

Abstract
Background
Establishing a genetic diagnosis for individuals with intellectual disability (ID) benefits patients and their families as it may inform the prognosis, lead to appropriate therapy, and facilitate access to medical and supportive services. Exome sequencing has been successfully applied in a diagnostic setting, but most clinical exome referrals are pediatric patients, with many adults with ID lacking a comprehensive genetic evaluation.
Methods
Our unique recruitment strategy involved partnering with service and education providers for individuals with ID. We performed exome sequencing and analysis, and clinical variant interpretation for each recruited family.
Results
All five families enrolled in the study opted‐in for the return of genetic results. In three out of five families exome sequencing analysis identified pathogenic or likely pathogenic variants in KANSL1, TUSC3, and MED13L genes. Families discussed the results and any potential medical follow‐up in an appointment with a board certified clinical geneticist.
Conclusion
Our study suggests high yield of exome sequencing as a diagnostic tool in adult patients with ID who have not undergone comprehensive sequencing‐based genetic testing. Research studies including an option of return of results through a genetic clinic could help minimize the disparity in exome diagnostic testing between pediatric and adult patients with ID.
Keywords: adults, clinical exome, intellectual disability
Exome sequencing has been successfully applied in a diagnostic setting, but most clinical exome referrals are pediatric patients, with many adults with intellectual disability lacking a comprehensive genetic evaluation. Our study points to high yield of exome sequencing as a diagnostic tool in adult patients who have not undergone comprehensive sequencing‐based genetic testing. Research studies including an option of return of results through a genetic clinic could help minimize the disparity in exome diagnostic testing between pediatric and adult patients.

1. INTRODUCTION
Intellectual disability (ID) is a lifelong condition that manifests early in childhood and is characterized by below‐average general intellectual function and limitations in learning, adaptive behavior and skills (Salvador‐Carulla et al., 2011). The prevalence of ID across the world varies but is estimated to be around 1% (Maulik, Mascarenhas, Mathers, Dua, & Saxena, 2011). ID can be caused by genetic or non‐genetic factors, such as congenital infections, perinatal asphyxia or trauma, or prenatal exposure to environmental toxins or teratogens. Establishing a genetic diagnosis of ID can benefit patients and their families, as it may inform the prognosis, preclude further unnecessary testing, and lead to appropriate therapy, change of care or monitoring strategies and facilitate access to appropriate medical and supportive services (Adams & Eng, 2018). In addition, such information may inform recurrence risk and play an important role in family planning.
At the beginning of the last decade routine genetic testing for individuals with ID comprised genome‐wide microarray analysis and phenotype guided single gene analysis (Miller et al., 2010). Utilizing these tests resulted in a genetic diagnosis in about 10%–20% of individuals (Baker, Raymond, & Bass, 2012). Targeted next‐generation sequencing (NGS) gene panels are commonly used in the clinic today due to their lower cost compared to exome and better insurance coverage. Gene panels typically include several hundred to thousands of genes and have a diagnostic yield around 21% (Pekeles et al., 2019). More recently exome sequencing has been applied in a research setting for novel ID gene discovery as well as diagnostic setting with great success. Diagnostic rates of up to 60% have been reported, dependent on setting, gender, and severity of the phenotype (Bowling et al., 2017; de Ligt et al., 2012; Mir & Kuchay, 2019; Willemsen & Kleefstra, 2014).
To date, the majority of clinical exome referrals are pediatric patients, with adults comprising only 12%–16% (Farwell et al., 2015; Yang et al., 2014) in reported cohorts. One reason for this is that many genetic ID conditions are first recognized in early life when a child is noted to be delayed or have dysmorphic features, for example. In contrast, adult medical training does not formally instruct clinicians on when a genetics evaluation would be appropriate (Maves, Williams, Williams, Levonian, & Josephson, 2007). Furthermore, in some individuals the causative disorder may not have been described until recently, long after any genetics workup was done. As a consequence, most adults with ID who have had access to genetic testing as children have likely had the less comprehensive older tests, for example, karyotype, microarray, or single‐gene analysis and are lacking a more comprehensive genetic evaluation. An illustrative case study of a 55‐year‐old adult recently diagnosed with Xia‐Gibbs syndrome, previously only described in pediatric patients, shows how exome sequencing can bring new knowledge to families, aiding both the diagnosed adult and younger individuals who can then develop more informed prognoses (Murdock et al., 2019). Recently studies of adults with epilepsy and comorbid ID reported the yield of diagnostic exome testing in the range of 25%–27% (Benson et al., 2020; Snoeijen‐Schouwenaars et al., 2019). We predicted that many other adults living with ID might now be able to be diagnosed with modern methods but are not readily accessing the tests due to a lack of access to care or knowledge of new opportunities.
To specifically reach undiagnosed adults, we designed a recruitment strategy to involve service and education providers for individuals with ID. Our study is at the intersection of research and clinical diagnostics and involves participants as engaged partners with the option of return of genetic results in a CAP/CLIA certified environment. Our aim was to determine the feasibility of this recruitment approach, and to utilize exome sequencing analysis to estimate the frequency of positive genetic diagnosis in adults with an ID of any severity, including mild ID.
2. METHODS
2.1. Editorial policies and ethical considerations
The study was approved by the institutional review board at Baylor College of Medicine (Institutional Review Board protocol # H‐42789), and written, informed consent was obtained from all participants in the study. For the participants with ID the informed consent was obtained from a parent or a legal authorized representative.
2.2. Recruitment and study participants
A brochure describing the study, enrollment criteria, and risks and benefits of participating was sent via email and postal mail to the clients of the Arc of Greater Houston, a community outreach, and service provider for individuals with ID. To be eligible for the study individuals had to be adults (≥18 yo), with a diagnosis of ID, and without an established genetic diagnosis. Individuals with all levels of ID severity, from mild to profound, were included. Initial screening interview was conducted to exclude individuals with an already established genetic diagnosis and individuals with known non‐genetic cause of ID (e.g. stroke, severe prematurity, prenatal or perinatal infections). Both parents were recruited when available for full parents‐child trios. Each family answered a questionnaire covering a detailed review of ID and related phenotypes, prenatal and perinatal history and exposures, information about any genetic testing conducted previously, and family history of ID. Each family had a choice to consent or decline return of genetic results. In cases with a genetic finding, the family was re‐contacted, the result confirmed in a CAP/CLIA certified laboratory and interpreted and shared with the family in a clinical setting by a board‐certified clinical geneticist. The pilot phase of the project included 11 participants (5 families): two full trios, two proband + one parent duos, and one proband.
2.3. Sample collection, exome sequencing, and variant interpretation
Saliva samples were collected using Oragene DNA saliva kits following manufacturer guidelines. Genomic DNA was extracted from saliva according to standard procedures. Exome sequencing was performed on all 11 participants according to previously described methods (Lupski et al., 2010). Briefly, genomic DNA samples underwent exome capture with Baylor College of Medicine VCRome 2.1pk whole exome design, and were then sequenced on the Illumina NovaSeqplatform. Sequencing reads were aligned and mapped to the human genome reference sequence (hg19) with the in‐house bioinformatics Mercury pipeline (Reid et al., 2014). Single nucleotide variants (SNVs) and short Indels were identified using Platypus (Rimmer et al., 2014). Variants were annotated using Annovar software (Yang & Wang, 2015), including variant type (stop gain or loss, amino acid changes, splice site variants), minor allele frequencies in gnomAD population databases (Karczewski et al., 2020), and multiple in silico predictions of variant deleteriousness (CADD, DANN, PolyPhen, SIFT, MutationTaster). Gene‐centric annotations included pLI scores from gnomAD (probability of being loss‐of‐function intolerant) (Genome Aggregation Database Consortium et al., 2020), and Domino database AD score (probability of a gene to be associated with autosomal dominant condition) (Quinodoz et al., 2017). Copy number variants (CNVs) were identified using XHMM (Fromer & Purcell, 2014) and additional 270 internal control samples, and annotated with overlapping gene information using custom in‐house scripts. Variant filtering and prioritization strategy utilized available trio information to prioritize de novo, rare (MAF < 0.1%) homozygous, and compound rare heterozygous protein altering variants (PAV: stop gain and loss, nonsynonymous and splice site SNVs, and frameshift indels). CNVs that overlapped coding exons and were identified in probands only were flagged as candidate variants. For families where complete trio was not available, we prioritized all rare homozygous PAVs, as well rare heterozygous PAV variants in genes with pLI score >0.9 or domino AD score >0.7.Findings were evaluated according to the American College of Medical Genetics and Genomics (ACMG) criteria for variant pathogenicity interpretation (Richards et al., 2015).
2.4. Candidate variants validation
Candidate SNVs and their inheritance were validated by an orthogonal DNA‐sequencing method. Target amplicons were amplified from genomic DNA using conventional PCR (HotStarTaqDNA polymerase, QIAGEN) and PCR amplification products were analyzed by Sanger sequencing using established methods. Candidate CNVs were validated using multiplex ligation‐dependent probe amplification (MLPA) by applying MLPA kit P443‐A1 (MRC‐Holland). MLPA tests were carried out according to the manufacturer's protocols, and analysis was done using the MLPA module of Coffalyser.Net software (MRC‐Holland).
3. RESULTS
3.1. Recruitment and study participants
The study brochure was sent to approximately 180 families by the Arc of Greater Houston staff. The research study staff was contacted by 13 families that were interested in participating in research. Five families met the eligibility criteria and were enrolled in the study. All 5 families opted‐in for the return of genetic results. Additional five families were not eligible for the study; two participants had a previously established genetic diagnosis, two had known non‐genetic causes of ID (severe prematurity and stroke) and one participant did not have an ID diagnosis. One family was eligible but declined to participate citing privacy concerns, and two families might be enrolled at a later date. Phenotype descriptions for individuals with ID are summarized in Table 1, and details of the molecular findings are outlined in Table 2. Pedigree charts are provided in Figure S1.
Table 1.
Family reported phenotype description for all participants in the study (PR = proband)
| PR #1 | PR #2 | PR #3 | PR #4 | PR#5 | |
|---|---|---|---|---|---|
| IQ | 67–69 | 30 | 55 | 64 | 68 |
| Significant problems with self‐feeding, toileting, dressing | ✓ | ✓ | |||
| Language disability | ✓ | ✓ | |||
| Social interaction problems | ✓ | ✓ | ✓ | ||
| Difficulties with memory and logical reasoning | ✓ | ✓ | ✓ | ✓ | |
| Inattention | ✓ | ✓ | |||
| Microcephaly | ✓ | ✓ | |||
| Sleep disturbances | ✓ | ✓ | |||
| Visual impairment | ✓ | ✓ | |||
| Hearing deficits | ✓ | ✓ | |||
| Hypotonia | ✓ | ✓ | ✓ | ✓ | |
| Skeletal abnormalities | ✓ | ✓ | ✓ |
Table 2.
Molecular findings for three participants with genetic diagnosis resulting from this study (PR = proband)
| PR #1 | PR #2 | PR #3 | |
|---|---|---|---|
| Gene | KANSL1 | TUSC3 | MED113L |
| Molecular finding | 17q21.31 deletion |
NM_006765.3:c.992C>A (p.Ser331Ter) |
NM_015335.4:c.263G>A (p.Trp88Ter) |
| Zygosity | Heterozygous | Homozygous | Heterozygous |
| de novo | Unknown | No | Yes |
| Genetic diagnosis | Koolen‐de Vries syndrome (OMIM # 610443) | Mental retardation, autosomal recessive 7 (OMIM # 611093) | Mental retardation and distinctive facial features with or without cardiac defects (OMIM # 616789) |
| Trio available | NO | YES | YES |
3.2. Family #1
Proband #1 was a 40‐year‐old Caucasian male with a diagnosis of ID. His mother reported an IQ score of 67–69, social interaction problems, difficulties with memory, problem‐solving, and logical reasoning, macrocephaly, mild visual impairment (strabismus and hyperopia), mild hearing deficit, and hypotonia. The proband had distinctive facial features including epicanthal folds, ptosis, mildly upslanted palpebral fissure, high nasal bridge, broad nasal root, bulbous nose (Figure 1a). Previous nondiagnostic testing included a normal male karyotype (46, XY) and negative Fragile X syndrome testing. Prenatal and perinatal history were unremarkable and there were no known exposures that could explain his ID. Both proband and his mother were recruited for the study.
Figure 1.
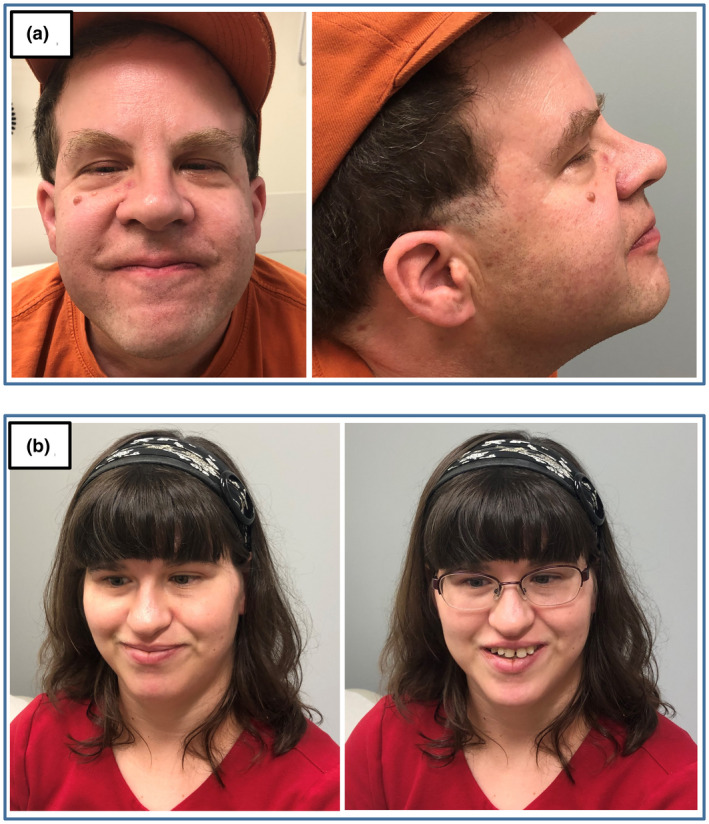
Facial features of proband #1 (a) and proband #3 (b)
The CNV analysis of exome sequencing data identified a heterozygous 310 kb deletion (chr17:43861911‐44172068, hg19) at 17q21.31 in the proband absent from his mother, other cases in the study, and 270 additional control samples. The deletion contained the coding exons of CRHR1, SPPL2C, MAPT, STH genes, and exons 3–15 (NM_001193466) of the KANSL1 (KAT8 regulatory NSL complex subunit 1) gene. The deletion was validated in the proband using MLPA probes specific for KANSL1 gene. The MLPA also confirmed that the mother did not carry the deletion, but the biological father was not available to ascertain de novo status. A subsequent chromosomal microarray (CMA) showed a slightly larger 505 kb deletion (chr17:43707119‐44212391) encompassing the same KANSL1 exons. Heterozygous deletions at 17q21.31 have been shown to cause Koolen‐de Vries syndrome (KdVS, OMIM #610443), a clinically heterogeneous disorder characterized by hypotonia, developmental delay, moderate ID, and characteristic facial dysmorphism (Koolen et al., 2012).
3.3. Family #2
Proband #2 was a 39‐year‐old Caucasian male with a diagnosis of ID. His mother reported an IQ score of 30, significant problems with toileting, language disability, social interaction problems, difficulties with memory, problem‐solving, and logical reasoning, attention deficit hyperactivity disorder, occasional aggression, microcephaly, hypotonia, high pain threshold, skeletal abnormalities, hypothyroidism, and rheumatoid arthritis. The proband had negative Fragile X syndrome test but no other genetic testing had been done. Prenatal and perinatal history were unremarkable without any known exposures. The proband had three healthy siblings. Both parents and the proband were recruited for the study. Our variant prioritization strategy identified a homozygous stop‐gain variant c.992C>A; p.Ser331Ter (NM_006765.3) in Tumor suppressor candidate 3 gene (TUSC3) in the proband. Both parents were heterozygous for the variant, and there was no evidence of consanguinity. The variant and its inheritance were confirmed by Sanger sequencing. This variant was observed at an ultra‐low frequency in the general population (gnomAD database MAF = 0.009%, 25/282,626, no homozygous). This variant has been reported previously in the hemizygous state in an individual with ID and autism where it was in trans with a TUSC3 gene deletion (Jones et al., 2013). Biallelic pathogenic variants in TUSC3 gene have been reported to cause autosomal recessive ID (OMIM # 611093) in multiple studies (Al‐Amri et al., 2016; Garshasbi et al., 2008; Molinari et al., 2008).
3.4. Family #3
Proband #3 was a 27‐year‐old Caucasian female with a diagnosis of ID (Figure 1b). Her mother reported an IQ score of 55, significant problems in activities such as self‐feeding, toileting, and dressing, language disability, social interaction problems, difficulties with memory, problem‐solving, and logical reasoning, inattention, self‐injurious behavior, sleep disturbances, cataracts, Duane syndrome, mild hearing deficit, hypotonia, spasticity, pronated ankles, compulsive skin picking, and sensory difficulties. Negative prior workup included karyotype, Fragile X, Prader‐Willi, Rett syndrome, and carnitine and biotin deficiency testing. Prenatal and perinatal history were unremarkable with no known exposures. Both parents and the proband were recruited for the study.
Exome sequencing analysis identified a de novo heterozygous stop‐gain variant c.263G>A, p.Trp88Ter (NM_015335.4) in mediator complex subunit 13‐like (MED13L) gene. The variant was confirmed in the proband and was not detected in the parents by Sanger sequencing. This novel variant has not been observed in the general population (gnomAD database). Multiple studies, reviewed in (Asadollahi et al., 2017) have reported on the role of loss of function variants in the MED13L gene in individuals with ID with or without heart defects (OMIM # 616789).
3.5. Family #4
Proband #4 was a 24‐year‐old Caucasian female with a diagnosis of ID. Her legal representative reported an IQ score of 64, sleep disturbances, hypotonia and mild scoliosis. The proband had a negative Fragile X syndrome test. The details of prenatal and perinatal history were not known. No relatives were available for the study.
3.6. Family #5
Proband #5 was a 35‐year‐old Caucasian female with a diagnosis of ID. Her mother reported an IQ score of 68, difficulties with memory, problem‐solving, and logical reasoning. The family reported that proband had negative genetic testing when she was a child (~6‐year‐old), but they could not recall the type of test. Prenatal and perinatal history was unremarkable, and there were no known exposures. Proband and the mother were recruited for the study.
In families #4 and #5 exome sequencing analysis did not identify any variants that would explain ID in the proband and meet the ACMG criteria for the return of pathogenic or likely pathogenic variant result.
4. DISCUSSION
Many adults with ID underwent less comprehensive genetic testing when they were pediatric patients (e.g. karyotype, single gene tests). The aim of this pilot study was to apply more comprehensive, exome sequencing‐based assays, and determine the rate of known Mendelian diseases in adults with ID of any severity. Our focus was to recruit participants and their families through non‐clinical organizations that provide education and support services to individuals with ID. Our recruitment was a one‐time‐only mailing of a study brochure which resulted in interest from 13 families and recruitment of 5 families. This demonstrates that there is interest in families with adults with ID in participating in genetic research. In addition, all 5 families recruited consented to receive a genetic diagnosis, indicating that this is an important part of participation for the families.
In three of five participating families exome sequencing identified pathogenic or likely pathogenic variant (according to ACMG standards) matching the indication of ID, indicating a high rate of genetic diagnosis in adults with ID. Each of the three families has been invited to an appointment with a board certified clinical geneticist for CAP/CLIA (College of American Pathologists accredited/Clinical Laboratory Improvement Amendments certified) confirmation of the results, and discussion of the findings and any potential medical follow‐up or change of care or disease management.
Proband #1 had distinctive facial features characteristic of individuals with Koolen de Vries syndrome (KdVS) including epicanthal folds, ptosis, mildly upslanted palpebral fissure, high nasal bridge, broad nasal root, and a bulbous nose. He also had mild ID, hypotonia, strabismus, and a friendly disposition that is often described in individuals with KdVS (Koolen, Morgan, & de Vries, 1993). The diagnosis had important management implications due to other body systems that are frequently involved in KdVS. In a recent review of the phenotypic spectrum of individuals with KdVS, 38.6% of individuals had a heart defect, and 45.2% had renal and urogenital anomalies (Koolen et al., 2016). In particular, cryptorchidism is found in 71% of males with KdVS and is associated with complications such as testicular cancer. A previous echocardiogram in this patient was normal, however, he had never undergone imaging of the kidneys or urogenital system. Thus, renal and scrotal ultrasounds were done after this new diagnosis was made and both were normal. The family was counseled that recurrence risk was low (<1%) for future pregnancies as all knownKANSL1 pathogenic variants and nearly all reported 17q21.31 deletions have been de novo (Koolen et al., 1993). However, the possibility of germline mosaicism could not be excluded.
Proband #2 had a homozygous stop‐gain variant in TUSC3, a gene involved in magnesium transport and homeostasis as well as glycosylation, with an important role in learning, memory and embryonic development, and recently recognized as a novel tumor suppressor gene (Yu et al., 2017). The TUSC3gene is mainly expressed in the brain, consistent with its association with ID (GTEx Consortium, 2013). Biallelic defects in TUSC3 cause a congenital disorder of N‐linked glycosylation (CDG) leading to ID and developmental delays and other systemic findings including microcephaly, dysmorphic features, and short stature. Like our patient, affected individuals may have significant speech delay and IQs in the moderate to severe ID ranges (Garshasbi et al., 2008; Khan et al., 2011). In terms of treatment, there is no known corrective therapies for TUSC3‐CDG and management is aimed at preventative/supportive measures. Regarding its suspected role as a tumor suppressor gene, TUSC3 has been proposed as a potential biomarker and therapeutic target for diagnosis and treatment of some cancers (Yu et al., 2017). The patient and family were also counseled to adhere to age‐appropriate cancer screening, including colonoscopy beginning at age 50 years. The patient's parents were confirmed to be heterozygous carriers of the TUSC3 variant and were advised of the 25% recurrence risk in another pregnancy and that their other unaffected children had a ~2/3rd's risk of also being carriers.
In proband #3 we identified a de novo heterozygous stop‐gain variant in MED13L gene. The MED13L gene is associated with an autosomal dominant ID syndrome with infantile onset, developmental delay, speech and language delay, and dysmorphic features (PMID: 25758992), all of which this patient featured. This diagnosis was especially important for management because of the increased risk of associated cardiac malformations including septal defects and/or transposition of the great arteries (Asadollahi et al., 2013; van Haelst et al., 2015). The parents of this patient did not report any cardiac issues in her, however we recommended a baseline echocardiogram that was done and did not reveal any structural heart defects. She was also advised to continue following with ophthalmology given her history of cataracts which notably have also been associated with the closely related MED13 gene (Boutry‐Kryza et al., 2012). As this was a de novo variant, the family was counseled that recurrence risk in another pregnancy was low (<1%) but not zero due to possible germline mosaicism.
It has been demonstrated before that complete trio facilitates genetic analysis, and results in higher diagnostic rates (Farwell et al., 2015). The two families in our study where exome sequencing failed to provide a genetic diagnosis did not have a complete trio. Two probands from these families also had milder phenotypes and higher IQ scores (Table 1). Also, exome sequencing analysis will identify only CNVs spanning multiple exons, while smaller CNVs and other types of structural variants would usually not be detected. Thus CMA testing would be the next logical test in these undiagnosed cases followed by whole genome sequencing that would enable the identification of a larger spectrum of sizes and types of SVs (Shashi et al., 2019), as well as identification of noncoding variants and might lead to a diagnosis.
This study is of small size, with only 5 families recruited and analyzed and indicates the need for larger genetic studies of adults with ID recruited in a non‐clinical setting. Currently, medical insurance providers rarely cover the expense of exome or genome sequencing testing for adults with ID due to perceived limited clinical utility. This has resulted in disparity in exome and whole‐genome diagnostic testing rates between the pediatric and adult patients. The benefits of exome sequencing in patients with ID has been established before (Benson et al., 2020; Bowling et al., 2017; de Ligt et al., 2012; Mir & Kuchay, 2019; Snoeijen‐Schouwenaars et al., 2019; Willemsen & Kleefstra, 2014). Our study shows that with minimal vetting we were able to identify participants most likely to benefit from exome sequencing. It also points to potentially high yield and value of exome and genome sequencing as a diagnostic tool in this population. In addition, these new diagnoses have had positive implications for these individuals’ medical insurance coverage and access to support services. Our study provides evidence that there is benefit in revisiting adult patients with ID who have not undergone newer comprehensive sequencing‐based genetic testing.
CONFLICT OF INTEREST
Baylor College of Medicine receives revenue from genetic testing via co‐ownership with Baylor Genetics Laboratories.
AUTHOR CONTRIBUTIONS
AS and DMur conducted participant recruitment. SD,QM, MG, JH, and DMuz performed exome sequencing and variant validation. AS, DMur, QM, and JH performed analyses of the genetic data. DMur conducted clinical visits. AS, DMur, and RG wrote the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig S1
ACKNOWLEDGMENTS
The authors thank all the families that participated in the study, as well as Janniece Sleigh and the staff of The Arc of Greater Houston, and Steve Vetrano and the staff of the Avondale House in Houston TX. Participant recruitment, exome sequencing and variant interpretation were supported by NIH/NHGRI Grant 1UM1HG008898 and a personal gift to the Human Genome Sequencing Center at Baylor College of Medicine.
Sabo A, Murdock D, Dugan S, et al. Community‐based recruitment and exome sequencing indicates high diagnostic yield in adults with intellectual disability. Mol Genet Genomic Med. 2020;8:e1439 10.1002/mgg3.1439
Funding information
National Human Genome Research Institute, National Institutes of Health Grant/Award Number 1UM1HG008898 and a personal gift to the Human Genome Sequencing Center at Baylor College of Medicine.
Contributor Information
Aniko Sabo, Email: sabo@bcm.edu.
Richard Gibbs, Email: agibbs@bcm.edu.
REFERENCES
- Adams, D. R. , & Eng, C. M. (2018). Next‐generation sequencing to diagnose suspected genetic disorders. New England Journal of Medicine, 379(14), 1353–1362. 10.1056/NEJMra1711801 [DOI] [PubMed] [Google Scholar]
- Al‐Amri, A. , Saegh, A. A. , Al‐Mamari, W. , El‐Asrag, M. E. , Ivorra, J. L. , Cardno, A. G. , … Ali, M. (2016). Homozygous single base deletion in TUSC3 causes intellectual disability with developmental delay in an Omani family. American Journal of Medical Genetics. Part A, 170(7), 1826–1831. 10.1002/ajmg.a.37690 [DOI] [PubMed] [Google Scholar]
- Asadollahi, R. , Oneda, B. , Sheth, F. , Azzarello‐Burri, S. , Baldinger, R. , Joset, P. , … Rauch, A. (2013). Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability. European Journal of Human Genetics: EJHG, 21(10), 1100–1104. 10.1038/ejhg.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadollahi, R. , Zweier, M. , Gogoll, L. , Schiffmann, R. , Sticht, H. , Steindl, K. , & Rauch, A. (2017). Genotype‐phenotype evaluation of MED13L defects in the light of a novel truncating and a recurrent missense mutation. European Journal of Medical Genetics, 60(9), 451–464. 10.1016/j.ejmg.2017.06.004 [DOI] [PubMed] [Google Scholar]
- Baker, K. , Raymond, F. L. , & Bass, N. (2012). Genetic investigation for adults with intellectual disability: Opportunities and challenges. Current Opinion in Neurology, 25(2), 150–158. 10.1097/WCO.0b013e328351820e [DOI] [PubMed] [Google Scholar]
- Benson, K. A. , White, M. , Allen, N. M. , Byrne, S. , Carton, R. , Comerford, E. , … Cavalleri, G. L. (2020). A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. European Journal of Human Genetics, 28(8). 10.1038/s41431-020-0610-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutry‐Kryza, N. , Labalme, A. , Till, M. , Schluth‐Bolard, C. , Langue, J. , Turleau, C. , … Sanlaville, D. (2012). An 800 kb deletion at 17q23.2 including the MED13 (THRAP1) gene, revealed by aCGH in a patient with a SMC 17p. American Journal of Medical Genetics. Part A, 158A(2), 400–405. 10.1002/ajmg.a.34222 [DOI] [PubMed] [Google Scholar]
- Bowling, K. M. , Thompson, M. L. , Amaral, M. D. , Finnila, C. R. , Hiatt, S. M. , Engel, K. L. , … Cooper, G. M. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Medicine, 9(1), 43 10.1186/s13073-017-0433-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulla, L. S. , Reed, G. M. , Vaez‐azizi, L. M. , Cooper, S.‐A. , Leal, R. M. , Bertelli, M. , … Saxena, S. (2011). Intellectual developmental disorders: Towards a new name, definition and framework for "mental retardation/intellectual disability" in ICD‐11. World Psychiatry, 10(3), 175–180. 10.1002/j.2051-5545.2011.tb00045.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt, J. , Willemsen, M. H. , van Bon, B. W. M. , Kleefstra, T. , Yntema, H. G. , Kroes, T. , … Vissers, L. E. L. M. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. New England Journal of Medicine, 367(20), 1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Farwell, K. D. , Shahmirzadi, L. , El‐Khechen, D. , Powis, Z. , Chao, E. C. , Tippin Davis, B. , … Tang, S. (2015). Enhanced utility of family‐centered diagnostic exome sequencing with inheritance model‐based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genetics in Medicine, 17(7), 578–586. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- Fromer, M. , & Purcell, S. M. (2014). Using XHMM software to detect copy number variation in whole‐exome sequencing data. Current Protocols in Human Genetics, 81, 7.23.1–7.23.21. 10.1002/0471142905.hg0723s81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garshasbi, M. , Hadavi, V. , Habibi, H. , Kahrizi, K. , Kariminejad, R. , Behjati, F. , … Kuss, A. W. (2008). A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. American Journal of Human Genetics, 82(5), 1158–1164. 10.1016/j.ajhg.2008.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genome Aggregation Database Consortium , Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium . (2013). The Genotype‐Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, M. A. , Rhodenizer, D. , da Silva, C. , Huff, I. J. , Keong, L. , Bean, L. J. H. , … Hegde, M. R. (2013). Molecular diagnostic testing for congenital disorders of glycosylation (CDG): Detection rate for single gene testing and next generation sequencing panel testing. Molecular Genetics and Metabolism, 110(1–2), 78–85. 10.1016/j.ymgme.2013.05.012 [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. A. , Rafiq, M. A. , Noor, A. , Ali, N. , Ali, G. , Vincent, J. B. , & Ansar, M. (2011). A novel deletion mutation in the TUSC3 gene in a consanguineous Pakistani family with autosomal recessive nonsyndromic intellectual disability. BMC Medical Genetics, 12, 56 10.1186/1471-2350-12-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen, D. A. , Kramer, J. M. , Neveling, K. , Nillesen, W. M. , Moore‐Barton, H. L. , Elmslie, F. V. , … de Vries, B. B. A. (2012). Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nature Genetics, 44(6), 639–641. 10.1038/ng.2262 [DOI] [PubMed] [Google Scholar]
- Koolen, D. A. , Morgan, A. , & de Vries, B. B. (1993). Koolen‐de Vries syndrome In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J., Stephens K., & Amemiya A. (Eds.), GeneReviews®. Seattle, WA: University of Washington; Retrieved from www.ncbi.nlm.nih.gov/books/NBK24676/ [Google Scholar]
- Koolen, D. A. , Pfundt, R. , Linda, K. , Beunders, G. , Veenstra‐Knol, H. E. , Conta, J. H. , … de Vries, B. B. A. (2016). The Koolen‐de Vries syndrome: A phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. European Journal of Human Genetics, 24(5), 652–659. 10.1038/ejhg.2015.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski, J. R. , Reid, J. G. , Gonzaga‐Jauregui, C. , Rio Deiros, D. , Chen, D. C. Y. , Nazareth, L. , … Gibbs, R. A. (2010). Whole‐genome sequencing in a patient with Charcot‐Marie‐Tooth neuropathy. New England Journal of Medicine, 362(13), 1181–1191. 10.1056/NEJMoa0908094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik, P. K. , Mascarenhas, M. N. , Mathers, C. D. , Dua, T. , & Saxena, S. (2011). Prevalence of intellectual disability: A meta‐analysis of population‐based studies. Research in Developmental Disabilities, 32(2), 419–436. 10.1016/j.ridd.2010.12.018 [DOI] [PubMed] [Google Scholar]
- Maves, S. N. , Williams, M. S. , Williams, J. L. , Levonian, P. J. , & Josephson, K. D. (2007). Analysis of 88 adult patients referred for genetics evaluation. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 145C(3), 232–240. 10.1002/ajmg.c.30141 [DOI] [PubMed] [Google Scholar]
- Miller, D. T. , Adam, M. P. , Aradhya, S. , Biesecker, L. G. , Brothman, A. R. , Carter, N. P. , … Ledbetter, D. H. (2010). Consensus statement: Chromosomal microarray is a first‐tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics, 86(5), 749–764. 10.1016/j.ajhg.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir, Y. R. , & Kuchay, R. A. H. (2019). Advances in identification of genes involved in autosomal recessive intellectual disability: A brief review. Journal of Medical Genetics, 56(9), 567–573. 10.1136/jmedgenet-2018-105821 [DOI] [PubMed] [Google Scholar]
- Molinari, F. , Foulquier, F. , Tarpey, P. S. , Morelle, W. , Boissel, S. , Teague, J. , … Colleaux, L. (2008). Oligosaccharyltransferase‐subunit mutations in nonsyndromic mental retardation. American Journal of Human Genetics, 82(5), 1150–1157. 10.1016/j.ajhg.2008.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdock, D. R. , Jiang, Y. , Wangler, M. , Khayat, M. M. , Sabo, A. , Juusola, J. , … Gibbs, R. A. (2019). Xia‐Gibbs syndrome in adulthood: A case report with insight into the natural history of the condition. Cold Spring Harbor Molecular Case Studies, 5(3), a003608. 10.1101/mcs.a003608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekeles, H. , Accogli, A. , Boudrahem‐Addour, N. , Russell, L. , Parente, F. , & Srour, M. (2019). Diagnostic yield of intellectual disability gene panels. Pediatric Neurology, 92, 32–36. 10.1016/j.pediatrneurol.2018.11.005 [DOI] [PubMed] [Google Scholar]
- Quinodoz, M. , Royer‐Bertrand, B. , Cisarova, K. , Di Gioia, S. A. , Superti‐Furga, A. , & Rivolta, C. (2017). DOMINO: Using machine learning to predict genes associated with dominant disorders. American Journal of Human Genetics, 101(4), 623–629. 10.1016/j.ajhg.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, J. G. , Carroll, A. , Veeraraghavan, N. , Dahdouli, M. , Sundquist, A. , English, A. , … Boerwinkle, E. (2014). Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics, 15, 30 10.1186/1471-2105-15-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmer, A. , Phan, H. , Mathieson, I. , Iqbal, Z. , Twigg, S. R. F. , Wilkie, A. O. M. , … Lunter, G. (2014). Integrating mapping‐, assembly‐ and haplotype‐based approaches for calling variants in clinical sequencing applications. Nature Genetics, 46(8), 912–918. 10.1038/ng.3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashi, V. , Schoch, K. , Spillmann, R. , Cope, H. , Tan, Q.‐G. , Walley, N. , … Goldstein, D. B. (2019). A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genetics in Medicine, 21(1), 161–172. 10.1038/s41436-018-0044-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeijen‐Schouwenaars, F. M. , van Ool, J. S. , Verhoeven, J. S. , van Mierlo, P. , Braakman, H. M. H. , Smeets, E. E. , … Willemsen, M. H. (2019). Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia, 60(1), 155–164. 10.1111/epi.14618 [DOI] [PubMed] [Google Scholar]
- van Haelst, M. M. , Monroe, G. R. , Duran, K. , van Binsbergen, E. , Breur, J. M. , Giltay, J. C. , & van Haaften, G. (2015). Further confirmation of the MED13L haploinsufficiency syndrome. European Journal of Human Genetics: EJHG, 23(1), 135–138. 10.1038/ejhg.2014.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, M. H. , & Kleefstra, T. (2014). Making headway with genetic diagnostics of intellectual disabilities. Clinical Genetics, 85(2), 101–110. 10.1111/cge.12244 [DOI] [PubMed] [Google Scholar]
- Yang, H. , & Wang, K. (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nature Protocols, 10(10), 1556–1566. 10.1038/nprot.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Muzny, D. M. , Xia, F. , Niu, Z. , Person, R. , Ding, Y. , … Eng, C. M. (2014). Molecular findings among patients referred for clinical whole‐exome sequencing. JAMA, 312(18), 1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Zhai, C. , Fan, Y. , Zhang, J. , Liang, N. , Liu, F. , … Du, J. (2017). TUSC3: A novel tumour suppressor gene and its functional implications. Journal of Cellular and Molecular Medicine, 21(9), 1711–1718. 10.1111/jcmm.13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1