

Abstract
Objective
This study aimed to identify the disease‐causing mutation of congenital cataract disease in a large northeastern Chinese family.
Materials and Methods
The subjects’ peripheral blood was collected, their genomic DNA was extracted, mutation screening of candidate genes was performed using polymerase chain reaction, and the amplified products were sequenced. Recombinant C‐terminal enhanced green fluorescent protein‐tagged wild‐type or mutant CRYGD was expressed in HEK293T cells, and the expression pattern was observed under a fluorescence microscope. The CRYGD protein mutation was analyzed via bioinformatics analysis.
Results
c.475delG, a novel frameshift mutation in CRYGD, was identified in the affected family members. This mutation causes premature termination of the polypeptide, resulting in truncated p.(Ala159ProfsTer9). According to the bioinformatics analysis results, compared with wild‐type CRYGD, p.(Ala159ProfsTer9) exhibits significantly decreased hydrophilicity. Fluorescence microscopy revealed that p.(Ala159ProfsTer9) aggregates in the cell in the form of granular deposits.
Conclusion
In this study, the novel frameshift mutation c.475delG, p.(Ala159ProfsTer9) in CRYGD was identified to cause congenital cataracts in a large Chinese family; increased hydrophobicity of p.(Ala159ProfsTer9) protein may be the underlying mechanism.
Keywords: bioinformatics analysis, c.475delG mutation, congenital cataract, CRYGD, hydrophobicity, p.(Ala159ProfsTer9)
A novel frameshift variant, c.475delG in CRYGD was identified in a large family with congenital cataract disease in the Northeast of China, which causes a premature termination of the polypeptide to become p.(Ala159ProfsTer9), decreased hydrophilicity, increased hydrophobicity.

1. INTRODUCTION
Congenital cataracts (complete or partial lens opacification) are the leading cause of visual impairment in early childhood. In general, a cataract is formed due to the aggregation of lens proteins, and it clinically presents as the clouding of the lens, disrupting the flow of light to the retina, thereby causing decreased clarity, dull color contrast, and impaired vision (Michael & Bron, 2011). The prevalence of cataracts is approximately 1–6 per 10,000 live births (Roshan et al., 2010), and approximately one‐third of all cataract cases are hereditary. Most congenital cataracts are autosomal dominant; however, autosomal recessive and X‐linked patterns have also been reported (Stephan et al., 1999). To date, approximately 28 genes have been identified to be associated with congenital cataracts; approximately 50% of the disease‐causing mutations belong to the crystallin family and these gene mutations impair lens development at different stages (Cui et al., 2017).
Crystallins, which exist as α‐, β‐, and γ‐types, are water‐soluble proteins, account for approximately 90% of all lens proteins, and are assumedly responsible for maintaining the lens transparency. Crystallin gamma D (CRYGD, [OMIM * 123690]) is the second most abundant γ‐crystallin in the lens nucleus; it is monomeric and contains 173 amino acids, and its structure contains two highly homologous, duplicated β‐sheet domains (Ji, Jung, Koharudin, & Gronenborn, 2013). Many mutations in CRYGD have been widely reported (Table S1 in Supporting Information), and some studies have shown that these mutations alter the stability and/or the solubility of γ‐crystallins and contribute to the loss of lens transparency (Banerjee, Puttamadappa, Pande, Shekhtman, & Pande, 2011).
In this study, the case of a large Chinese family with congenital cataracts, with 18 affected individuals across five generations, has been reported. c.475delG mutant was identified in exon 3 of CRYGD after Sanger sequencing of candidate crystallin genes. This change led to a frameshift and a truncated protein p.(Ala159ProfsTer9). According to the bioinformatics analysis results and cell culture and transfection, the increased hydrophobicity of the p.(Ala159ProfsTer9) protein may be the mechanism underlying congenital cataracts.
2. MATERIALS AND METHODS
In this study, the congenital cataracts were autosomal dominant, and there were 18 affected and 13 unaffected members in the entire family (Figure 1a), and fourteen individuals of this family were recruited, the characteristics of them were described (Table S4 in Supporting Information). The proband (Ⅲ12) was a 60‐year‐old male with poor eyesight from very early childhood, all the affected family individuals underwent different surgical procedures (Table S4 in Supporting Information). A total of 150 unrelated control subjects with no family history of congenital cataracts were also recruited. Informed consent was obtained from all participants, and the study protocol was approved by the Institutional Review Board of the Key Laboratory of Reproductive Health of the Liaoning Province (Shenyang, China).
Figure 1.

Pedigree of the Chinese family with congenital cataracts. (a) Across five generations, 18 members were affected with bilateral congenital nuclear cataracts. (b) Sanger sequence of exon 3 of CRYGD in the proband (Ⅲ12) and an unaffected member (Ⅳ9). The heterozygous c.475delG in the affected member. (c) HRM showed different plots of wild‐type samples and the various mutants; the affected individuals were represented by red lines and control by gray lines. (d) Protein alignment of mammalian samples showing that the regions surrounding the mutation are highly conserved. Numbers on the left and right indicate the position of this fragment. The position of the mutant is marked by an asterisk (*). The reference sequence of the CRYGD gene was NM_006891.3
2.1. Mutation screening
Peripheral blood of all of the participants was collected, and genomic DNA was extracted using QuickGene DNA whole blood kits with QuickGene‐810 (GX‐10060209, FUJIFILM, Japan). The exons and flanking intronic regions of the candidate genes (CRYGA, CRYGB, CRYGC, and CRYGD) were amplified using polymerase chain reaction (PCR). Primers were designed according to a previous study (Roshan et al., 2010); the specific primer sequences for CRYGD (NM_006891.3) have been listed (Table S3 in Supporting Information). Sanger sequencing of the PCR products was performed by SangonBiotech (Shanghai, China).
2.2. Bioinformatics analysis
CRYGD amino acid and nucleotide sequences were obtained from the NCBI Gene Database (http://www.ncbi.nlm.nih.gov/gene/). The effect of the c.475delG mutation was predicted using MutationTaster (http://www.mutationtaster.org/), conservative analysis of the mutation site among many mammals was performed using MEGA6, whereas the physical and chemical parameters of the CRYGD protein were analyzed using ProtParam (http://web.expasy.org/protparam/). The secondary structures of CRYGD were predicted using the GOR IV, secondary structure prediction using FirstGlance in Jmol (http://bioinformatics.org/firstglance/fgij/), and the protein structure change in the CRYGD mutant was predicted using the Protein Data Bank (PDB) database and FirstGlance in Jmol.
2.3. High‐resolution melting
Primers for high‐resolution melting (HRM) were designed based on guidelines from Wojdacz, Hansen, and Dobrovic (2008). An 88‐bp region containing c.475(G/‐) sites was amplified using the primers (Table S3 in Supporting Information). The PCR amplification protocol was as follows: 95°C for 1 min, 95°C for 40 s, 58°C for 45 s, 72°C for 30 s, and 72°C for 10 min, a total of 30 cycles. HRM analysis was conducted using the Light Cycler 480 System (Roche), as described previously (Turner, Sasse, & Varadi, 2016), under the following conditions: 95°C for 1 min, 40°C for 1 min, 74°C for 5 s, and continuous acquisition to 90°C at the rate of 25 acquisitions per 1°C.
2.4. Plasmid constructs
The human CRYGD (GenBank, NM_006891.3) cDNA plasmid PEGFP‐N1‐CRYGD and mutant plasmid PEGFP‐N1‐CRYGD c.475delG were purchased from YouBio company (http://www.youbio.cn/).
2.5. Cell culture and transfection
HEK293 T cells were routinely grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C with 5% CO2. When cells reached 89%–90% confluency, they were passaged to 35‐mm dishes with 0.3 × 105 cells/dish and cultured overnight. HEK293 T cells were transfected with PEGFP‐N1‐CRYGD and PEGFP‐N1‐CRYGD 475delG plasmids using Lipofectamine 2000 (Invitrogen); after 48 h, the cells were observed under a fluorescence microscope.
3. RESULTS
3.1. Mutation confirmation in CRYGD
Mutation screening in four candidate crystallin genes (CRYGA, CRYGB, CRYGC, CRYGD) was performed, and a heterozygous c.475delG mutant was identified in exon 3 of CRYGD (NM_006891.3) in the proband (Figure 1b), which resulted in the substitution of a newly formed stop codon and the loss of 167‐174 C‐terminal residues of CRYGD protein in the fourth ‘‘Greek key motif.’’ The variant c.475delG was confirmed in all affected individuals but was not detected in unaffected family members or in the 150 unrelated controls. The c.475delG mutation was only detected in the affected individuals in this family by HRM (Figure 1c). The region surrounding the mutation site was highly conserved among many mammals (Figure 1d). No phenotype of congenital cataract was observed in Ⅲ9 at the time of this study; however, mutation analysis showed that she possessed the disease‐causing mutation.
3.2. Bioinformatics analysis
The pathogenicity of c.475delG was predicted using MutationTaster; the score was 0.99999999, suggesting that the mutation was “damaging.” This deletion variant was neither found in ExAC databases (http://exac.broadinstitute.org/) nor in the 1000 Genomes Project (http://www.1000genomes.org/). The physical and chemical parameters of the CRYGD protein were analyzed using ProtParam; the mutant protein p.(Ala159ProfsTer9) was found to be eight amino acids shorter than the wild type, and it had an acidic theoretical isoelectric point and rendered CRYGD more unstable and decreased its solubility (Table S2 in Supporting Information). The secondary structure analysis showed that the fourth C‐terminus GKM (129‐171) was partially absent in the mutant (Figure 2a). Both the extended strand and the random coil were reduced, resulting in the destruction of the secondary structure (Figure 2c).The secondary structure was also analyzed by FirstGlance in Jmol (http://bioinformatics.org/firstglance/fgij/), which showed that the ration of alpha‐helices and beta‐strands of the p.(Ala159ProfsTer9) changed largely compared with that of the wild type (Figure 2d).
Figure 2.
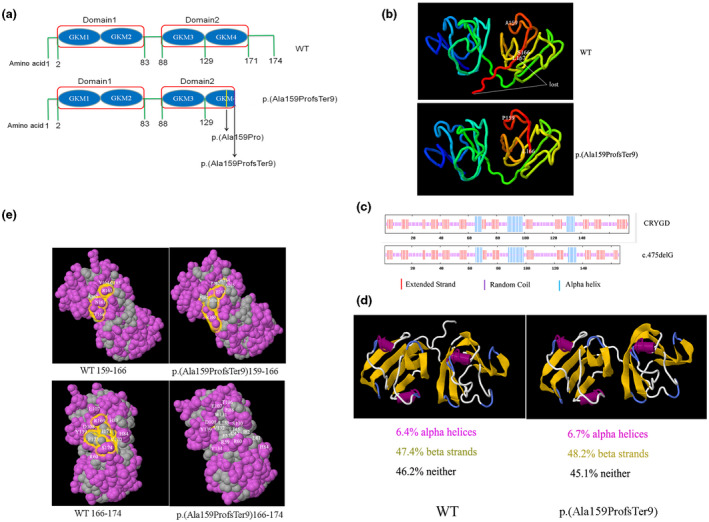
Bioinformatics analysis of the mutant CRYGD p.(Ala159ProfsTer9). (a) Diagrammatic scheme of the functional domain of CRYGD and p.(Ala159ProfsTer9) protein. (b) The tertiary structure of CRYGD and p.(Ala159ProfsTer9) protein. (c) The predicted secondary structure showing the reduced extended strand and random coil in the mutant. (d) The predicted secondary structure showing the changed alpha‐helices and beta‐strands in the mutant. (e) The predicted hydrophobic surface and amino changes in the mutant
The crystal structure of the mutant protein p.(Ala159ProfsTer9) was generated by Swiss‐Model (https://www.swissmodel.expasy.org/) and compared with the structure of the wild type, which was retrieved from the Protein Data Bank (ID 1HK0) (ftp://ftp.wwpdb.org). In the truncated CRYGD protein, the C‐terminal tail of CRYGD, which includes amino acids 167–174, was lost, and amino acids 159–166 and the direction of the C‐terminal tail changed (Figure 2b).
The tertiary structure and surface hydrophobicity were studied using FirstGlance in Jmol. According to the hydrophobic/polar model, the polar acids 161Asn and 165Gly changed to the hydrophobic amino acids 161Met and 165Ala and the hydrophobic surface increased in the p.(Ala159ProfsTer9) protein. Besides, in the p.(Ala159ProfsTer9) protein, amino acids 167–174 were lost and more hydrophobic amino acids, such as 131Trp, 133Leu, 132Val, 145Leu, 57Phe, 42Cys, 82Ile, and 81Leu, were exposed; further, some polar amino acids such as 169Arg and 174Ser were lost (Figure 2e).
3.3. Cellular distribution
To confirm the pathogenicity of the mutation p.(Ala159ProfsTer9), an immunofluorescence assay was performed to detect the subcellular localization of mutant CRYGD. pEGFP‐N1‐CRYGD and PEGFP‐N1‐CRYGD c.475delG plasmids were separately transfected to HEK293T cells. Immunofluorescence analysis showed that compared with the wild‐type CRYGD, the mutant p.(Ala159ProfsTer9) aggregated in the cell in the form of granular deposits (Figure 3).
Figure 3.

Localization of EGFP‐tagged wild‐type or p.(Ala159ProfsTer9) CRYGD in HEK293T cells. Green fluorescence showed that mutant CRYGD was mainly localized in the cell in the form of granular deposits
4. DISCUSSION
In this study, a c.475delG mutant in CRYGD in a large Chinese family was identified by Sanger sequencing. No phenotype of congenital cataract was observed in Ⅲ9; however, mutation analysis showed that she had the c.475delG mutation, and so Ⅲ9 may be germ cell chimera with the c.475delG.
To date, approximately 21 mutations of CRYGD, including 19 missense, 1 insert, and 1 deletion, have been reported to cause different types of congenital cataracts. In this study, the deletion mutant c.475delG was reported, which resulted in the frameshift and truncation of the wild‐type CRYGD protein to mutant p.(Ala159ProfsTer9).
Crystallins are the structural proteins that play essential roles in lens development and transparency maintenance. γD‐crystallin is a member of the β/γ‐crystallin family, which shares common features of antiparallel β‐sheets in the protein, called the “Greek key motif.” The CRYGD protein has two functional domains, and each domain consists of two GKMs. The c.475delG mutation in the present study is located in GKM4 of CRYGD proteins and is highly conserved in different species. The deletion of G at c.475 in exon 3 of CRYGD has been predicted to cause a frameshift and a premature stop codon, leading to the deletion of C‐terminal residues 167–174.
In this study, the c.475delG mutation resulted in the changes in amino acids 156–166 and deletion of C‐terminal residues 167–174. The C‐terminal residues 159–174 of the wild‐type CRYGD protein comprise four polar amino acids (160Thr, 161ASN, 166Ser, and 174Ser) and seven hydrophobic amino acids (159Ala, 162Ala, 164Val, 167Leu,170Val, 171Ile, and 173Phe). The polar amino acids 160Thr, 161Asn, and 166Ser changed to hydrophobic amino acids 161Met, 166Leu, and neutral amino acid 160Arg in the mutant CRYGD p.(Ala159ProfsTer9).
In wild‐type CRYGD protein, only the residues (162Ala, 164Val, 170Val, 171Ile, and 173Phe) were exposed to the surface. In the mutant p.(Ala159ProfsTer9), 161Met, 162Ala, 164Val, and 165Ala were also exposed, so the surface area was larger than that of the wild‐type CRYGD protein. Even more so, the hydrophobic amino acids buried in the wild‐type protein, such as 131Trp, 133Leu, 132Val, 145Leu, 42Cys, 57Phe, 82Ile, and 81Leu, were exposed in the mutant, and so the p.(Ala159ProfsTer9) had a larger hydrophobic surface which may have resulted in the reduction of solubility.
The previous study showed that the C‐terminal residues of CRYGD protein play a great role in solubility and intermolecular interactions, and the residues 156Asp, 163Arg, and 168Arg were involved in charge interactions; the residue 173Phe was crucial in the amine–aromatic and hydrophobic interactions (Basak et al., 2003). In the mutant p.(Ala159ProfsTer9), amino acid 163Arg changed to 163Glu and amino acids 167Leu and 173Phe were lost, and so the amine–aromatic and hydrophobic interactions were disturbed. In conclusion, disturbance in the hydrophobic interactions and increase in exposure to hydrophobic surfaces may lead to the aggregation of p.(Ala159ProfsTer9) and the loss of transparency.
CONFLICT OF INTEREST
No conflict of interest exists in the submission of this manuscript, and the manuscript is approved by all authors for publication.
AUTHOR CONTRIBUTIONS
M. Lin contributed in the literature search, experimental studies, and drafting the manuscript; Y. Jin contributed in the clinical examination of the eyes of related individuals; Y. Sui and Y. Li contributed in blood collection; N. Zhao, X. Ni, H. Li, and X. Chen contributed in clinical data acquisition; Y. Lu contributed in data analysis and manuscript editing and review; M. Jiang contributed in conception and design of the study, manuscript editing, and review.
Supporting information
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENT
We thank all the individuals that participated in this study.
Lin M, Jin Y, Chen X, et al. Increased hydrophobicity of CRYGD p.(Ala159ProfsTer9): Suspected cause of congenital cataracts in a large Chinese family. Mol Genet Genomic Med. 2020;8:e1436 10.1002/mgg3.1436
Meina Lin, Yongping Lu and Miao Jiang contributed equally to this work as corresponding authors.
Funding information
The doctor starting fund of Liaoning province, No. 201601386; Social development and industrialization in Liaoning province, No. 2017225019; Shenyang science and technology planning project, No. 18‐014‐4‐53; Training discipline project of China Medical University: Clinical Genetics (Ophthalmology), No. 3110118049; Provincial Nature Fund guidance plan, No. 20170540492, 20180551240, 20180551252.
Contributor Information
Meina Lin, Email: lincmu@126.com.
Yongping Lu, Email: Valensu423@163.com.
Miao Jiang, Email: jiangmiaocpt@aliyun.com.
REFERENCES
- Banerjee, P. R. , Puttamadappa, S. S. , Pande, A. , Shekhtman, A. , & Pande, J. (2011). Increased hydrophobicity and decreased backbone flexibility explain the lower solubility of a cataract‐linked mutant of gammaD‐crystallin. Journal of Molecular Biology, 412, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak, A. , Bateman, O. , Slingsby, C. , Pande, A. , Asherie, N. , Ogun, O. , … Pande, J. (2003). High‐resolution X‐ray crystal structures of human gammaD crystallin (1.25 A) and the R58H mutant (1.15 A) associated with aculeiform cataract. Journal of Molecular Biology, 328, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Cui, X. K. , Zhu, K. K. , Zhou, Z. , Wan, S. M. , Dong, Y. , Wang, X. C. , … Hu, Y. Z. (2017). A novel frameshift mutation in CX46 associated with hereditary dominant cataracts in a Chinese family. International Journal of Ophthalmology, 10, 684–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, F. , Jung, J. , Koharudin, L. M. , & Gronenborn, A. M. (2013). The human W42R gammaD‐crystallin mutant structure provides a link between congenital and age‐related cataracts. Journal of Biological Chemistry, 288, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, R. , & Bron, A. J. (2011). The ageing lens and cataract: A model of normal and pathological ageing. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 366, 1278–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan, M. , Vijaya, P. H. , Lavanya, G. R. , Shama, P. K. , Santhiya, S. T. , Graw, J. , … Satyamoorthy, K. (2010). A novel human CRYGD mutation in a juvenile autosomal dominant cataract. Molecular Vision, 16, 887–896. [PMC free article] [PubMed] [Google Scholar]
- Stephan, D. A. , Gillanders, E. , Vanderveen, D. , Freas‐Lutz, D. , Wistow, G. , Baxevanis, A. D. , … Brownstein, M. J. (1999). Progressive juvenile‐onset punctate cataracts caused by mutation of the gammaD‐crystallin gene. Proceedings of the National Academy of Sciences of the United States of America, 96, 1008–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, A. , Sasse, J. , & Varadi, A. (2016). Rapid detection of pathological mutations and deletions of the haemoglobin beta gene (HBB) by High Resolution Melting (HRM) analysis and Gene Ratio Analysis Copy Enumeration PCR (GRACE‐PCR). BMC Medical Genetics, 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdacz, T. K. , Hansen, L. L. , & Dobrovic, A. (2008). A new approach to primer design for the control of PCR bias in methylation studies. BMC Research Notes, 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4