann et al. (2006) | F1(7) | + | + | +/+ | − | + | + | Facial dysmorphism | FGFR2 |
c.1947‐AGA‐1949 del (p.R649S_D650del) |
| F2(6) | + | − | +/+ | − | + | + | FGFR2 | c.1942G>A (p.A648T) | ||
| F3(6) | + | + | +/+ | + | + | + | FGFR2 | c.1942G>A (p.A648T) | ||
| F4(1) | − | + | +/+ | + | + | + | FGFR2 | c.1882G>A (p.A628 T) | ||
| F5(4) | + | + | +/NR | NR | + | + | FGF10 | c.317G>T (p.C106F) | ||
| F6(3) | + | + | +/+ | − | + | + | Facial dysmorphism | FGFR3 | c.1537G>A (p.D513N) | |
| Milunsky et al. (2006) | F1(1) | |||||||||
| 3 years | − | + | +/− | + | + | + | Facial dysmorphism | FGF10 | c.467 \T>G (p. I156R) | |
| F2(2) | ||||||||||
| 19 years | + | + | −/− | + | + | − | ALSG | FGF10 | c.409A>T (p.K137X) | |
| 2 years | + | − | −/− | + | + | + | LADD | FGF10 | c.409A>T (p.K137X) | |
| F1(8) | ||||||||||
| Inan et al. (2006) | 13 years | + | + | +/+ | + | + | + | Facial dysmorphism, choanal atresia, ptosis, nail dysplasia, epilepsy, hypocalcemia | ||
| NR | + | − | − | − | + | − | ||||
| NR | + | − | − | + | − | − | ||||
| 19 years | + | − | −/+ | + | + | + | Epilepsy | |||
| 21 years | + | + | −/− | + | NR | NR | ||||
| NR | + | NR | −/− | NR | NR | + | ||||
| NR | + | NR | −/− | NR | + | NR | Divergent exotropia, anosmia | |||
| 4 years | + | − | −/− | − | + | + | ||||
| Guven et al. (2008) | F1(3) | |||||||||
| 17 years | + | + | +/+ | − | + | + | ||||
| 10 years | + | + | +/+ | − | + | + | ||||
| 45 years | + | + | −/+ | NR | + | + | ||||
| McKenna et al. (2009) | 12 years | − | − | +/− | − | + | + | Ptosis, chin deviation | ||
| Mathrawala and Hegde (2011) | 7 years | − | + | +/+ | + | + | + | Hydrocele, hypoplastic first rib, palpitations | ||
| Moses (2013) | 1.4 years | − | + | +/+ | + | + | + | Cervical vertebra fusion, hemivertebra, wide foramen magnum, inner ear dysplasia | ||
| Pathivada et al. (2016) | 7 years | NR | + | +/− | + | + | − | |||
| Hajianpour et al. (2017) | F1(3) | |||||||||
| 9 years | + | + | −/− | + | + | + | Facial dysmorphism, FTT, joint hypermotility | FGFR2 | Exon 15 and 16 del | |
| 2 years | + | + | −/− | + | − | − | Hypospadias, hypotonia, gross motor delay | FGFR2 | Exon 15 and 16 del | |
| NR | + | + | −/− | + | + | + | Strabismus, joint hypermobility | FGFR2 | Exon 15 and 16 del | |
| F2(3) | ||||||||||
| 3 years | + | + | +/− | + | + | + | Dolichocephaly, facial dysmorphism, micrognathia | FGF10 | c.401T>A (p.M134K) | |
| NR | + | + | −/− | + | + | NR | ||||
| NR | + | + | −/− | + | + | NR | Eczematous dermatitis, toenail dysplasia | FGF10 | c.401T>A (p.M134K) | |
| Talebi et al. (2017) | F1(2) | |||||||||
| 23 years | + | + | +/+ | NR | + | + | FGFR3 | c.1882G>A (p.D628N) | ||
| NR | + | + | +/+ | NR | NR | + | FGFR3 | c.1882G>A (p.D628N) | ||
| Present study (2020) | F1(2) | |||||||||
| 10 years | + | + | +/+ | + | + | + | FGFR2 | c.1547C>T (p.A516V) | ||
| NR | + | NR | NR | + | + | NR | FGFR2 | c.1547C>T (p.A516V) | ||
Abbreviations: F#, family number; FGF, fibroblast growth factor;FGFR, fibroblast growth factor receptors; ALSG, aplasia of the lacrimal and salivary glands; FTT, failure to thrive; ind, number of individuals; NR, not recorded; PDA, patent ductus arteriosus.
FGFR2 is a tyrosine kinase receptor with two alternative gene products: KGFR (keratinocyte growth factor receptor) and BEK (bacterially expressed kinase). Allelic variants of FGFR2 are associated with various phenotypes depending on the location of the mutation. Variants may result in phenotypes such as Crouzon syndrome, Jackson–Weiss syndrome, Apert syndrome, Pfeiffer syndrome, Beare–Stevenson Cutis Gyrata syndrome, Saethre–Chotzen syndrome, various cancers, and LADD syndrome (Lew, Bae, Rohmann, Wollnik, & Schlessinger, 2007).
Previous LADD syndrome reports have identified pathogenic FGFR2 variants which led to missense mutations in exon 16 and deletion mutations (Hajianpour et al., 2017; Rohmann et al., 2006). Our study reveals a novel variant in the first genetically analyzed Korean family with LADD syndrome. The proband and his father carried a single base substitution c.1547C>T (p.Ala516Val) in exon 11 of FGFR2 that resulted in a missense mutation inherited in an autosomal dominant pattern. Our study suggests that heterozygous FGFR2 c.1547C>T (p.Ala516Val) is a pathogenic variant.
LADD syndrome patients with FGFR2 mutations are characterized by lacrimal and salivary system abnormalities, auricular anomalies and hearing difficulties, dental and digital malformations, and variable facial dysmorphism and gastrointestinal involvement. Neither the proband nor his father in our study had associated facial dysmorphism or gastrointestinal symptoms.
Table 2 summarizes the genetically proven LADD syndromes in previous reports and our study. There were six proven pathologic variants for FGFR2, two for FGFR3, and four for FGF10. All the proven cases had dental and digital involvement. Three of the six variants of FGFR2 and both mutations of FGFR3 had no definite salivary gland involvement. In FGFR2 mutations, patients with deletion mutations had many anomalies in other organs compared to individuals with missense mutations. None of the patients with FGF10 mutations had hearing loss. Due to the small number of cases, it is difficult to prove a definite association between genotype and phenotype. There was also a range of phenotypes in families with the same genetic mutation, implying that additional factors influence the genotype to phenotype process.
Table 2.
Genotype phenotype correlation in LADD syndrome
| Gene | Pathologic variant | Inheritance pattern | Phenotype a | Reference | |
|---|---|---|---|---|---|
| L() A/H(/) S() De() Di() | Other | ||||
| FGFR2 | c.1947‐AGA‐1949 del (p.R649S_D650del) | AD | L(+) A/H(+/+)S(−)De(+)Di(+) | Facial dysmorphism | Rohmann et al. (2006) |
| c.1942G>A (p.A648T) | AD | L(−) A/H(+/+)S(−)De(+)Di(+) | Rohmann et al. (2006) | ||
| c.1942G>A (p.A648T) | AD | L(+) A/H(+/+)S(+)De(+)Di(+) | Rohmann et al. (2006) | ||
| c.1882G>A (p.A628T) | De novo | L(+) A/H(+/+)S(+)De(+)Di(+) | Rohmann et al. (2006) | ||
| Exon 15 and 16 del | AD | L(+) A/H(−/−)S(+)De(+)Di(+) | Facial dysmorphism, FTT, hypotonia, joint hypermotility, hypospadias, gross motor delay, strabismus | Hajianpour et al. (2017) | |
| c.1547C>T (p.A516V) | AD | L(+) A/H(+/+)S(−)De(+)Di(+) | Present study (2020) | ||
| FGFR3 | c.1537G>A (p.D513N) | AD | L(+) A/H(+/+)S(−)De(+)Di(+) | Facial dysmorphism | Rohmann et al. (2006) |
| c.1882G>A (p.D628N) | AD | L(+) A/H(+/+)S(?)De(+)Di(+) | Talebi et al. (2017) | ||
| FGF10 | c.317G>T (p.C106F) | AD | L(+) A/H(+/?)S(?)De(+)Di(+) | Rohmann et al. (2006) | |
| c.467T>G (p. I156R) | De novo | L(+) A/H(+/−)S(+)De(+)Di(+) | Facial dysmorphism | Milunsky et al. (2006) | |
| c.409A>T (p.K137X) | AD | L(+) A/H(−/−)S(+)De(+)Di(+) | Milunsky et al. (2006) | ||
| c.401T>A (p.M134K) | AD | L(+) A/H(+/−)S(+)De(+)Di(+) | Dolichocephaly, facial dysmorphism, micrognathia, eczematous dermatitis, toenail dysplasia | Hajianpour et al. (2017) | |
Abbreviations: ?, not recorded; A, auricular; AD, autosomal dominant; De, dental; Di, digital; H, hearing; L, lacrimal; S, salivary.
For pathologic variants with many affected individuals, if at least one individual had the phenotype, it was recorded as positive.
The combination of xerostomia and xerophthalmia, seen in patients with LADD syndrome, can simulate Sjogren syndrome (Hajianpour et al., 2017) (Ostuni et al., 1995). While Sjogren syndrome is an immune disorder targeting tear glands and salivary glands, LADD syndrome is a genetic disorder causing dysgenesis or agenesis of the lacrimal duct/glands and salivary glands. The incidence or prevalence of pediatric Sjogren syndrome patients has not been reported but is very low compared to that of the adult population. This highlights the importance of considering other disorders such as LADD syndrome when diagnosing patients suspected of Sjogren syndrome, especially in children because of the low prevalence rate of pediatric Sjogren, 0.098% to 3.6% (Petty et al., Textbook of Pediatric Rheumatology 7th edition).
When LADD syndrome is clinically suspected, sequencing and del/dup analysis of the three known genes associated with this condition may identify the pathogenic mutation. Otherwise, whole exome sequencing (WES) can be utilized when a clinical diagnosis of LADD syndrome is not made. WES can be used to diagnose patients with a suspected genetic condition who are not clinically diagnosed with a defined genetic syndrome. Utilizing genetic analysis such as WES may ascertain the affected gene for purposes of genetic counseling, identification of novel variants, decisions regarding therapy, and prevention of unnecessary treatment. In this case, we initially performed candidate sequencing of the proband's FGF10. However, this targeted approach did not reveal a mutation in FGF10. With a strong clinical impression of LADD syndrome, we performed WES on the proband and identified a novel variant in FGFR2. This variant was verified by Sanger sequencing and revealed a heterozygous autosomal dominant inheritance pattern between father and son. In silico computational prediction analysis with Varsome marked it as likely pathogenic (Kopanos et al., 2019). WES may prove to be an important genetic diagnostic tool for patients with a suspected genetic condition who are not clinically diagnosed with a defined genetic syndrome.
4. CONCLUSION
We report the first LADD syndrome in a Korean family with a proven mutation. This novel, most likely pathogenic, variant in FGFR2: c.1547C>T (p.Ala516Val) may be used to identify patients with this genetic condition. For patients referred under the impression of Sjogren syndrome, rheumatologists are recommended to examine the patient for other anomalies carefully. If multiple systemic involvements are present, LADD syndrome should be considered as a possible diagnosis. WES may help identify patients with a suspected genetic condition who are not clinically diagnosed with a defined genetic syndrome. Computational prediction analysis tools may further assist validating novel genetic variants as pathogenic.
Ryu YH, Kyun Chae J, Kim J-W, Lee S. Lacrimo‐auriculo‐dento‐digital syndrome: A novel mutation in a Korean family and review of literature. Mol Genet Genomic Med. 2020;8:e1412 10.1002/mgg3.1412
[Correction added on 05 August 2020, after first online publication: in Correspondence section, Jung‐Wook Kim has been added as co‐corresponding author.]
Contributor Information
Jung‐Wook Kim, Email: pedoman@snu.ac.kr.
Soyoung Lee, Email: leadethme@naver.com.
REFERENCES
- Azar, T. , Arnold, J. E. , Scott, J. A. , & Robin, N. H. (2000). Epiglottic hypoplasia associated with lacrimo‐auriculo‐dental‐digital syndrome. Annals of Otology, Rhinology & Laryngology, 109, 779–781. 10.1177/000348940010900814 [DOI] [PubMed] [Google Scholar]
- Guven, Y. , Rosti, R. O. , Tuna, E. B. , Kayserili, H. , & Aktoren, O. (2008). Orodental findings of a family with lacrimo‐auriculo‐dento digital (LADD) syndrome. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology, 106, e33–e44. 10.1016/j.tripleo.2008.07.019 [DOI] [PubMed] [Google Scholar]
- Hajianpour, M. J. , Bombei, H. , Lieberman, S. M. , Revell, R. , Krishna, R. , Gregorsok, R. , … Milunsky, J. M. (2017). Dental issues in lacrimo‐auriculo‐dento‐digital syndrome: An autosomal dominant condition with clinical and genetic variability. The Journal of the American Dental Association, 148, 157–163. 10.1016/j.adaj.2016.11.016 [DOI] [PubMed] [Google Scholar]
- Haktanir, A. , Degirmenci, B. , Acar, M. , Albayrak, R. , & Yucel, A. (2005). CT findings of head and neck anomalies in lacrimo‐auriculo‐dento‐digital (LADD) syndrome. Dentomaxillofacial Radiology, 34, 102–105. 10.1259/dmfr/65931528 [DOI] [PubMed] [Google Scholar]
- Hollister, D. W. , Klein, S. H. , De Jager, H. J. , Lachman, R. S. , & Rimoin, D. L. (1973). The lacrimo‐auriculo‐dento‐digital syndrome. The Journal of Pediatrics, 83, 438–444. 10.1016/s0022-3476(73)80268-9 [DOI] [PubMed] [Google Scholar]
- Hollister, D. W. , Klein, S. H. , de Jager, H. J. , Lachman, R. S. , & Rimoin, D. (1974). Lacrimo‐auriculo‐dento‐digital (LADD) syndrome. Birth Defects Original Article Series, 10, 153. [PubMed] [Google Scholar]
- Inan, U. U. , Yilmaz, M. D. , Demir, Y. , Degirmenci, B. , Ermis, S. S. , & Ozturk, F. (2006). Characteristics of lacrimo‐auriculo‐dento‐digital (LADD) syndrome: Case report of a family and literature review. International Journal of Pediatric Otorhinolaryngology, 70, 1307–1314. 10.1016/j.ijporl.2005.12.015 [DOI] [PubMed] [Google Scholar]
- Kopanos, C. , Tsiolkas, V. , Kouris, A. , Chapple, C. E. , Albarca Aguilera, M. , Meyer, R. , & Massouras, A. (2019). VarSome: The human genomic variant search engine. Bioinformatics, 35, 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koruyucu, M. , Kang, J. , Kim, Y. J. , Seymen, F. , Kasimoglu, Y. , Lee, Z. H. , … Kim, J. W. (2018). Hypoplastic AI with highly variable expressivity caused by ENAM mutations. Journal of Dental Research, 97, 1064–1069. 10.1177/0022034518763152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutz, J. M. , & Hoyme, H. E. (1988). Levy‐Hollister syndrome. Pediatrics, 82, 96–99. [PubMed] [Google Scholar]
- Lacombe, D. , Serville, F. , Marchand, D. , & Battin, J. (1993). Split hand/split foot deformity and LADD syndrome in a family: Overlap between the EEC and LADD syndromes. Journal of Medical Genetics, 30, 700–703. 10.1136/jmg.30.8.700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehotay, M. , Kunkel, M. , & Wehrbein, H. (2004). Lacrimo‐auriculo‐dento‐digital syndrome. Journal of Orofacial Orthopedics/Fortschritte Der Kieferorthopädie, 65, 425–432. 10.1007/s00056-004-0347-6 [DOI] [PubMed] [Google Scholar]
- Levy, W. J. (1967). Mesoectodermal dysplasia: A new combination of anomalies. American Journal of Ophthalmology, 63, 978–982. 10.1016/0002-9394(67)90043-8 [DOI] [PubMed] [Google Scholar]
- Lew, E. D. , Bae, J. H. , Rohmann, E. , Wollnik, B. , & Schlessinger, J. (2007). Structural basis for reduced FGFR2 activity in LADD syndrome: Implications for FGFR autoinhibition and activation. Proceedings of the National Academy of Sciences of the United States of America, 104, 19802–19807. 10.1073/pnas.0709905104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathrawala, N. R. , & Hegde, R. J. (2011). Lacrimo‐auriculo‐dento‐digital syndrome. Journal of Indian Society of Pedodontics and Preventive Dentistry, 29, 168 10.4103/0970-4388.84693 [DOI] [PubMed] [Google Scholar]
- McKenna, G. , Burke, F. , & Mellan, K. (2009). Presentation of lacrimo‐auriculo‐dento‐digital (LADD) syndrome in a young female patient. European Archives of Paediatric Dentistry, 10, 35–39. 10.1007/BF03262698 [DOI] [PubMed] [Google Scholar]
- Meuschel‐Wehner, S. , Klingebiel, R. , & Werbs, M. (2002). Inner ear dysplasia in sporadic lacrimo‐auriculo‐dento‐digital syndrome. ORL; Journal for Oto‐Rhino‐Laryngology and Its Related Specialties, 64, 352–354. 10.1159/000066077 [DOI] [PubMed] [Google Scholar]
- Milunsky, J. , Zhao, G. , Maher, T. , Colby, R. , & Everman, D. (2006). LADD syndrome is caused by FGF10 mutations. Clinical Genetics, 69, 349–354. 10.1111/j.1399-0004.2006.00597.x [DOI] [PubMed] [Google Scholar]
- Moses, J. (2013). Lacrimo‐auriculo‐dento‐digital syndrome with unilateral inner ear dysplasia and craniocervical osseous abnormalities: Case report and review of literature. Clinical Neuroradiology, 23, 221–224. 10.1007/s00062-012-0170-1 [DOI] [PubMed] [Google Scholar]
- Ostuni, P. A. , Modolo, M. , Revelli, P. , Secchi, A. , Battista, C. , Tregnaghi, A. , … Todesco, S. (1995). Lacrimo‐auricolo‐dento‐digital syndrome mimicking primary juvenile Sjogren's syndrome. Scandinavian Journal of Rheumatology, 24, 55–57. 10.3109/03009749509095158 [DOI] [PubMed] [Google Scholar]
- Pathivada, L. , Krishna, M. K. , & Rallan, M. (2016). A case of lacrimo‐auriculo‐dento‐digital syndrome with multiple congenitally missing teeth. Case Reports in Dentistry, 2016, 8563961 10.1155/2016/8563961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, D. , & Lammer, E. J. (2004). Lacrimoauriculodentodigital syndrome with cleft lip/palate and renal manifestations. The Cleft Palate‐Craniofacial Journal, 41(5), 501–506. 10.1597/03-080.1 [DOI] [PubMed] [Google Scholar]
- Rohmann, E. , Brunner, H. G. , Kayserili, H. , Uyguner, O. , Nürnberg, G. , Lew, E. D. , … Wollnik, B. (2006). Mutations in different components of FGF signaling in LADD syndrome. Nature Genetics, 38, 414–417. 10.1038/ng1757 [DOI] [PubMed] [Google Scholar]
- Roodhooft, A. , Brussaard, C. , Elst, E. , & Van Acker, K. (1990). Lacrimo‐auriculo‐dento‐digital (LADD) syndrome with renal and foot anomalies. Clinical Genetics, 38, 228–232. 10.1111/j.1399-0004.1990.tb03574.x [DOI] [PubMed] [Google Scholar]
- Seymen, F. , Koruyucu, M. , Toptanci, I. R. , Balsak, S. , Dedeoglu, S. , Celepkolu, T. , … Kim, J.‐W. (2017). Novel FGF10 mutation in autosomal dominant aplasia of lacrimal and salivary glands. Clinical Oral Investigations, 21, 167–172. 10.1007/s00784-016-1771-x [DOI] [PubMed] [Google Scholar]
- Shams, I. , Rohmann, E. , Eswarakumar, V. P. , Lew, E. D. , Yuzawa, S. , Wollnik, B. , … Lax, I. (2007). Lacrimo‐auriculo‐dento‐digital syndrome is caused by reduced activity of the Fibroblast growth factor 10 (FGF10)‐FGF receptor 2 signaling pathway. Molecular and Cellular Biology, 27, 6903–6912. 10.1128/MCB.00544-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang, E. L. , & Holmes, L. B. (1977). The lacrimo‐auriculo‐dento‐digital syndrome. Pediatrics, 59, 927–930. [PubMed] [Google Scholar]
- Talebi, F. , Mardasi, F. G. , Asl, J. M. , Bavarsad, A. H. , & Tizno, S. (2017). Identification of a novel missence mutation in FGFR3 gene in an Iranian family with LADD syndrome by Next‐Generation Sequencing. International Journal of Pediatric Otorhinolaryngology, 97, 192–196. 10.1016/j.ijporl.2017.04.016 [DOI] [PubMed] [Google Scholar]
- Temtamy, S. A. (1974). Letter: On the nomenclature of "new" syndrome. The Journal of Pediatrics, 84, 608–609. 10.1016/s0022-3476(74)80693-1 [DOI] [PubMed] [Google Scholar]
- Thompson, E. , Pembrey, M. , & Graham, J. (1985). Phenotypic variation in LADD syndrome. Journal of Medical Genetics, 22, 382–385. 10.1136/jmg.22.5.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann, H.‐R. , & Drescher, J. (1986). LADD syndrome: Report of new cases and review of the clinical spectrum. European Journal of Pediatrics, 144, 579–582. 10.1007/BF00496040 [DOI] [PubMed] [Google Scholar]