

Abstract
Background
To date, several studies have suggested that genes involved in monogenic forms of Parkinson's disease (PD) contribute to unrelated sporadic cases, but there is limited evidence in the Chinese population.
Methods
We performed a systematic analysis of 12 autosomal‐dominant PD (AD‐PD) genes (SNCA, LRRK2, GIGYF2, VPS35, EIF4G1, DNAJC13, CHCHD2, HTRA2, NR4A2, RIC3, TMEM230, and UCHL1) using panel sequencing and database filtration in a case‐control study of a cohort of 391 Chinese sporadic PD patients and unrelated controls. We evaluated the association between candidate variants and sporadic PD using gene‐based analysis.
Results
Overall, 18 rare variants were discovered in 18.8% (36/191) of the index patients. In addition to previously reported pathogenic mutations (LRRK2 p.Arg1441His and p.Ala419Val), another four unknown variants were found in LRRK2, which also contribute to PD risk (p = 0.002; odds ratio (OR) = 7.83, 95% confidence intervals (CI) = 1.76–34.93). The cumulative frequency of undetermined rare variants was significantly higher in PD patients (14.1%) than in controls (3.5%) (p = 0.0002; OR=4.54, 95% CI = 1.93‐10.69).
Conclusion
Our results confirm the strong impact of LRRK2 on the risk of sporadic PD, and also provide considerable evidence of the existence of additional undetermined rare variants in AD‐PD genes that contribute to the genetic etiology of sporadic PD in a Chinese cohort.
Keywords: autosomal‐dominant genes, gene‐based analysis, Parkinson's disease, rare variants, sporadic
Our results confirm the strong impact of LRRK2 on the risk of sporadic PD, and also provide considerable evidence of the existence of additional undetermined rare variants in AD‐PD genes that contribute to the genetic etiology of sporadic PD in a Chinese cohort.

1. INTRODUCTION
Parkinson's disease (PD) is the most common movement disorder (Alves, Forsaa, Pedersen, Dreetz Gjerstad, & Larsen, 2008), with a global prevalence of 6.2 million cases in 2015 that is estimated to rise to 12.9 million by 2040 (Dorsey & Bloem, 2018). PD is defined by two major neuropathological hallmarks: loss of dopaminergic neurons in the substantia nigra pars compacta of the midbrain and the presence of Lewy bodies (LB) (Postuma et al., 2015). However, the etiology of PD has not been fully elucidated. In addition to increasing age and exposure to environmental risk factors, genetics also play an important role in the pathology of PD (Blauwendraat, Nalls, & Singleton, 2019).
Several genome‐wide association studies (GWASs) have revealed a considerable number of candidate variants associated with PD, although some were first discovered in family‐based linkage studies; however, many of these genes were successfully replicated in common sporadic PD patients (Simon‐Sanchez et al., 2009; Spataro et al., 2015), indicating that genes involved in monogenic forms of the disease also act as susceptibility factors in the unrelated sporadic form of PD. Despite these discoveries, much of the genetic contribution to PD remains unexplained (Spataro et al., 2015), in part, because when GWASs began, the field was dominated by the simple common disease–common variant hypothesis. GWAS effectively represent common genetic variants with a typical minor allele frequency (MAF) >1%–5% (Lee, Abecasis, Boehnke, & Lin, 2014). Analyses of rare variants (MAF <1%) may explain additional disease risk or trait variability (Gibson, 2012) especially in diseases with complex traits such as PD.
Several studies have indicated an association between rare variants of Mendelian genes and sporadic PD in different populations (Benitez et al., 2016; Lesage & Brice, 2012; Spataro et al., 2015; Tan et al., 2019); however, there is limited evidence in the Chinese population. Recently, Yang et al. (2019) identified several rare variants of seven autosomal‐dominant PD (AD‐PD) genes consisting of SNCA (OMIM No. 163890), LRRK2 (OMIM No. 609007), GIGYF2 (OMIM No. 612003), VPS35 (OMIM No. 601501), EIF4G1 (OMIM No. 600495), DNAJC13 (OMIM No. 614334), and CHCHD2 (OMIM No. 616244) in a case‐control study with Chinese ethnic background, indicating the possible contribution of other AD‐PD genes. In this study, we investigated these seven AD‐PD genes and five additional previously reported monogenic AD‐PD genes, including HTRA2 (OMIM No. 606441), NR4A2 (OMIM No. 601828), RIC3 (OMIM No. 610509), TMEM230 (OMIM No. 617019), and UCHL1 (OMIM No. 191342), (Blauwendraat et al., 2019), to further illustrate the association between monogenic genes and sporadic PD patients in Chinese population.
2. METHODS
2.1. Ethical compliance
This study was conducted in accordance with the Declaration of Helsinki with formal approval obtained from the ethics review boards of the Second Affiliated Hospital of Zhejiang University. All participants provided written informed consent to genetic analysis and disclosure of medical information.
2.2. Subjects
In total, 191 sporadic PD cases (aged 16–82 years) and 200 ethnicity‐matched controls were recruited from the outpatient neurology clinics of the Second Affiliated Hospital of Zhejiang University (China) between January 2016 and June 2019. All subjects were examined by at least two neurology physicians specializing in movement disorders. Inclusion criterion referred to the diagnosis of PD based on the clinical criteria defined by the Movement Disorder Society (Postuma et al., 2015). Patients with secondary Parkinsonism and other forms of atypical Parkinsonism or with a family history of PD were excluded.
2.3. Sample preparation and sequencing
The quality and concentration of genomic DNA extracted from peripheral whole blood samples using standard procedures were assessed with a Qubit 3.0 Fluorometer (Life Invitrogen). Fragmented genomic DNA was captured by a customized array designed to target all exons, splicing sites, and flanking intronic sequences of 12 selected genes (as shown in Table S1). Sequencing was conducted as 150‐bp paired‐end runs on an Illumina NovaSeq 5000 system to an average depth of coverage >300‐fold. Sequence reads were mapped to the human assembly GRCh37/hg19 (GCA_000001405.1) using a Burrows–Wheeler–Aligner (BWA) (Li & Durbin, 2009), and variant calling was conducted using SAMtools (Li, 2011), followed by variant annotation using ANNOVAR (Yang & Wang, 2015).
2.4. Variant filtration
To identify candidate rare variants, we adopted a three‐level filtration algorithm. First, we required a MAF <1% or “not available” for variants in the Genome Aggregation Database (gnomAD), the 1000 Genomes Project (May 2019), and the Exome Aggregation Consortium (ExAC) (Genomes Project et al., 2015); otherwise, variants with unbalanced reads (variant allele <25%) and regions covered by <5× reads were eliminated. Second, we selected non‐synonymous substitutions including missense and nonsense mutations as well as small insertions and deletions, which are considered to be the most likely to cause loss‐of‐function of the encoded protein (Adzhubei et al., 2010; Fu et al., 2013). Third, for further prioritization, all selected non‐synonymous variants were analyzed with dbNSFP (X. Liu, Wu, Li, & Boerwinkle, 2016) (version 3.5) and CADD (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019). We retained only variants previously reported to be pathogenic in ClinVar (Landrum & Kattman, 2018) (www.ncbi.nlm.nih.gov/clinvar/) as well as rare variants that had CADD phred scores >15 or REVEL scores >0.5 or MetaL ranking score >0.75 or predicted to be damaging in the three most commonly used in silico pathogenicity prediction applications (SIFT, PolyPhen‐2, and mutationTaster) according to previous studies (Richards et al., 2015; Tian et al., 2018). All candidate variants were validated by Sanger sequencing, with primers designed using Primer3 (Koressaar et al., 2018). Carriers of candidate variants were screened using Multiplex Ligation‐Dependent Probe Amplification to exclude confounders from common gross deletions or duplications. The three‐dimensional (3D) protein structures of the wild‐type and variant‐type proteins were predicted using Phyre2 (Kelley, Mezulis, Yates, Wass, & Sternberg, 2015) and visualized by PyMOL (The PyMOL Molecular Graphics System, Version 2.3, Schrödinger, LLC).
2.5. Statistical analysis
Demographic characteristics were depicted as the means ± standard deviation (SD) and compared using Student's t‐tests. Sex‐related variables were assessed using Chi‐square tests. For all candidate rare variants, we calculated the proportion of carriers and assessed Hardy–Weinberg equilibrium with the Chi‐square test in both cases and controls. The subgroup of candidate variants categorized as unclear pathogenicity was evaluated with gene‐based burden analysis. Associations between rare variants and sporadic PD were analyzed using Chi‐square tests or Fisher's exact test, odd ratios (OR), and 95% confidence intervals (CI) (Li & Leal, 2008). All analyses were conducted using SPSS version 26 (IBM, Armonk, NY, USA). A two‐tailed p‐value of 0.05 was set as a nominal significance threshold.
3. RESULTS
3.1. Summary of demographic data
We screened for 12 AD‐PD genes consisting of three genes previously reported to contain mutations robustly associated with PD (SNCA, LRRK2, and VPS35) and nine genes associated with PD with low confidence (GIGYF2, EIF4G1, DNAJC13, CHCHD2, HTRA2, NR4A2, RIC3, TMEM230, and UCHL1) in 191 Chinese sporadic PD patients and 200 unrelated controls. Our cohort comprised 55% early onset PD (EOPD) with the age at onset of 43.72 ± 7.13 years and 45% late‐onset PD (LOPD) with the age at onset of 60.32 ± 7.96 years. Overall, the mean age of onset at enrollment was 51.15 ± 11.16 years for cases (55% males) and there was no significant difference compared with the mean age at onset of controls at 49.49 ± 9.86 years (53% males) (Table 1).
Table 1.
Summary of demographic data
| Series | N | AAO mean ± SD (range) in years | Male:Female ratio |
|---|---|---|---|
| Total PD | 191 | 51.15 ± 11.16 | 106:85 |
| EOPD (AAO ≤ 50) | 106 | 43.72 ± 7.13 | 56:50 |
| LOPD (AAO > 50) | 85 | 60.32 ± 7.96 | 50:35 |
| control | 200 | 49.49 ± 9.86 | 106:94 |
Abbreviations: AAO, age at onset; EOPD, early onset Parkinson's disease; LOPD, late‐onset Parkinson's disease; N, number.
3.2. Overview of candidate rare variants
We validated candidate variants after the three‐level filtration in this cohort. A total of 18 rare non‐synonymous coding variants were validated in 18.8% (36/191) of sporadic patients. Among them, 33.3% (6/18) of the variants were found in LRRK2 with the locations presented in Figure 1, and 55.6% (10/18) in CHCHD2, DNAJC13, GIGYF2, NR4A2, and SNCA, all of which had two candidate variants. The rest of these variants were singletons, located in HTRA2 and EIF4G1. Despite high sequencing coverage, no rare variants were found in RIC3, TMEM230, UCHL1, and VPS35 in our cohort (Table 2). Remarkably, none of the cases of candidate variant carriers were affected by common gross deletions and duplications.
Figure 1.
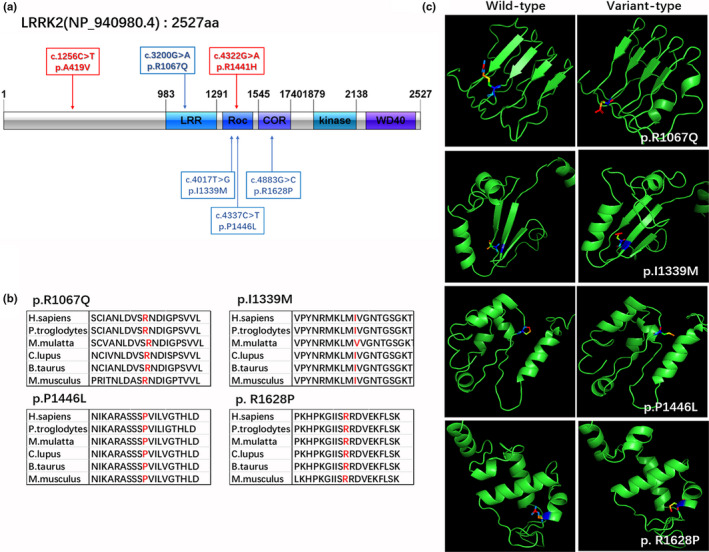
Schematic representation of LRRK2 and its variations. (a) LRRK2 contains 51 exons encoding a 2527‐amino acid protein with a leucine‐rich repeat domain, a Ras/GTPase (ROC) domain, and a protein kinase domain. The previously identified pathogenic variations in our cohort are indicated by red arrows and the other candidate variations are indicated by blue arrows. (b) The position and surroundings of unclear variants were highly conserved across different species. (c) The 3D structures of wild‐type and variant‐type proteins. Protein models are shown as secondary structures. Variant sites are shown as amino acid sequences
Table 2.
Summary of candidate variants after three‐level filtration in our cohort
| Gene | Chromosome position a | dbsnp150 | Amino Acid Change | Freq.GnomAD | Freq.1000G | Freq.ExAC | CADD‐phred | REVEL_score | MetaLR_rankscore | In silico tools | GERP++ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | PolyPhen‐2 | MutationTaster | |||||||||||
| CHCHD2 | 56172037 | rs864309650 | T61I | NA | NA | NA | 28.1 | 0.73 | 0.72 | D | D | D | 5.62 |
| 56174102 | rs142444896 | P2L | 9.70E‐03 | 4.39E‐03 | 7.50E‐03 | 24.7 | 0.28 | 0.57 | D | D | D | 4.83 | |
| DNAJC13 | 132181345 | novel | L583S | 4.47E‐05 | NA | 7.42E‐05 | 28.3 | 0.47 | 0.66 | D | D | D | 5.96 |
| 132222104 | rs145101163 | R1588H | 8.12E‐06 | NA | NA | 26.6 | 0.31 | 0.47 | D | D | D | 5.5 | |
| EIF4G1 | 184044759 | novel | R1139H | 1.63E‐05 | NA | 3.31E‐05 | 34 | 0.2 | 0.75 | D | D | D | 5.72 |
| GIGYF2 | 233684598 | rs757005602 | E811A | 2.80E‐04 | 2.00E‐04 | 3.00E‐04 | 22.8 | 0.47 | 0.85 | T | D | D | 5.25 |
| 233712060 | rs200216092 | P1155T | 3.26E‐05 | NA | 3.30E‐05 | 24.4 | 0.82 | 0.84 | D | D | D | 5.54 | |
| HTRA2 | 74757881 | rs200036604 | T215M | 1.22E‐05 | NA | 8.31E‐06 | 22.7 | 0.28 | 0.84 | D | P | D | 4.94 |
| LRRK2 | 40646786 | rs34594498 | A419V | 4.80E‐04 | 1.40E‐03 | 5.00E‐04 | 24.3 | 0.33 | 0.18 | D | D | A | 5.12 |
| 40704252 | rs74681492 | P1446L | 3.24E‐05 | NA | NA | 34 | 0.83 | 0.84 | D | D | D | 5.63 | |
| 40704237 | rs34995376 | R1441H | 1.99E‐04 | 9.98E‐04 | 2.00E‐04 | 28.8 | 0.64 | 0.82 | D | D | A | 5.64 | |
| 40702326 | rs773070538 | I1339M | 1.76E‐03 | 6.39E‐03 | 1.70E‐03 | 24.8 | 0.59 | 0.76 | D | D | D | 5.46 | |
| 40713845 | rs33949390 | R1628P | 4.06E‐06 | NA | 8.24E‐06 | 27.8 | 0.55 | 0.69 | D | D | D | 5.54 | |
| 40692148 | rs111341148 | R1067Q | 9.76E‐05 | 5.99E‐04 | 1.00E‐04 | 34 | 0.28 | 0.49 | D | D | D | 5.85 | |
| NR4A2 | 157182438 | rs201003462 | V539M | 8.14E‐06 | NA | NA | 24.8 | 0.63 | 0.97 | T | D | D | 6.06 |
| 157182309 | rs753783927 | V582M | 1.63E‐05 | NA | 1.69E‐05 | 26.8 | 0.4 | 0.76 | D | D | D | 6.07 | |
| SNCA | 90756775 | novel | V15D | 8.14E‐05 | 2.00E‐04 | 9.90E‐05 | 30 | 0.93 | 0.91 | D | D | D | 4.28 |
| 90650354 | rs191055637 | M127I | NA | NA | NA | 25.4 | 0.49 | 0.85 | T | P | D | 4.31 | |
ExAC, Exome Aggregation Consortium; GnomAD, Genome Aggregation Database; PolyPhen‐2, SIFT, and MutationTaster: The in silico tools more commonly used for missense variant interpretation.
A, disease‐causing automatic; D, damaging or disease‐causing; NA, not available; P, possibly damaging; T, tolerated. GERP++ is a score for the conservation of the amino acid: scores >3 can be considered as highly conserved.
Position on Genome Reference Consortium human genome build 37 (GenBank assembly accession: GCA_000001405.1).
3.3. Rare non‐synonymous variants
Among the candidate variants, 16.7% (3/18) were previously reported and known as pathogenic in the ClinVar databases; these variants consisted of LRRK2 p.Arg1441His, LRRK2 p.Ala419Val, and CHCHD2 p.Thr61Ile. In our cohort, LRRK2 p.Ala419Val was the most common pathogenic mutation, accounting for 4.2% (8/191) of cases. Regarding CHCHD2 p.Thr61Ile, the carrier of this mutation also carried the LRRK2 p.Ala419Val mutation.
A total of 66.7% (12/18) of the candidate variants have been reported previously, although their pathogenicity is unknown; and 16.7% (3/18) were novel with unknown significance, which were located in DNAJC13 p.Leu583Ser, SNCA p.Val15Asp, and EIF4G1 p.Arg1139His (Table 2). To eliminate interference from known mutations and investigate whether additional rare variants in specific genes contribute collectively to PD risk, we performed a gene‐based analysis of the unclear variants. The cumulative frequency of these variants was significantly higher in PD patients (14.1%) than that in controls (3.5%) (p = 0.0002; OR = 4.54, 95% CI = 1.93–10.69), suggesting that most of these variants are likely to be true risk factors for PD (Table 3).
Table 3.
Gene‐based analysis for rare non‐synonymous variants
| Gene | All rare variants involved | Remove known mutations | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| patients/191 | controls/200 | p value | OR (95%CI) | Power | patients/191 | controls/200 | p value | OR (95%CI) | Power | |
| LRRK2 | 23 | 2 | 0.000* | 13.55 (3.15‐58.33) | 0.94 | 14 | 2 | 0.002* | 7.83 (1.76‐34.93) | 0.77 |
| CHCHD2 | 2 | 1 | 0.54 | — | — | 1 | 1 | 0.97 | — | — |
| DNAJC13 | 3 | 0 | 0.12 | — | — | 3 | 0 | 0.12 | — | — |
| EIF4G1 | 1 | 2 | 0.59 | — | — | 1 | 2 | 0.59 | — | — |
| GIGYF2 | 2 | 0 | 0.24 | — | — | 2 | 0 | 0.24 | — | — |
| HTRA2 | 1 | 0 | 0.49 | — | — | 1 | 0 | 0.49 | — | — |
| NR4A2 | 3 | 1 | 0.29 | — | — | 3 | 1 | 0.29 | — | — |
| SNCA | 2 | 1 | 0.54 | — | — | 2 | 1 | 0.54 | — | — |
| All genes | 36 | 7 | 0.0002* | 6.40 (2.77‐14.79) | 0.99 | 27 | 7 | 0.0002* | 4.54 (1.93‐10.69) | 0.93 |
Abbreviation: 95% CI, 95% confidence intervals.
p < 0.05 was considered significant and marked in bold font.
Of the unclear variants, 26.7% (4/15) were located in LRRK2 (p.Pro1446Leu, p.Ile1339Met, p. Arg1628Pro, and p.Arg1067Gln). The overall frequency of LRRK2 undetermined variants was much higher in PD patients (7.3%) than in controls (1%) (p = 0.002; OR = 7.83, 95% CI = 1.76‐34.93) and remained statistically significant after the Bonferroni correction (α = 0.05/8 = 0.0063). Among these variants, LRRK2 p.Arg1628Pro was the most common risk variant and was significantly enriched in cases (5.2%) when compared to controls (0.5%) (p = 0.005; OR = 10.99, 95% CI = 1.39–86.74, Table S2). Remarkably, two variants located in DNAJC13 (p.Leu583Ser and p.Arg1588His), two variants located in GIGYF2 (p.Glu811Ala and p.Pro1155Thr), and one variant located in HTRA2 (p.Thr215Met) were present in 3.1% (6/191) of the PD individuals but absent in the controls. However, some candidate variants including p.Pro2Leu of CHCHD2, p.Arg1139His of EIF4G1, p.Val582Met of NR4A2, and p.Val15Asp of SNCA were present on both cases and controls. It should be noted that there was a greater number of carriers of EIF4G1 p.Arg1139His in the control group than in the PD patients (Table 3).
4. DISCUSSION
The phenotype of autosomal‐dominant inheritance familial PD, which is similar to sporadic PD, suggests that monogenic and sporadic forms of the disease are etiologically related (Simon‐Sanchez et al., 2009). In this study, we found 18 rare variants in 18.8% of sporadic PD patients. One third of these variants located in the LRRK2 gene were involved in 12.0% cases, which showed the strong effect of LRRK2 on the risk of sporadic PD. Additionally, there were significantly more carriers of candidate variants in PD cases than in controls, implicating these variants as true risk factors for PD.
LRRK2 was first identified in an autosomal‐dominant inheritance in late‐onset Parkinsonian families in 2004 (Zimprich et al., 2004). Variants in different domains of LRRK2 have been identified in both familial and sporadic PD in different ethnic populations (Berg et al., 2005; Di Fonzo et al., 2005; Gilks et al., 2005). In our study, six candidate variants of LRRK2 were found to be located mainly in functional domains. In addition, 12.0% (23/191) of patients found to carry those variants exhibited typical Parkinsonian symptoms, with most showing initial motor features of slowly progressive asymmetric tremor at rest or bradykinesia (Table 2). As previously reported, the LRRK2 p.Gly2019Ser mutation is thought to be the most frequent (Lunati, Lesage, & Brice, 2018); however, we did not find any carrier of this mutation in our cases. The most likely reason for this is that p.Gly2019Ser exists as a founder variant mainly in Eastern European Jews and North African Berbers, but not in Asian populations (Hulihan et al., 2008; Tan et al., 2010; Thaler, Ash, Gan‐Or, Orr‐Urtreger, & Giladi, 2009). We found only one case carrying the p.Arg1441His mutation, a pathogenic variant first identified in a Taiwanese PD family (Mata et al., 2005). LRRK2 P.Arg1441His occurred adjacent to two previously reported pathogenic mutations, p.Arg1441Cys and p.Arg1441Gly, identified as a 4322G‐A transition in exon 31 of LRRK2. This mutation resulted in an Arg1441His substitution in the Ras/GTPase (ROC) domain, which may impair the regulation of kinase activity (Gilsbach & Kortholt, 2014). Since Mata et al. (2005) identified the first p.Arg1441His carrier with an Asian ethnic background among 100 affected probands with a family history of Parkinsonism, familial PD carriers of diverse ethnicity have been identified in follow‐up studies (Ferreira et al., 2007; Spanaki, Latsoudis, & Plaitakis, 2006; Zabetian et al., 2005). Subsequently, a large case‐control study also confirmed one Asian PD carrier of p.Arg1441His (Ross et al., 2011). To date, the association of LRRK2 p.Arg1441His in sporadic PD is supported by limited data; thus, our study provides further evidence in support of this. Regarding LRRK2 P.Ala419Val, the most common pathogenic variant in this cohort was classified as pathogenic in ClinVar. Ross et al. reported a significant difference in the prevalence of p.Ala419Val between PD patients and controls in an Asian population (Ross et al., 2011), and this was confirmed by Guo et al. (Li et al., 2015) in a Chinese population. Guo and colleagues also found that p.Ala419Val especially affected patients with EOPD, which is consistent with our data (Table 2).
The most common risk variant was also found in LRRK2 p.Arg1628Pro, located in the COR domain. The substitution of a highly basic polar arginine (R) with a neutral nonpolar proline (P) is likely to cause a conformational change in the secondary structure of the LRRK2 protein. In a study of 1986 individuals from Taiwan and Singapore, Wu et al. (Ross et al., 2008) demonstrated that p.Arg1628Pro increased the risk for PD, although non‐Asian carriers were not identified in previous studies, indicating an important Asian genetic specificity. Later, Wu showed that this variant was also associated with PD in Chinese patients (Tan et al., 2010), although a subsequent study by Deng et al. (Yuan et al., 2016) failed to find statistically significant differences in either genotypic or allelic frequency of p.Arg1628Pro between patients and controls in a Chinese population; our results corroborated Wu's findings. The discrepancies in the association between genetic variants and the presence of PD in the same or different populations may explain, to a large extent, the inconsistency in the results of these studies. Overall, our study further confirmed the association between LRRK2 and idiopathic PD in the Chinese population.
SNCA, which was the first recognized AD‐PD gene (Golbe, Di Iorio, Bonavita, Miller, & Duvoisin, 1990), encodes α‐synuclein, the primary component of LB (Goedert, 2001). Although we did not find any known pathogenic mutations in SNCA in this study, we identified one novel variant, p.Val15Asp, and one unclear variant, p.Met127Ile, located between two phosphorylation sites with high conservation across variable species. None of the controls carried p.Met127Ile, suggesting that this variant is associated with susceptibility to PD.
In CHCHD2, we discovered one unknown variant, p.Pro2Leu, and one pathogenic variant, p.Thr61Ile, previously reported by Funayama et al. (2015) in two unrelated Japanese families segregated with disease. Although almost all the subsequent studies in the Chinese population suggested that CHCHD2 mutations are not a common cause of PD in Chinese familial or sporadic cases (Gao et al., 2017; Liu et al., 2015; Shi et al., 2016), our findings provide new evidence of the role of this gene in susceptibility to sporadic PD in China. Nevertheless, this association needs further confirmation in additional series.
In our study, we found a novel variant, DNAJC13 p.Leu583Ser, carried by one patient and an unclear variant, DNAJC13 p.Arg1588His, carried by two patients. Although these two variants are not located in the functional regions of DNAJC13, neither were carried by controls, suggesting these variants may contribute to the risk of disease by regulating gene function. Vilarino‐Guell et al.9, 2014) identified a heterozygous missense variant of DNAJC13 not only in both familial and sporadic PD patients, but also asymptomatic carriers. In addition, Tan et al. (Foo et al., 2014) found that coding variants of DNAJC13 were extremely rare and present in healthy controls without enrichment in PD cases in a Chinese population. Since the data in the Chinese population are limited, the contribution of DNAJC13 variants to the risk of PD remains plausible. However, considering the rare frequency of DNAJC13 variants and the inconsistency of previous reports, it can be speculated that the contribution of this gene to sporadic PD is very limited in the Chinese population.
Despite adopting a three‐level filtration of candidate variants designed to identify the most pathogenic sites, the novel variant EIF4G1 p.Arg1139His identified in one of the patients was also present in two of the control individuals; therefore, the importance of this variant should be considered with caution. This paradoxical result may be due to the limited sample size and the low frequency of EIF4G1 in Asian populations (Zhao et al., 2013). It is also interesting to note that loss‐of‐function variants of known pathogenic AD‐PD genes, such as LRRK2 and SNCA, were also identified in healthy controls (Hernandez, Reed, & Singleton, 2016). The most likely explanation for this phenomenon is that these variants cause disease when inherited in the form of compound heterozygotes or homozygotes or with risk variants of other genes, which will confound risk prediction.
We discovered two NR4A2 variants (p.Val539Met and p.Val582Met) with unknown pathogenicity. NR4A2 has been widely studied in the Chinese population. Xu et al. (2002) and Zheng, Heydari, and Simon (2003) reported associations between homozygosity or heterozygosity for a variant in the intron of this gene and PD. Many follow‐up studies (Le et al., 2003; Liu et al., 2013; Tan et al., 2004) revealed the presence of NR4A2 variants in both familial and sporadic cases of PD among various populations. Our study provided more evidence for the association between rare variants of NR4A2 and sporadic PD in Chinese population. Other selected genes, GIGYF2 and HTRA2, were found to have one or two unclear variants carried by only PD patients but not by controls. Since no rare variants were found in RIC3, TMEM230, UCHL1, and VPS35 in our cohort, further investigations are required to clarify the role of those genes in susceptibility to PD in the Chinese population.
Some limitations of our study should be noted. Due to the moderate sample size, we detected only a small number of rare variants in these genes. Therefore, the value of individual single‐nucleotide polymorphisms (SNPs) in predicting risks is limited and combination with multiple low‐penetrance SNPs may increase the predictive power. Although we performed gene‐based analysis in candidate variants, some genes present in a limited number of carriers do not meet the requirements for statistical analysis and studies with larger samples are needed to clarify our findings.
5. CONCLUSIONS
In our study, 18 rare non‐synonymous coding variants were validated in 18.8% (36/191) of index patients. Among them, most of the variants were found in LRRK2, indicating the strong impact of LRRK2 on sporadic PD risk in the Chinese population. Unclear rare variants in DNAJC13, GIGYF2, and HTRA2 may also confer susceptibility to PD risk since none of the controls were affected. In summary, our findings validate the etiological relationship between rare variants of AD‐PD genes and sporadic PD cases in an eastern Chinese population. These results must be interpreted with caution considering our moderate sample size and insufficient power, and further studies in larger samples are required to clarify this relationship.
CONFLICT OF INTEREST
The authors disclose no conflict of interest regarding this manuscript.
AUTHOR CONTRIBUTIONS
Project conception and organization: RZ, JLP, and BRZ. Acquisition of data and statistical analysis: RZ, CYJ, YC, YR, TG, ZHL, and JXD. First draft writing: RZ and JLP. Manuscript review and critique: RZ, CYJ, JLP, and BRZ. Approval of article and agreement for submission: RZ, JLP, and BRZ. All the co‐authors listed above gave their final approval of this manuscript version.
Supporting information
Table S1‐S2
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China [grant numbers 81520108010 and 81771216] and the Key Research and Development Program of Zhejiang Province [grant number 2020C03020].
Zheng R, Jin C‐Y, Chen Y, et al. Analysis of rare variants of autosomal‐dominant genes in a Chinese population with sporadic Parkinson’s disease. Mol Genet Genomic Med. 2020;8:e1449 10.1002/mgg3.1449
Contributor Information
Jia‐Li Pu, Email: jialipu@zju.edu.cn.
Bao‐Rong Zhang, Email: brzhang@zju.edu.cn.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves, G. , Forsaa, E. B. , Pedersen, K. F. , Dreetz Gjerstad, M. , & Larsen, J. P. (2008). Epidemiology of Parkinson's disease. Journal of Neurology, 255(Suppl 5), 18–32. 10.1007/s00415-008-5004-3 [DOI] [PubMed] [Google Scholar]
- Benitez, B. A. , Davis, A. A. , Jin, S. C. , Ibanez, L. , Ortega‐Cubero, S. , Pastor, P. , … Cruchaga, C. (2016). Resequencing analysis of five Mendelian genes and the top genes from genome‐wide association studies in Parkinson's Disease. Molecular Neurodegeneration, 11, 29 10.1186/s13024-016-0097-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, D. , Schweitzer, K. J. , Leitner, P. , Zimprich, A. , Lichtner, P. , Belcredi, P. , … Gasser, T. (2005). Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease*. Brain, 128(Pt 12), 3000–3011. 10.1093/brain/awh666 [DOI] [PubMed] [Google Scholar]
- Blauwendraat, C. , Nalls, M. A. , & Singleton, A. B. (2019). The genetic architecture of Parkinson's disease. The Lancet Neurology, 19(2), 170–178. 10.1016/s1474-4422(19)30287-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo, A. , Rohé, C. F. , Ferreira, J. , Chien, H. F. , Vacca, L. , Stocchi, F. , … Bonifati, V. (2005). A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. The Lancet, 365(9457), 412–415. 10.1016/s0140-6736(05)17829-5 [DOI] [PubMed] [Google Scholar]
- Dorsey, E. R. , & Bloem, B. R. (2018). The Parkinson pandemic‐A call to action. JAMA Neurology, 75(1), 9–10. 10.1001/jamaneurol.2017.3299 [DOI] [PubMed] [Google Scholar]
- Ferreira, J. J. , Guedes, L. C. , Rosa, M. M. , Coelho, M. , van Doeselaar, M. , Schweiger, D. , … Bonifati, V. (2007). High prevalence of LRRK2 mutations in familial and sporadic Parkinson's disease in Portugal. Movement Disorders, 22(8), 1194–1201. 10.1002/mds.21525 [DOI] [PubMed] [Google Scholar]
- Foo, J. N. , Liany, H. , Tan, L. C. , Au, W. L. , Prakash, K. M. , Liu, J. , & Tan, E. K. (2014). DNAJ mutations are rare in Chinese Parkinson's disease patients and controls. Neurobiology of Aging, 35(4), 935.e1–935.e2. 10.1016/j.neurobiolaging.2013.09.018 [DOI] [PubMed] [Google Scholar]
- Fu, W. , O’Connor, T. D. , Jun, G. , Kang, H. M. , Abecasis, G. , Leal, S. M. , … Akey, J. M. (2013). Analysis of 6,515 exomes reveals the recent origin of most human protein‐coding variants. Nature, 493(7431), 216–220. 10.1038/nature11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama, M. , Ohe, K. , Amo, T. , Furuya, N. , Yamaguchi, J. , Saiki, S. , … Hattori, N. (2015). CHCHD2 mutations in autosomal dominant late‐onset Parkinson's disease: A genome‐wide linkage and sequencing study. The Lancet Neurology, 14(3), 274–282. 10.1016/s1474-4422(14)70266-2 [DOI] [PubMed] [Google Scholar]
- Gao, C. , Chen, Y.‐M. , Sun, Q. , He, Y.‐C. , Huang, P. , Wang, T. , … Chen, S.‐D. (2017). Mutation analysis of CHCHD2 gene in Chinese Han familial essential tremor patients and familial Parkinson's disease patients. Neurobiology of Aging, 49, 218.e9–218.e11. 10.1016/j.neurobiolaging.2016.10.001 [DOI] [PubMed] [Google Scholar]
- Genomes Project, C. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Garrison, E. P. , Kang, H. M. , … Abecasis, G. R. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, G. (2012). Rare and common variants: Twenty arguments. Nature Reviews Genetics, 13(2), 135–145. 10.1038/nrg3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks, W. P. , Abou‐Sleiman, P. M. , Gandhi, S. , Jain, S. , Singleton, A. , Lees, A. J. , … Wood, N. W. (2005). A common LRRK2 mutation in idiopathic Parkinson's disease. The Lancet, 365(9457), 415–416. 10.1016/s0140-6736(05)17830-1 [DOI] [PubMed] [Google Scholar]
- Gilsbach, B. K. , & Kortholt, A. (2014). Structural biology of the LRRK2 GTPase and kinase domains: Implications for regulation. Frontiers in Molecular Neuroscience, 7, 32 10.3389/fnmol.2014.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert, M. (2001). Alpha‐synuclein and neurodegenerative diseases. Nature Reviews Neuroscience, 2(7), 492–501. 10.1038/35081564 [DOI] [PubMed] [Google Scholar]
- Golbe, L. I. , Di Iorio, G. , Bonavita, V. , Miller, D. C. , & Duvoisin, R. C. (1990). A large kindred with autosomal dominant Parkinson's disease. Annals of Neurology, 27(3), 276–282. 10.1002/ana.410270309 [DOI] [PubMed] [Google Scholar]
- Hernandez, D. G. , Reed, X. , & Singleton, A. B. (2016). Genetics in Parkinson disease: Mendelian versus non‐Mendelian inheritance. Journal of Neurochemistry, 139(Suppl 1), 59–74. 10.1111/jnc.13593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulihan, M. M. , Ishihara‐Paul, L. , Kachergus, J. , Warren, L. , Amouri, R. , Elango, R. , … Farrer, M. J. (2008). LRRK2 Gly2019Ser penetrance in Arab‐Berber patients from Tunisia: A case‐control genetic study. The Lancet Neurology, 7(7), 591–594. 10.1016/s1474-4422(08)70116-9 [DOI] [PubMed] [Google Scholar]
- Kelley, L. A. , Mezulis, S. , Yates, C. M. , Wass, M. N. , & Sternberg, M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10(6), 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar, T. , Lepamets, M. , Kaplinski, L. , Raime, K. , Andreson, R. , & Remm, M. (2018). Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics, 34(11), 1937–1938. 10.1093/bioinformatics/bty036 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , & Kattman, B. L. (2018). ClinVar at five years: Delivering on the promise. Human Mutation, 39(11), 1623–1630. 10.1002/humu.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, W. D. , Xu, P. , Jankovic, J. , Jiang, H. , Appel, S. H. , Smith, R. G. , & Vassilatis, D. K. (2003). Mutations in NR4A2 associated with familial Parkinson disease. Nature Genetics, 33(1), 85–89. 10.1038/ng1066 [DOI] [PubMed] [Google Scholar]
- Lee, S. , Abecasis, G. R. , Boehnke, M. , & Lin, X. (2014). Rare‐variant association analysis: Study designs and statistical tests. American Journal of Human Genetics, 95(1), 5–23. 10.1016/j.ajhg.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage, S. , & Brice, A. (2012). Role of Mendelian genes in “sporadic” Parkinson's disease. Parkinsonism & Related Disorders, 18, S66–S70. 10.1016/s1353-8020(11)70022-0 [DOI] [PubMed] [Google Scholar]
- Li, B. , & Leal, S. M. (2008). Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. American Journal of Human Genetics, 83(3), 311–321. 10.1016/j.ajhg.2008.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27(21), 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Tang, B.‐S. , Liu, Z.‐H. , Kang, J.‐F. , Zhang, Y. , Shen, L. U. , … Guo, J.‐F. (2015). LRRK2 A419V variant is a risk factor for Parkinson's disease in Asian population. Neurobiology of Aging, 36(10), 2908.e11–2908.e15. 10.1016/j.neurobiolaging.2015.07.012 [DOI] [PubMed] [Google Scholar]
- Liu, H. , Tao, Q. , Deng, H. , Ming, M. , Ding, Y. , Xu, P. , … Le, W. (2013). Genetic analysis of NR4A2 gene in a large population of Han Chinese patients with Parkinson's disease. European Journal of Neurology, 20(3), 584–587. 10.1111/j.1468-1331.2012.03824.x [DOI] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E. (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Guo, J. , Li, K. , Qin, L. , Kang, J. , Shu, L. I. , … Tang, B. (2015). Mutation analysis of CHCHD2 gene in Chinese familial Parkinson's disease. Neurobiology of Aging, 36(11), 3117.e7–3117.e8. 10.1016/j.neurobiolaging.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Lunati, A. , Lesage, S. , & Brice, A. (2018). The genetic landscape of Parkinson's disease. Revue Neurologique, 174(9), 628–643. 10.1016/j.neurol.2018.08.004 [DOI] [PubMed] [Google Scholar]
- Mata, I. F. , Kachergus, J. M. , Taylor, J. P. , Lincoln, S. , Aasly, J. , Lynch, T. , … Farrer, M. J. (2005). Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics, 6(4), 171–177. 10.1007/s10048-005-0005-1 [DOI] [PubMed] [Google Scholar]
- Postuma, R. B. , Berg, D. , Stern, M. , Poewe, W. , Olanow, C. W. , Oertel, W. , … Deuschl, G. (2015). MDS clinical diagnostic criteria for Parkinson's disease. Movement Disorders, 30(12), 1591–1601. 10.1002/mds.26424 [DOI] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, O. A. , Soto‐Ortolaza, A. I. , Heckman, M. G. , Aasly, J. O. , Abahuni, N. , Annesi, G. , … Farrer, M. J. (2011). Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: A case–control study. The Lancet Neurology, 10(10), 898–908. 10.1016/s1474-4422(11)70175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, O. A. , Wu, Y.‐R. , Lee, M.‐C. , Funayama, M. , Chen, M.‐L. , Soto, A. I. , … Wu, R.‐M. (2008). Analysis of Lrrk2 R1628P as a risk factor for Parkinson's disease. Annals of Neurology, 64(1), 88–92. 10.1002/ana.21405 [DOI] [PubMed] [Google Scholar]
- Shi, C.‐H. , Mao, C.‐Y. , Zhang, S.‐Y. , Yang, J. , Song, B. O. , Wu, P. , … Xu, Y.‐M. (2016). CHCHD2 gene mutations in familial and sporadic Parkinson's disease. Neurobiology of Aging, 38, 217 217.e9–217.e13. 10.1016/j.neurobiolaging.2015.10.040 [DOI] [PubMed] [Google Scholar]
- Simón‐Sánchez, J. , Schulte, C. , Bras, J. M. , Sharma, M. , Gibbs, J. R. , Berg, D. , … Gasser, T. (2009). Genome‐wide association study reveals genetic risk underlying Parkinson's disease. Nature Genetics, 41(12), 1308–1312. 10.1038/ng.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanaki, C. , Latsoudis, H. , & Plaitakis, A. (2006). LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology, 67(8), 1518–1519. 10.1212/01.wnl.0000239829.33936.73 [DOI] [PubMed] [Google Scholar]
- Spataro, N. , Calafell, F. , Cervera‐Carles, L. , Casals, F. , Pagonabarraga, J. , Pascual‐Sedano, B. , … Bosch, E. (2015). Mendelian genes for Parkinson's disease contribute to the sporadic forms of the disease. Human Molecular Genetics, 24(7), 2023–2034. 10.1093/hmg/ddu616 [DOI] [PubMed] [Google Scholar]
- Tan, E.‐K. , Chung, H. , Chandran, V. R. , Tan, C. , Shen, H. , Yew, K. , … Zhao, Y. I. (2004). Nurr1 mutational screen in Parkinson's disease. Movement Disorders, 19(12), 1503–1505. 10.1002/mds.20246 [DOI] [PubMed] [Google Scholar]
- Tan, E. K. , Peng, R. , Teo, Y. Y. , Tan, L. C. , Angeles, D. , Ho, P. , … Wu, R. M. (2010). Multiple LRRK2 variants modulate risk of Parkinson disease: A Chinese multicenter study. Human Mutation, 31(5), 561–568. 10.1002/humu.21225 [DOI] [PubMed] [Google Scholar]
- Tan, M. M. X. , Malek, N. , Lawton, M. A. , Hubbard, L. , Pittman, A. M. , Joseph, T. , … Morris, H. R. (2019). Genetic analysis of Mendelian mutations in a large UK population‐based Parkinson's disease study. Brain, 142(9), 2828–2844. 10.1093/brain/awz191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler, A. , Ash, E. , Gan‐Or, Z. , Orr‐Urtreger, A. , & Giladi, N. (2009). The LRRK2 G2019S mutation as the cause of Parkinson's disease in Ashkenazi Jews. Journal of Neural Transmission, 116(11), 1473–1482. 10.1007/s00702-009-0303-0 [DOI] [PubMed] [Google Scholar]
- Tian, J. , Vemula, S. R. , Xiao, J. , Valente, E. M. , Defazio, G. , Petrucci, S. , … LeDoux, M. S. (2018). Whole‐exome sequencing for variant discovery in blepharospasm. Molecular Genetics & Genomic Medicine, 6, 601–626. 10.1002/mgg3.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilariño‐Güell, C. , Rajput, A. , Milnerwood, A. J. , Shah, B. , Szu‐Tu, C. , Trinh, J. , … Farrer, M. J. (2014). DNAJC13 mutations in Parkinson disease. Human Molecular Genetics, 23(7), 1794–1801. 10.1093/hmg/ddt570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P.‐Y. , Liang, R. , Jankovic, J. , Hunter, C. , Zeng, Y.‐X. , Ashizawa, T. , … Le, W.‐D. (2002). Association of homozygous 7048G7049 variant in the intron six of Nurr1 gene with Parkinson's disease. Neurology, 58(6), 881–884. 10.1212/wnl.58.6.881 [DOI] [PubMed] [Google Scholar]
- Yang, H. , & Wang, K. (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nature Protocols, 10(10), 1556–1566. 10.1038/nprot.2015.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Zhao, Y. , Liu, Z. , Zhang, R. , He, Y. , Zhou, Y. , … Tang, B. (2019). Systematically analyzing rare variants of autosomal‐dominant genes for sporadic Parkinson's disease in a Chinese cohort. Neurobiology of Aging, 76, 215.e1–215.e7. 10.1016/j.neurobiolaging.2018.11.012 [DOI] [PubMed] [Google Scholar]
- Yuan, L. , Song, Z. , Deng, X. , Zheng, W. , Guo, Y. , Yang, Z. , & Deng, H. (2016). Systematic analysis of genetic variants in Han Chinese patients with sporadic Parkinson's disease. Scientific Reports, 6, 33850 10.1038/srep33850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabetian, C. P. , Samii, A. , Mosley, A. D. , Roberts, J. W. , Leis, B. C. , Yearout, D. , … Griffith, A. (2005). A clinic‐based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology, 65(5), 741–744. 10.1212/01.wnl.0000172630.22804.73 [DOI] [PubMed] [Google Scholar]
- Zhao, Y. I. , Ho, P. , Prakash, K.‐M. , Foo, J.‐N. , Liu, J.‐J. , Au, W.‐L. , … Tan, E.‐K. (2013). Analysis of EIF4G1 in Parkinson's disease among Asians. Neurobiology of Aging, 34(4), 1311 e1315–1316 10.1016/j.neurobiolaging.2012.09.003 [DOI] [PubMed] [Google Scholar]
- Zheng, K. , Heydari, B. , & Simon, D. K. (2003). A common NURR1 polymorphism associated with Parkinson disease and diffuse Lewy body disease. Archives of Neurology, 60(5), 722–725. 10.1001/archneur.60.5.722 [DOI] [PubMed] [Google Scholar]
- Zimprich, A. , Biskup, S. , Leitner, P. , Lichtner, P. , Farrer, M. , Lincoln, S. , … Gasser, T. (2004). Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron, 44(4), 601–607. 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2