

Abstract
Background
Congenital scoliosis (CS) is a spinal deformity due to vertebral malformations. Although insufficiency of TBX6 dosage contributes to a substantial proportion of CS, the molecular etiology for the majority of CS remains largely unknown. TBX6‐mediated genes involved in the process of somitogenesis represent promising candidates.
Methods
Individuals affected with CS and without a positive genetic finding were referred to this study. Proband‐only exome sequencing (ES) were performed on the recruited individuals, followed by analysis of TBX6‐mediated candidate genes, namely MEOX1, MEOX2, MESP2, MYOD1, MYF5, RIPPLY1, and RIPPLY2.
Results
A total of 584 patients with CS of unknown molecular etiology were recruited. After ES analysis, protein‐truncating variants in RIPPLY1 and MYF5 were identified from two individuals, respectively. In addition, we identified five deleterious missense variants (MYOD1, n = 4; RIPPLY2, n = 1) in TBX6‐mediated genes. We observed a significant mutational burden of MYOD1 in CS (p = 0.032) compared with the in‐house controls (n = 1854). Moreover, a potential oligogenic disease‐causing mode was proposed based on the observed mutational co‐existence of MYOD1/MEOX1 and MYOD1/RIPPLY1.
Conclusion
Our study characterized the mutational spectrum of TBX6‐mediated genes, prioritized core candidate genes/variants, and provided insight into a potential oligogenic disease‐causing mode in CS.
Keywords: congenital scoliosis, exome sequencing, mutational burden, TBX6‐mediated genes
Our study characterized the mutational spectrum of TBX6‐mediated genes, prioritized core candidate genes/variants, and provided insight into a potential oligogenic disease‐causing mode in CS.

1. INTRODUCTION
Congenital scoliosis (CS) is a spinal deformity caused by malformations of vertebrae, which include the failure of formation (CS type I), failure of segmentation (CS type II), or a combination of the two (CS type III) (Hedequist & Emans, 2007). With an incidence of 0.5–1 per 1000 live births, CS is a major contributor to childhood and adolescent disability (Shen, Wang, Liu, Xue, & Qiu, 2013).
In our previous studies, we found that TBX6 gene contributes to about 10% of CS patients with a compound inheritance disease‐causing mode, that is, a TBX6 null mutation or 16p11.2 deletion in trans with a common T‐C‐A (rs2289292, rs3809624, and rs3809627) haplotype (Liu et al., 2019; Wu et al., 2015; Yang et al., 2019). We defined this entity of patients as TBX6‐associated congenital scoliosis (TACS) (Liu et al., 2019). In addition to null mutations and copy number deletions, missense variants in TBX6 which lead to a functional null effect also lead to TACS in combination with the risk T‐C‐A allele (Chen et al., 2020). In contrast to this mutation + polymorphism combination, biallelic loss‐of‐function or dominant‐negative mutations in TBX6 cause Spondylocostal Dysostosis (MIM#122600), a severe skeletal dysplasia syndrome characterized by multiple segmentation defects of the spine and costal dysplasia(Sparrow et al., 2013). Although TBX6 mutations contribute to a substantial proportion of CS, the molecular etiology for the majority of CS remains largely unknown, and more candidate genes/loci warrant investigation.
During the process of somite segment determination, TBX6 activates mesoderm posterior 2 (MESP2) (Morimoto, Takahashi, Endo, & Saga, 2005; Oginuma, Niwa, Chapman, & Saga, 2008; Takahashi et al., 2000; Yasuhiko et al., 2006). MESP2 then promotes the formation of somite boundary via activation of RIPPLY1/RIPPLY2, which negatively regulates TBX6 expression (Nakajima, Morimoto, Takahashi, Koseki, & Saga, 2006; Takahashi et al., 2010). MESP2 also promotes the expression of MYOD1 (Bondue et al., 2008; Stamataki, Kastrinaki, Mankoo, Pachnis, & Karagogeos, 2001; Windner et al., 2015), which then activates several pre‐myogenic mesoderm factors essential for myogenesis and somitogenesis including MYF5, MEOX1, and MEOX2 (Gianakopoulos et al., 2011; Stamataki et al., 2001).
Of these TBX6‐mediated genes, RIPPLY2, MESP2, and MYF5 are associated with known Mendelian syndromes involving skeletal abnormalities. Similar with TBX6, biallelic loss‐of‐function variants in RIPPLY2 or MESP2 cause autosomal recessive Spondylocostal Dysostosis (MIM#616566 for RIPPLY2 and MIM#608681 for MESP2). Recessive MYF5 variants are recently reported to be associated with External Ophthalmoplegia, Rib, and Vertebral Anomalies (MIM#618155), whose spine phenotype highly resembles that of Spondylocostal Dysostosis (Di Gioia et al., 2018). Therefore, these TBX6‐mediated genes together represent a promising candidate gene set for CS. However, due to the milder symptom of CS compared with the above‐mentioned recessive Mendelian syndromes, the effect of the candidate variants in CS probably vary and their disease‐causing modes are presumably complicated.
To explore the mutational landscape of TBX6‐mediated genes in CS, and to give insight into the potential multi‐factorial disease‐causing mode, we performed exome sequencing (ES) on 584 individuals with congenital scoliosis without a prior molecular diagnosis, and then, studied both heterozygous variants and combinations of variant alleles.
2. MATERIALS AND METHODS
2.1. Ethical compliance
Approval for the study was obtained from the ethics committee at Peking Union Medical College Hospital (JS‐098). Written informed consent was provided by each participant.
2.2. Patient recruitment
We consecutively recruited individuals who are affected with CS in Peking Union Medical College Hospital from 2009 to 2016, as part of the Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) study (http://www.discostudy.org/).
2.3. Exome sequencing
Proband‐only exome sequencing was performed on patients (1) without a molecular diagnosis of TACS, that is, not caused by defects of TBX6 gene; (2) without a molecular diagnosis by any other known CS‐causing gene. In brief, genomic DNA was extracted from the peripheral blood. Illumina paired‐end libraries were prepared from DNA samples and subjected to exome capture using one of four capture kits (xGEN targeted capture kit [IDT], seqcap pure capture kit [Nimblegen], VCRome SeqCap EZ Choice HGSC 96 Reactions [Roche], and All Exon V6+UTR r2 core design [91 Mb, Agilent]), depending on the time of enrollment in our cohort, followed by sequencing on an Illumina HiSeq 2000/4000 platform.
2.4. Variant calling and annotation
Raw data from exome sequencing were processed using PUMCH Pipeline (PUMP) (Wang et al., 2018; Zhao et al., 2020). GRCh37 or hg19 (GenBank accession number: GCA_000001405.1) was used as the reference sequence. HaplotypeCaller function of Genome Analysis Toolkit (GATK), version 3.4.0. was used for calling of single‐nucleotide variants and small insertion/deletions (indels). Combined Annotation Dependent Depletion (CADD) (Kircher et al., 2014) were used to predict the conservation and pathogenicity of the variants. Minor allele frequency (MAF) of each variant was obtained from public databases including the 1000 Genomes Project (http://www.internationalgenome.org/), the Exome variant server, NHLBI GO Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), and the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/).
2.5. Analytic strategy
TBX6‐mediated genes were curated through literature review. Only genes regulated by TBX6 during somitogenesis were selected, including MEOX1, MEOX2, MESP2, MYOD1, MYF5, RIPPLY1, and RIPPLY2. Variants in candidate genes were filtered against population databases. Because a MAF of 0.01 is generally used to distinguish a variant from a polymorphism, we define a rare variant as having a MAF lower than 0.01. Taken into consideration the disease incidence of congenital scoliosis (0.0005–0.001) (Shen et al., 2013), we suggest that a variant with MAF ≥0.001 should not be pathogenic in a dominant disease trait. Therefore, a MAF cutoff of 0.001 was used to filter for ultrarare variants. In our analysis, ultrarare variants (MAF ≤0.001) were selected for monogenic analysis and rare variants (MAF ≤0.01) were selected for potential oligogenic analysis. Protein‐truncating variants (including stop‐gain, frameshift, splice donor, splice acceptor, and start‐loss variant), predicted deleterious missense variants (CADD score ≥20) and in‐frame indels were prioritized. Sanger sequencing was performed on protein‐truncating variants, deleterious missense variants, and variants presenting an oligogenic disease‐causing mode.
2.6. In‐house controls
In‐house controls are consisted of 1854 unrelated individuals without apparent skeletal deformities. ES were performed on these individuals using the same protocol as employed in CS cases.
2.7. Statistics
R (version 3.6.1) was used for the statistical analysis. Fisher's Exact Test was used to compare the burden of deleterious missense variants with MAF ≤0.001 between cases and controls.
3. RESULTS
3.1. Mutational spectrum of ultrarare variants in TBX6‐mediated genes
We recruited 584 individuals affected with CS that could not be explained by TBX6 or other CS‐causing genes (Figure 1). After ES data processing and variant filtering, we identified a total of 28 ultrarare (MAF ≤0.001) variants in TBX6‐mediated genes (Figures 2 and 4). Of them, seven variants are protein‐truncating or predicted to be deleterious (CADD >15), presenting a tendency toward a significant mutational burden as compared with the in‐house controls (nine ultrarare deleterious variants in 1854 control samples, p = 0.08, Fisher's Exact Test).
Figure 1.
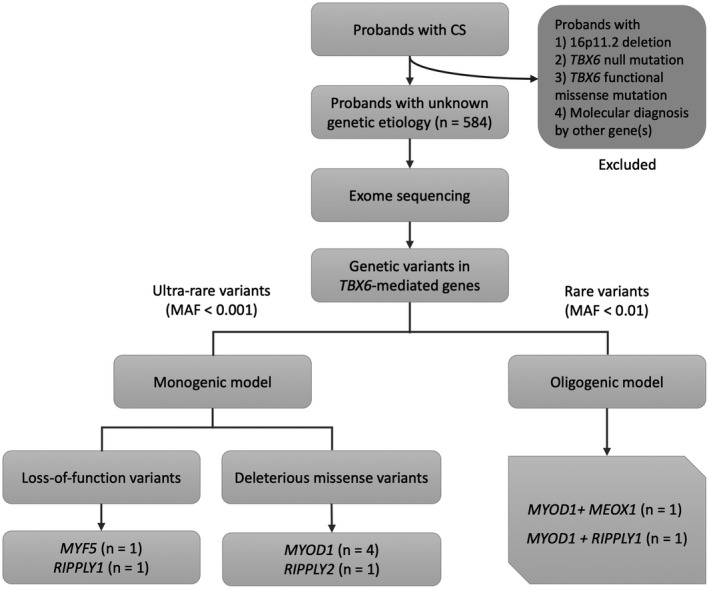
Workflow and main findings of the study
Figure 2.
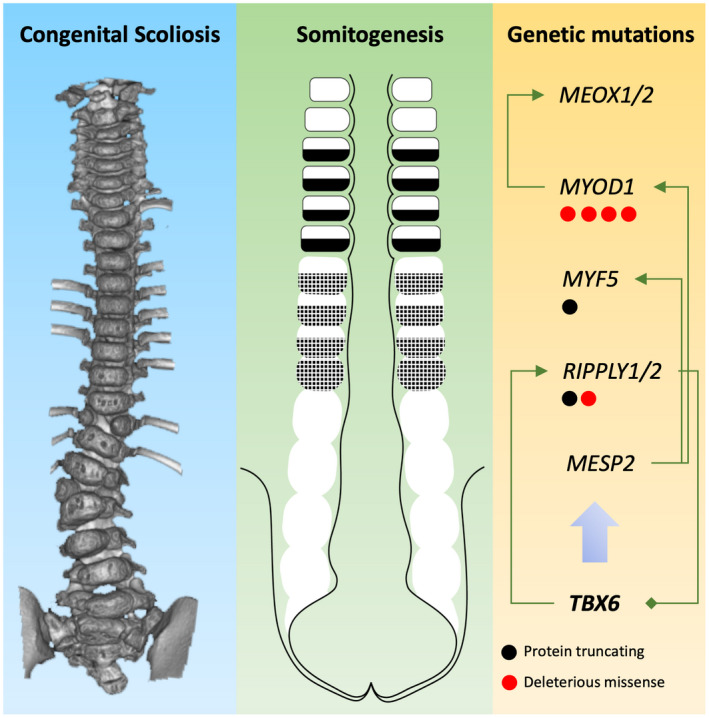
Developmental and molecular etiology of congenital scoliosis. The illustration demonstrates how genetic mutations identified in TBX6‐mediated genes may alter the process of somitogenesis and whereby cause congenital scoliosis. Arrows between gene symbols indicate activation. The diamond between RIPPLY1/2 and TBX6 indicates a negative feedback between the molecules
Figure 4.
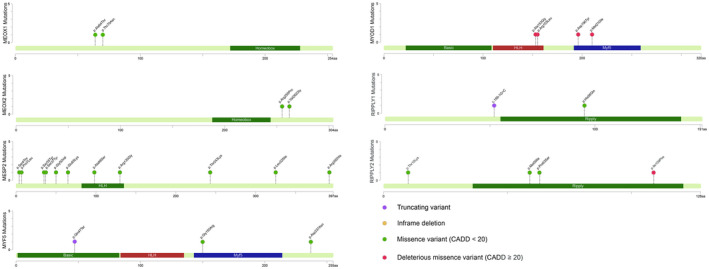
Mutational spectrum of the candidate genes in CS with unknown molecular etiology. Mapping of genetic variants in candidate genes to protein sequences annotated with functional domains
3.2. Identification of protein‐truncating variants in RIPPLY1 and MYF5
We first analyzed variants predicted to cause complete loss‐of‐function of the proteins. As a result, two protein‐truncating variants in TBX6‐mediated genes were identified from two patients (Table 1).
Table 1.
Protein‐truncating and deleterious missense variants
| Case ID | Zygosity | Chr | Pos | VAF | Mutation type | Gene | Variant | gnomAD‐MAF | gnomAD‐EAS‐MAF | ExAC‐MAF | ExAC‐EAS‐MAF | Gerp++ | CADD | pLI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSS161458 | Hem | X | 106145446 | 100 | Splice acceptor | RIPPLY1 | c.156‐1G>C | 0 | 0 | 0 | 0 | 4.33 | — | 0.01 |
| CSS160633 | Het | 12 | 81110981 | 48.39 | Nonsense | MYF5 | c.139C>T(p.Gln47Ter) | 0 | 0 | 0 | 0 | 6.17 | 36 | 0 |
| CSS161458 | Het | 11 | 17741793 | 50 | Missense | MYOD1 | c.464G>T(p.Arg155Leu) | 0 | 0 | 0.000026 | 0.00036 | 4.74 | 35 | 0.15 |
| CSS161565 | Het | 11 | 17741787 | 52 | Missense | MYOD1 | c.458C>G(p.Ala153Gly) | 0 | 0 | 0.000026 | 0.00036 | 4.88 | 34 | 0.15 |
| CSS161580 | Het | 11 | 17741915 | 52.63 | Missense | MYOD1 | c.586G>T(p.Asp196Tyr) | 0 | 0 | 0 | 0 | 4.74 | 23 | 0.15 |
| CSS170323 | Het | 11 | 17741959 | 53.49 | Missense | MYOD1 | c.630G>C(p.Met210Ile) | 3.23E‐05 | 0.0006 | 0.000015 | 0.00017 | 4.55 | 22.2 | 0.15 |
| CSS170010 | Het | 6 | 84567046 | 44.44 | Missense | RIPPLY2 | c.325A>T(p.Ile109Phe) | 0 | 0 | 0 | 0 | 5.64 | 21.5 | 0.01 |
GRCh37 or hg19 (GenBank accession number: GCA_000001405.1) was used as the reference sequence.
Abbreviations: CADD, Combined Annotation Dependent Depletion; Chr, chromosome; EAS, East Asian population; MAF, minor allele frequency; pLI, probability of loss‐of‐function intolerance; Pos, position; VAF, variant allele frequency.
A hemizygous splice‐donor variant c.156‐1G>C in RIPPLY1 was identified in CSS161458, a 6‐year‐old boy affected with CS type II. CSS161458 had a right thoracic curve (magnitude of 40°, apex located at T9) with multiple segmentation failures from T8 to L1 (Figure 3). Although RIPPLY1 has not been associated with a human developmental disease, its paralog, RIPPLY2, causes Spondylocostal Dysostosis (MIM#616566) in an autosomal recessive mode. In addition, knock‐out of Ripply1/Ripply2 in mouse results in disrupted somitogenesis and causes gross spinal deformity (J. Takahashi et al., 2010), suggesting the important role of RIPPLY1 in the normal development of the spine. Our data suggest that truncating variants in RIPPLY1, in a hemizygous state, could be associated with vertebrae malformations in human.
Figure 3.
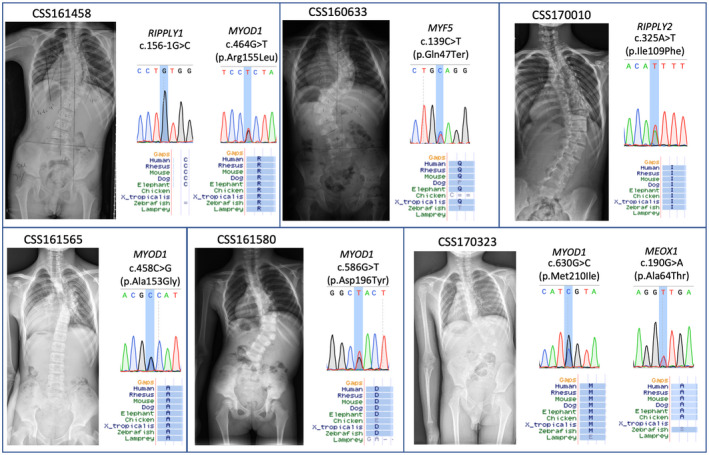
Images and mutational information of patients with protein‐truncating or deleterious missense variants. Spine X‐rays of six patients with truncating variants or deleterious missense variants are presented. Sanger sequencing results and residue conservation of these variants are also shown
A heterozygous stop‐gain variant in MYF5 was identified in CSS160633, a 3‐year‐old boy affected with type I CS. He had two curves located at thoracic spine and lumbar spine, with wedge vertebrae located at apex region separately (Figure 3). Recently, biallelic frameshift and deleterious missense variants in MYF5 has been associated with External Ophthalmoplegia, Rib, and Vertebral Anomalies (MIM#618155). The spinal phenotypes of this syndrome include cervical and thoracic scoliosis, cervical fusions, clivus malformations, basilar invagination, and narrow disc spaces, which represents a more complex condition than that in our patient. Therefore, we propose that heterozygous loss‐of‐function of MYF5 might be associated with a non‐syndromic form of CS.
3.3. Deleterious missense variants
In addition to protein‐truncating variants, we identified five deleterious missense variants (n = 4 for MYOD1, n = 1 for RIPPLY2) in TBX6‐mediated genes (Table 1).
Notably, we observed an excess of ultrarare deleterious missense variants in MYOD1 as compared with the in‐house control (4/584 in CS cases vs. 2/1854 in controls, p = 0.032, Fisher's Exact Test). These findings suggest the possible involvement of MYOD1 in the pathogenesis of CS. MYOD1 encodes an early transcriptional factor during somitogenesis, and is required for MYF5 expression in the early mesoderm (Maguire, Isaacs, & Pownall, 2012). Intriguingly, two out of four deleterious missense variants in MYF5 were adjacently located in the HLH domain (Figure 4), which is essential for the transcriptional activity of MYF5 (Hamamori, Wu, Sartorelli, & Kedes, 1997). Therefore, these two variants c.464G>T(p.Arg155Leu) and c.458C>G(p.Ala153Gly) represent promising candidates in our analysis, and suggest the perturbation of the HLH domain as a potential etiology of CS.
3.4. Potential oligogenic disease‐causing mode
To give further insight into the complex molecular mechanism of CS, we analyzed variant combinations of TBX6‐mediated genes. As a result, we found two patients with a potential oligogenic inheritance (Table 2).
Table 2.
Potential oligogenic variants identified in two probands
| Case ID | Zygosity | Chr | Pos | VAF | Mutation type | Gene | Variant | gnomAD‐AF | gnomAD‐EAS‐AF | ExAC‐AF | ExAC‐EAS‐AF | Gerp++ | CADD | pLI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSS170323 | Het | 11 | 17741959 | 53.49 | Missense | MYOD1 | c.630G>C(p.Met210Ile) | 3.23E‐05 | 0.0006 | 0.000015 | 0.00017 | 4.55 | 22.2 | 0.15 |
| CSS170323 | Het | 17 | 41738713 | 50.43 | Missense | MEOX1 | c.190G>A(p.Ala64Thr) | 3.24E‐05 | 0.0006 | 0.000075 | 0.001 | 2.64 | 7.499 | 0 |
| CSS161458 | Het | 11 | 17741793 | 50 | Missense | MYOD1 | c.464G>T(p.Arg155Leu) | 0 | 0 | 0.000026 | 0.00036 | 4.74 | 35 | 0.15 |
| CSS161458 | Hom | X | 106145446 | 100 | Splice acceptor | RIPPLY1 | c.156‐1G>C | 0 | 0 | 0 | 0 | 4.33 | 10.73 | 0.01 |
GRCh37 or hg19 (GenBank accession number: GCA_000001405.1) was used as the reference sequence.
Abbreviations: CADD, Combined Annotation Dependent Depletion; Chr, chromosome; EAS, East Asian population; MAF, minor allele frequency; pLI, probability of loss‐of‐function intolerance;Pos, position; VAF, variant allele frequency.
CSS170323 carries a heterozygous missense variant c.630G>C(p.Met210Ile) in MYOD1 and a heterozygous missense variant c.190G>A(p.Ala64Thr) in MEOX1 (Table 2). CSS170323 presented with L2 hemivertebra and fused ribs (the right 11th rib and 12th rib). During mesoderm development, the expression of MEOX1 is increased by MYOD1 (Gianakopoulos et al., 2011), suggesting that these two variant potentially result in the cumulative perturbation of TBX6‐mediated pathway.
CSS161458 had a heterozygous splicing variant c.156‐1G>C in RIPPLY1, as described above, and a heterozygous missense variant c.464G>T(p.Arg155Leu) in MYOD1 was also identified. Although no direct interaction between RIPPLY1 and MYOD1 has been reported, they may together dysregulate the TBX6 pathway given the deleterious nature of both variants (Table 2).
4. DISCUSSION
In this study, we performed exome sequencing on 584 patients with CS and without a molecular diagnosis. Variants in seven TBX6‐mediated genes involved in somitogenesis were selected for analysis. Protein‐truncating variants, in‐frame indels and deleterious missense variants were prioritized. Potential oligenic disease‐causing modes were also identified.
The candidate gene strategy has been widely used in parallel sequencing studies on presumptive genetic disorders (Lin et al., 2020; Wu et al., 2019). The biological relevance and the rationality of the candidate gene set are critical for the success of this strategy. For example, focusing on genes encoding ion channels could promote the identification of novel genes in epilepsy (Epi & Epilepsy Phenome/Genome, 2017); the use of a systemic multiple candidate gene approach also explains a substantial proportion of the heritability in complex traits such as hypertension (Ji et al., 2017).
In our disease context of CS, the candidate gene set was selected based on a developmental biologic aspect: the skeletal system of a vertebrate embryo originates from the pre‐somitic mesoderm, which segments into somite during early embryo development. During this process, TBX6 is expressed in the entire pre‐somitic mesoderm, and regulates a series of genes to enable the normal somitogenesis and subsequent development of the skeletal system (Oginuma et al., 2008). As TBX6 has been revealed as a core disease gene of CS (Chen et al., 2020; Liu et al., 2019; Wu et al., 2015), we conducted this study based on a hypothesis that TBX6‐mediated genes may as well contribute to CS. The rationality of this hypothesis is supported by disrupted spinal development in animal models depleted of the candidate genes (Takahashi et al., 2010; Windner et al., 2015), and by several autosomal recessive syndromes resulting in spinal malformation cause by these genes (Di Gioia et al., 2018; Karaca et al., 2015).
Due to the fact that CS represents a relatively milder phenotype spectrum than the autosomal recessive syndromes caused by biallelic loss‐of‐function of the candidate genes, the variant spectrum and disease‐causing mode of these genes in CS are assumed to be more complicated. More recently, there has been a surge of interest in digenic/oligogenic inheritance mode in complex disease (Schaffer, 2013). For example, mutational combination of PCDH15 and USH1G have been identified in non‐syndromic hearing loss; oligogenic inheritance of three cardiomyopathy‐associated genes has been reported in a family with cardiac anomaly and supported by experimental evidence (Gifford et al., 2019). In our study, potential oligogenic inheritance mode were identified in two cases. Mutation combinations between MYOD1/MEOX1 and MYOD1/RIPPLY1 were observed. Given the relatively low probability of loss‐of‐function intolerance (pLI) of the candidate genes in the gnomAD database, a deleterious variant alone in any of these genes might not be disease causing. However, the combined effect of deleterious variants in multiple genes might synergistically lead to the disease. Although experimental validation of the oligogenic model still needs to be conducted, our data give insight into the complex disease‐causing mode of CS and suggest the combined effect of mutations in TBX6‐mediated genes as an important mechanism in the pathogenesis of CS.
In conclusion, our study characterized the mutational spectrum of TBX6‐mediated genes in CS, prioritized core candidate genes/variants, and provided insight into a potential oligogenic disease‐causing mode.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Jianguo Zhang, Nan Wu, Zhihong Wu, and Xisheng Weng conceived of the project and designed the study, Yang Yang, Sen Zhao, Yuanqiang Zhang, Shengru Wang, Jiashen Shao, Bowen Liu, Yaqi Li, and Zihui Yan collected and interpreted the data, Lianlei Wang, Yongyu Ye, Jiachen L, Hengqiang Z, Zihui Y, Zefu C, and Gang L conducted the statistical analysis. Yuchen Niu and Ziaoxin Li conducted the bioinformatic analyses. Yangyang, Yuanqiang Zhang, and Lianlei Wang recruited patients. Sen Zhao, Yangyang, and Nan Wu wrote the first draft of the manuscript, Jianguo Zhang, Zhihong Wu, and Xisheng Weng critically revised the work for important intellectual content.
ACKNOWLEDGMENT
We thank Drs. Jianxiong Shen, Yipeng Wang, Yu Zhao, Ye Tian, Hong Zhao, Shugang Li, and Jianhua Hu from Peking Union Medical College Hospital, Beijing, China for their help in patient collection. We thank Ms. Yufei Dong from Architectural Association School of Architecture, London, UK for drawing Figure 2. We thank our patients and their families for participating this study.
Yang Y, Zhao S, Zhang Y, et al. Mutational burden and potential oligogenic model of TBX6‐mediated genes in congenital scoliosis. Mol Genet Genomic Med. 2020;8:e1453 10.1002/mgg3.1453
Yang Yang, Sen Zhao and Yuanqiang Zhang have contributed equally to this study.
FUNDING INFORMATION
This research was funded in part by the National Natural Science Foundation of China (81822030 to N.W., 81930068 and 81772299 to Z.W., 81672123 and 81972037 to J.Z., 81902178 to S.W), National Key Research & Development Program of China (2017YFC1104902 to J.Z.), the Fundamental Research Funds for the Central Universities (3332019021 for S.W.), Beijing Natural Science Foundation (7191007 to Z.W.), CAMS Initiative Fund for Medical Sciences (2016‐I2M‐3‐003 to N.W., 2016‐I2M‐2‐006 and 2017‐I2M‐2‐001 to Z.W.), Tsinghua University‐Peking Union Medical College Hospital Initiative Scientific Research Program (to N.W.), the National Undergraduates Innovation and Training Program of Peking Union Medical College (202010023022 to Sen. Z.), and CAMS Innovation Fund for Graduates (2018‐1002‐01‐09 to Yuan. Z.).
Contributor Information
Jianguo Zhang, Email: jgzhang_pumch@yahoo.com.
Nan Wu, Email: dr.wunan@pumch.cn.
REFERENCES
- Bondue, A. , Lapouge, G. , Paulissen, C. , Semeraro, C. , Iacovino, M. , Kyba, M. , & Blanpain, C. (2008). Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell, 3(1), 69–84. 10.1016/j.stem.2008.06.009 [DOI] [PubMed] [Google Scholar]
- Chen, W. , Lin, J. , Wang, L. , Li, X. , Zhao, S. , Liu, J. , … Wu, N. (2020). TBX6 missense variants expand the mutational spectrum in a non‐Mendelian inheritance disease. Human Mutation, 41(1), 182–195. 10.1002/humu.23907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gioia, S. A. , Shaaban, S. , Tüysüz, B. , Elcioglu, N. H. , Chan, W.‐M. , Robson, C. D. , … Engle, E. C. (2018). Recessive MYF5 mutations cause external ophthalmoplegia, rib, and vertebral anomalies. American Journal of Human Genetics, 103(1), 115–124. 10.1016/j.ajhg.2018.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi, K. c., & Epilepsy Phenome/Genome, P. (2017). Ultra‐rare genetic variation in common epilepsies: a case‐control sequencing study. The Lancet Neurology, 16(2), 135–143. 10.1016/S1474-4422(16)30359-3 [DOI] [PubMed] [Google Scholar]
- Gianakopoulos, P. J. , Mehta, V. , Voronova, A. , Cao, Y. I. , Yao, Z. , Coutu, J. , … Skerjanc, I. S. (2011). MyoD directly up‐regulates premyogenic mesoderm factors during induction of skeletal myogenesis in stem cells. Journal of Biological Chemistry, 286(4), 2517–2525. 10.1074/jbc.M110.163709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford, C. A. , Ranade, S. S. , Samarakoon, R. , Salunga, H. T. , de Soysa, T. Y. , Huang, Y. U. , … Srivastava, D. (2019). Oligogenic inheritance of a human heart disease involving a genetic modifier. Science, 364(6443), 865–870. 10.1126/science.aat5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamori, Y. , Wu, H. Y. , Sartorelli, V. , & Kedes, L. (1997). The basic domain of myogenic basic helix‐loop‐helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein. Twist. Mol Cell Biol, 17(11), 6563–6573. 10.1128/mcb.17.11.6563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedequist, D. , & Emans, J. (2007). Congenital scoliosis: A review and update. Journal of Pediatric Orthopedics, 27(1), 106–116. 10.1097/BPO.0b013e31802b4993 [DOI] [PubMed] [Google Scholar]
- Ji, L. D. , Li, J. Y. , Yao, B. B. , Cai, X. B. , Shen, Q. J. , & Xu, J. (2017). Are genetic polymorphisms in the renin‐angiotensin‐aldosterone system associated with essential hypertension? Evidence from genome‐wide association studies. Journal of Human Hypertension, 31(11), 695–698. 10.1038/jhh.2017.29 [DOI] [PubMed] [Google Scholar]
- Karaca, E. , Yuregir, O. O. , Bozdogan, S. T. , Aslan, H. , Pehlivan, D. , Jhangiani, S. N. , … Lupski, J. R. (2015). Rare variants in the notch signaling pathway describe a novel type of autosomal recessive Klippel‐Feil syndrome. American Journal of Medical Genetics. Part A, 167A(11), 2795–2799. 10.1002/ajmg.a.37263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, M. , Zhao, S. , Liu, G. , Huang, Y. , Yu, C. , Zhao, Y. , … Wu, N. (2020). Identification of novel FBN1 variations implicated in congenital scoliosis. Journal of Human Genetics, 65(3), 221–230. 10.1038/s10038-019-0698-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Wu, N. , Yang, N. , Takeda, K. , Chen, W. , Li, W. , … Qiu, G. (2019). TBX6‐associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genetics in Medicine, 21(7), 1548–1558. 10.1038/s41436-018-0377-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, R. J. , Isaacs, H. V. , & Pownall, M. E. (2012). Early transcriptional targets of MyoD link myogenesis and somitogenesis. Developmental Biology, 371(2), 256–268. 10.1016/j.ydbio.2012.08.027 [DOI] [PubMed] [Google Scholar]
- Morimoto, M. , Takahashi, Y. , Endo, M. , & Saga, Y. (2005). The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature, 435(7040), 354–359. 10.1038/nature03591 [DOI] [PubMed] [Google Scholar]
- Nakajima, Y. , Morimoto, M. , Takahashi, Y. , Koseki, H. , & Saga, Y. (2006). Identification of Epha4 enhancer required for segmental expression and the regulation by Mesp2. Development, 133(13), 2517–2525. 10.1242/dev.02422 [DOI] [PubMed] [Google Scholar]
- Oginuma, M. , Niwa, Y. , Chapman, D. L. , & Saga, Y. (2008). Mesp2 and Tbx6 cooperatively create periodic patterns coupled with the clock machinery during mouse somitogenesis. Development, 135(15), 2555–2562. 10.1242/dev.019877 [DOI] [PubMed] [Google Scholar]
- Schaffer, A. A. (2013). Digenic inheritance in medical genetics. Journal of Medical Genetics, 50(10), 641–652. 10.1136/jmedgenet-2013-101713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Wang, Z. , Liu, J. , Xue, X. , & Qiu, G. (2013). Abnormalities associated with congenital scoliosis: a retrospective study of 226 Chinese surgical cases. Spine (Phila Pa 1976), 38(10), 814–818. 10.1097/BRS.0b013e31827ed125 [DOI] [PubMed] [Google Scholar]
- Sparrow, D. B. , McInerney‐Leo, A. , Gucev, Z. S. , Gardiner, B. , Marshall, M. , Leo, P. J. , … Dunwoodie, S. L. (2013). Autosomal dominant spondylocostal dysostosis is caused by mutation in TBX6. Human Molecular Genetics, 22(8), 1625–1631. 10.1093/hmg/ddt012 [DOI] [PubMed] [Google Scholar]
- Stamataki, D. , Kastrinaki, M. , Mankoo, B. S. , Pachnis, V. , & Karagogeos, D. (2001). Homeodomain proteins Mox1 and Mox2 associate with Pax1 and Pax3 transcription factors. FEBS Letters, 499(3), 274–278. 10.1016/s0014-5793(01)02556-x [DOI] [PubMed] [Google Scholar]
- Takahashi, J. , Ohbayashi, A. , Oginuma, M. , Saito, D. , Mochizuki, A. , Saga, Y. , & Takada, S. (2010). Analysis of Ripply1/2‐deficient mouse embryos reveals a mechanism underlying the rostro‐caudal patterning within a somite. Developmental Biology, 342(2), 134–145. 10.1016/j.ydbio.2010.03.015 [DOI] [PubMed] [Google Scholar]
- Takahashi, Y. , Koizumi, K. , Takagi, A. , Kitajima, S. , Inoue, T. , Koseki, H. , & Saga, Y. (2000). Mesp2 initiates somite segmentation through the Notch signalling pathway. Nature Genetics, 25(4), 390–396. 10.1038/78062 [DOI] [PubMed] [Google Scholar]
- Wang, K. , Zhao, S. , Liu, B. , Zhang, Q. , Li, Y. , Liu, J. , … Wu, N. (2018). Perturbations of BMP/TGF‐beta and VEGF/VEGFR signalling pathways in non‐syndromic sporadic brain arteriovenous malformations (BAVM). Journal of Medical Genetics, 55(10), 675–684. 10.1136/jmedgenet-2017-105224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windner, S. E. , Doris, R. A. , Ferguson, C. M. , Nelson, A. C. , Valentin, G. , Tan, H. , … Devoto, S. H. (2015). Tbx6, Mesp‐b and Ripply1 regulate the onset of skeletal myogenesis in zebrafish. Development, 142(6), 1159–1168. 10.1242/dev.113431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, N. , Ming, X. , Xiao, J. , Wu, Z. , Chen, X. , Shinawi, M. , … Zhang, F. (2015). TBX6 null variants and a common hypomorphic allele in congenital scoliosis. New England Journal of Medicine, 372(4), 341–350. 10.1056/NEJMoa1406829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, N. , Wang, L. , Hu, J. , Zhao, S. , Liu, B. , Li, Y. , … Qiu, G. (2019). A recurrent rare SOX9 variant (M469V) is associated with congenital vertebral malformations. Current Gene Therapy, 19(4), 242–247. 10.2174/1566523219666190924120307 [DOI] [PubMed] [Google Scholar]
- Yang, N. , Wu, N. , Zhang, L. , Zhao, Y. , Liu, J. , Liang, X. , … Zhang, F. (2019). TBX6 compound inheritance leads to congenital vertebral malformations in humans and mice. Human Molecular Genetics, 28(4), 539–547. 10.1093/hmg/ddy358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhiko, Y. , Haraguchi, S. , Kitajima, S. , Takahashi, Y. , Kanno, J. , & Saga, Y. (2006). Tbx6‐mediated Notch signaling controls somite‐specific Mesp2 expression. Proceedings of the National Academy of Sciences of the United States of America, 103(10), 3651–3656. 10.1073/pnas.0508238103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Zhang, Y. , Chen, W. , Li, W. , Wang, S. , Wang, L. , … Wu, N. (2020). Diagnostic yield and clinical impact of exome sequencing in early‐onset scoliosis (EOS). Journal of Medical Genetics. 10.1136/jmedgenet-2019-106823 [DOI] [PMC free article] [PubMed] [Google Scholar]