

Abstract
Background
The Oculo‐Auriculo‐Vertebral Spectrum (OAVS) or Goldenhar Syndrome is an embryonic developmental disorder characterized by hemifacial microsomia associated with auricular, ocular and vertebral malformations. The clinical heterogeneity of this spectrum and its incomplete penetrance limited the molecular diagnosis. In this study, we describe a novel causative gene, ZYG11B.
Methods
A sporadic case of OAVS was analyzed by whole exome sequencing in trio strategy. The identified candidate gene, ZYG11B, was screened in 143 patients by next generation sequencing. Overexpression and immunofluorescence of wild‐type and mutated ZYG11B forms were performed in Hela cells. Moreover, morpholinos were used for transient knockdown of its homologue in zebrafish embryo.
Results
A nonsense de novo heterozygous variant in ZYG11B, (NM_024646, c.1609G>T, p.Glu537*) was identified in a single OAVS patient. This variant leads in vitro to a truncated protein whose subcellular localization is altered. Transient knockdown of the zebrafish homologue gene confirmed its role in craniofacial cartilages architecture and in notochord development. Moreover, ZYG11B expression regulates a cartilage master regulator, SOX6, and is regulated by Retinoic Acid, a known developmental toxic molecule leading to clinical features of OAVS.
Conclusion
Based on genetic, cellular and animal model data, we proposed ZYG11B as a novel rare causative gene for OAVS.
Keywords: craniofacial anomalies, etiology, genetics, Goldenhar, hemifacial microsomia, OAVS, ubiquitine ligase, wavy notochord, ZYG11B
The Oculo‐Auriculo‐Vertebral Spectrum or Goldenhar Syndrome is an embryonic developmental disorder characterized by hemifacial microsomia associated with auricular, ocular and vertebral malformations. Its etiology is poorly characterized. In this study, we describe a novel causative gene, ZYG11B.

1. INTRODUCTION
The Oculo‐Auriculo‐Vertebral Spectrum (OAVS) or Goldenhar Syndrome (GS) or Hemifacial Microsomia [MIM: 164210] is a rare developmental disease whose incidence is around 1/26 500 (Barisic et al., 2014). It is the second most frequent embryonic malformative spectrum of head and neck. The OAVS includes various malformations involving structures derived from the first and second branchial arches, mainly ears, eyes, mandible and vertebrae. The clinical phenotype is highly heterogeneous and characterized by hemifacial microsomia and/or asymmetric ear anomalies (microtia, preauricular tags, external auditory canal atresia, conductive and/or sensorineural hearing loss…) and/or ocular defects (epibulbar dermoids, upper eyelid coloboma, microphtalmia…), and/or vertebral malformations (fused cervical vertebrae, vertebral puzzle…). Other features are cleft lip and/or palate, cardiac, renal or cerebral malformations, and rarely mental deficiency (Gorlin, Cohen, & Hennekam, 2001). OAVS was first described in 1952 by Maurice Goldenhar (Goldenhar, 1952) and the clinical spectrum of this disorder was then expanded (Gorlin et al., 2001; Gorlin, Jue, Jacobsen, & Goldschmidt, 1963).
Nongenetic causes are hypothesized for this spectrum, including maternal diabetes (Wang, Martínez‐Frías, & Graham, 2002), placental vascular disruption (Poswillo, 1975) and exposure to toxic substances during pregnancy such as retinoic acid (RA). Indeed, RA human embryopathies present features overlapping with OAVS phenotypes (Coberly, Lammer, & A. M., 1996; Glineur et al., 1999; Lammer et al., 1985; Mondal, Shenoy, & Mishra, 2017). The genetic origin of OAVS is supported by the description of familial cases (Stoll, Viville, Treisser, & Gasser, 1998; Vendramini‐Pittoli & Kokitsu‐Nakata, 2009), and the identification of diverse chromosomal aneuploidies and few recurrent CNVs (Copy Number Variants) such as deletion 22q11.2 (Derbent et al., 2003; Dos Santos et al., 2014; Xu, Fan, & Siu, 2008) and duplication 4p16.1 (Barber, 2018; Beleza‐Meireles et al., 2015; Bragagnolo et al., 2018) in OAVS patients. Moreover, the identification of MYT1 as the first causative gene in OAVS confirms the genetic etiology (Berenguer et al., 2017; Lopez et al., 2016). Of note, recently in AMIGO2, one de novo nonsense variant was found associated with OAVS. These previously described cases are in favor of a dominant autosomal transmission with incomplete penetrance.
Identification of new genes associated with OAVS is a crucial step in understanding the physiopathology of the disease. In this aim, we performed exome sequencing in other OAVS patients and identified a novel causative gene named ZYG11B (zyg‐11 family member B, cell cycle regulator).
2. METHODS
2.1. Patient
The proband is a male patient. He is the first child of an unrelated couple without medical history. He was born at term after an uneventful pregnancy. No psychomotor delay was observed along his development. He presented with right hemifacial microsomia involving mandibular hypoplasia. Auricular anomalies included left microtia (grade 3), bilateral preauricular tags and bilateral dysplasia with polyotia. Audiometric tests were normal. Ocular anomalies were left epibulbar dermoid and right eyelid coloboma (Figure 1a–c). Vertebral fusion of hypoplastic C7‐D1 vertebrae was observed (Figure 1d,e). In addition, costal agenesis, right nostril hypoplasia with pit and tag, bilateral cheek pits, high arched palate and 2–3 toe syndactylies were noted. Renal ultrasonography and echocardiography were normal. Neither transfrontal echography nor magnetic resonance imaging of the brain showed cerebral anomalies.
Figure 1.
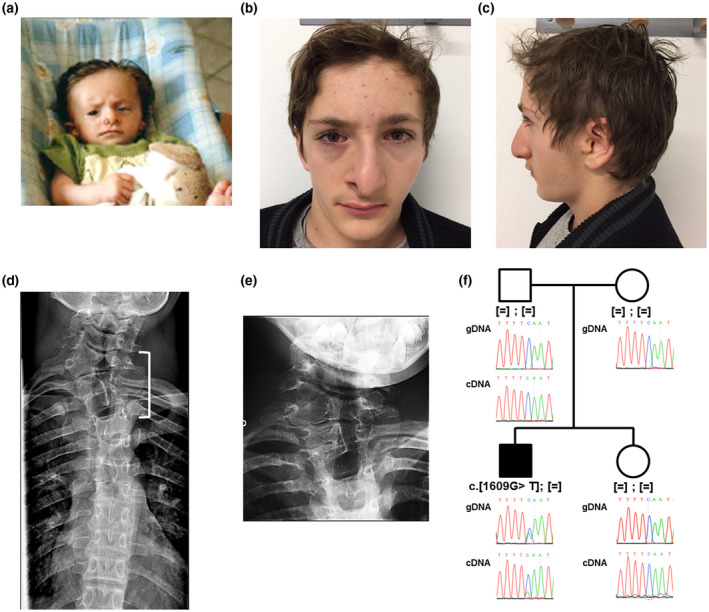
Photographs and pedigree of the patient carrying the c.1609G>T, p.(Gly537*) variant in ZYG11B. (a–c) Photographs showed the proband at infant stage (a) and at 15 years (b and c). (d) X‐ray imaging of patient vertebrae showing vertebral fusion of hypoplastic C7‐D1 vertebrae (white bracket). (e) Enlarged of C7‐D1 vertebrae. (f) Family tree and electrophoregrams showing the de novo heterozygous nonsense variant in the proband. Sequencing of RT‐PCR amplicons revealed that the transcript escapes to nonsense‐mediated RNA decay
Exome sequencing. Written informed consent for genetic studies was obtained prior to collecting blood samples for DNA extraction. Moreover, consent was obtained from the patient's parent for the publication of identifying images in an online open‐access publication. The local ethics committee (Comité de Protection des Personnes: DC2012/76) approved this study. Library preparation, exome capture, sequencing and data analysis were performed by IntegraGen SA, using SureSelect Human All Exon kit V2 in‐solution enrichment methodology (Agilent), followed by paired‐end 75 bases massively parallel sequencing on Illumina HiSeq2000 (Illumina). Variants were filtered using Eris software (IntegraGen), with a minimal read depth of 10x. Only rare variants (allele frequency <0.1% in public databases GnomAD, Exac and EVS), with a potential effect on proteins (nonsynonym, and synonym or intronic variants within 5 bp of a splice site), and segregating with a possible Mendelian inheritance in the trio were kept for analysis.
Cohort screening for ZYG11B mutations (ZYG11B Ref Seq: NM_024646/NP_078922). We performed screening for other nucleotide variants of ZYG11B in 143 OAVS patients. Exons, exon–intron boundaries, 5′UTR and 3′‐UTR (transcript ENST00000294353.6, http://www.ensembl.org/) were amplified using a standard protocol. Seventeen primer pairs were designed (under request). PCR fragments were sequenced by NGS (GS Junior, Roche or MiSeq, Illumina) and Sanger sequencing. The online tool SureDesign provided by Agilent was used for RNA probes design targeting ZYG11B exons, exon–intron junctions, 5′‐UTR and 3′‐UTR (ENSG00000162378, http://www.ensembl.org). After library preparation, the SureSelect QXT kit (Agilent) was used for the capture step following the manufacturer's instructions. Sequencing was performed on MiSeq sequencer from Illumina using the MiSeq Reagent Kit v3 (Agilent) for paired‐end sequencing. Alignment and variant calling was processed with the MiSeq Reporter Software (Illumina).
2.2. Expression plasmid
All primers used for cloning experiments are listed in Table S1. The construct including ZYG11B wild‐type cDNA in pCS2+ vector was named ZYG11B‐WT and the one carrying the variant c.1609G>T was named ZYG11B‐E537*.
2.3. Cell culture
For All‐Trans RA (ATRA, Sigma Aldrich) experiments, cells were treated for 48 hr with 0.1 mM of ATRA. For siRNA experiments, cells were transfected with 20nM of control nonspecific siRNA (EHUEGFP, Sigma) or with specific siRNA targeting ZYG11B (EHU022091, Sigma) using DharmaFECT™ Transfection Reagent following manufacturer's instructions (Dharmacon Horizon Discovery). Cell proliferation was assessed by Crystal Violet staining.
2.4. Quantitative real‐time PCR
Total RNA extraction and reverse‐transcription were processed as previously described (Lopez et al., 2016). Primers used are listed in Table S1. The relative transcript level was calculated as fold change using the 2−ΔΔCt method.
2.5. Immunodetection
Immunocytochemistry and Western blot were performed with primary antibody targeting ZYG11B (HPA028156, SIGMA, diluted 1/300).
2.6. Zebrafish zyg11 knockdown
Zebrafish were produced in our facilities, in accordance with the French Directive (Ministère de l'Agriculture) and in conformity with the European Communities Council Directive (2010/63/EU). Morpholino oligonucleotides (MOs) were designed as complementary to the sequence flanking the translation initiation codon of zyg11 (MO‐ATG: 5′‐TTGAGAAAGATGGATCATAACGGCC‐3′). At 5 dpf, embryos were stained using Alcian blue/Alizarin red stain according to Walker and Kimmel (https://wiki.zfin.org).
3. RESULTS
WES was performed in trio strategy, identifying a heterozygous nonsense de novo variant (chr.1 g.52801942G>T, c.1609G>T, p.(Gly537*)) in ZYG11B not reported in any database. A total of 30 reads (16+, 14−) out of 59 identified the variant which was confirmed by Sanger sequencing (Figure 1f). The pLI factor of ZYG11B is 1 according to GnomAD database, with an observed/expected loss‐of‐function variants score of 0.11 (90% CI at 0.05–0.26), in favor of an essential biological function. After exome filtering, six additional variants were considered: due to their segregation, frequencies in GnomAD (>1%), genes’ pLI, and genes function, all these variants were discarded. Nonsense RNA decay was excluded as patient's leucocytes expressed mutated transcripts (Figure 1f). ZYG11B was screened in 143 OAVS patients, without identifying any other pathogenic variant (Supplementary methods).
Although ZYG11B transcripts were detected in HeLa cells, Western blot analysis was not sensitive enough to detect endogenous ZYG11B protein. However, overexpression of ZYG11B‐WT or ZYG11B‐E537* was strongly detected in transfected cells. As expected, the nonsense variation produced a truncated protein of lower molecular weight, ≈50kDa instead of ≈75kDa (Figure 2a). Immunodetection of overexpressed ZYG11B‐WT revealed a vesicular, concentric and perinuclear signal whereas ZYG11B‐E537* presented also a vesicular signal but diffuse in all the cytoplasm (Figure 2b).
Figure 2.
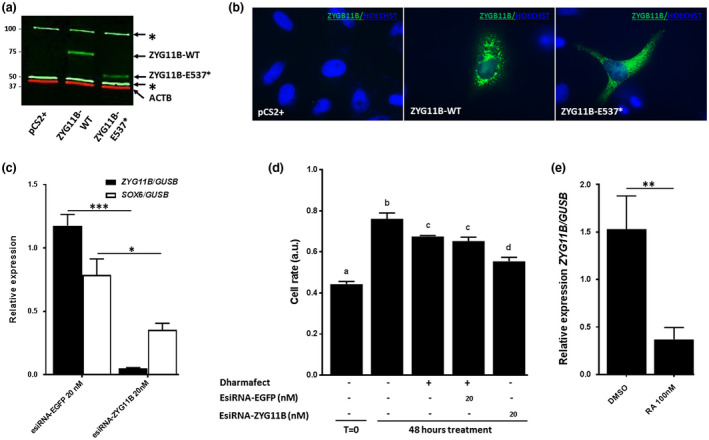
The nonsense variant encodes a mislocalized truncated ZYG11B protein. (a) Western blot validated overexpression experiments. (b) Immunocytochemistry detection of ZYG11B‐WT and ZYG11B‐E537* in HeLa cells. (c) RT‐qPCR experiments validated ZYG11B expression knockdown by the specific siRNA‐ZYG11B which also induced a significant down‐regulation of SOX6 expression, t test p < .05. (d) Cell proliferation is significantly decreased when treated with siRNA‐ZYG11B (Kruskal–Wallis test followed by Dunns’ post hoc multiple comparison test (p < .05)). (d) RT‐qPCR analysis of ZYG11B expression showed a down‐regulation following RA treatment in HeLa cells (t test p < .05)
Based on haplo‐insufficiency hypothesis, inhibition of ZYG11B expression was tested in cell and animal models. We then performed siRNA experiments targeting specifically endogenous ZYG11B transcripts as demonstrated by RT‐qPCR experiments (Figure 2c). We investigated if the regulation of ZYG11B expression would alter SOX6 expression, as it is a master regulator involved in cartilage development. Indeed, a strong down‐regulation of SOX6 gene expression was observed (Figure 2c). Choosing cell proliferation as an endpoint to highlight potential cell cycle deregulation, cell rate was measured at t = 0, just before transfection, and after 48 hr of transfection. A significant lower proliferation rate was observed with the specific siRNA versus control siRNA (Figure 2d).
T‐qPCR experiments demonstrated that ZYG11B endogenous expression in HeLa cells can be down‐regulated by RA treatment for 48 hr (Figure 2e). However, MYT1 overexpression for 24 hr did not affect ZYG11B endogenous expression (Figure S1).
We then investigated the developmental role of ZYG11B in zebrafish model. Two ZYG11B homologues were identified, zyg11 and si:ch1073‐82l19.1 consistent with the known whole genome duplication in teleost (Figure 3a). Developmental expression study showed that zyg11 was significantly expressed from early stages and throughout zebrafish embryogenesis whereas si:ch1073‐82l19.1 was not (Figure 3b). Transient knockdown was then only performed for zyg11 with MO‐ATG at 2 ng per embryo. In order to assess the involvement of zyg11 in cartilage development, embryos were stained with Alcian Blue at 5dpf. Of note, preliminary experiments to discard nonspecific effects due to activation of p53‐dependent apoptosis were performed through coinjection of MO specifically targeting p53 transcripts (data not shown). At this stage, four phenotypes were defined (Figure 3c): (a) phenotype 0 was defined as wild‐type embryo; (b) phenotype 1 was defined as embryo presenting altered architecture of craniofacial cartilages and microphtalmia; (c) phenotype 2 was defined as phenotype 1 plus a wavy notochord in the anterior part; (d) phenotype 3 was defined as globally strongly altered phenotype with major curved tail probably corresponding to nonspecific phenotype. Contrary to control groups, in the group injected with MO 2 ng/embryo, 25% of embryos presented phenotype 1 and 25% phenotype 2. A very small number of embryos presented with phenotype 3. These results confirmed a specific role of zyg11 in head development and specifically craniofacial cartilages and eye development. In phenotypes 2 and 3, craniofacial cartilage alterations include flattening of ceratohyal cartilage, abnormal distance between the ceratohyal and meckel's cartilages, abnormal angulation between the ceratohyal and palatoquadrate cartilages and abnormal angulation between the two ceratohyal cartilages in phenotype 3, craniofacial cartilages were almost absent (Figure 3c). Interestingly, those animals with phenotype 3 from injected group at MO 2 ng/embryo also presented partial wavy notochord as for phenotype 2 embryos (Figure 3c). Significant higher rates of phenotypes 2 and 3 were observed on MO‐AUG injected group.
Figure 3.
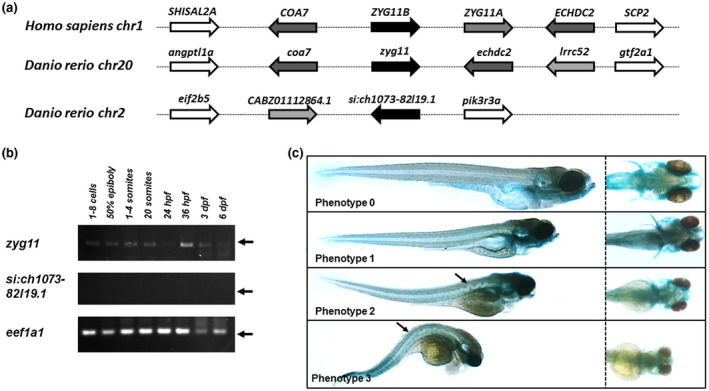
Zebrafish zyg11 gene knockdown impaired craniofacial cartilages and notochord development. (a) Conserved synteny between human ZYG11B genomic environment and zebrafish homologues. (b) expression pattern of zyg11 and si:ch1073‐82l19.1 genes in zebrafish by RT‐PCR. (c) At 120 hpf, embryos were classified in 4 phenotypes
We intended to perform rescue experiments in zebrafish model with human ZYG11B‐WT and ZYG11B‐E537* forms by coinjecting their cRNA and MO‐ATG targeting zyg11. Unfortunately, recovery of the MO‐induced phenotype by wild‐type form was not significantly observed. It could be explained by evolutionary differences. Indeed, in human, tandem duplication arose leading to ZYG11A and ZYG11B whereas in zebrafish genome zyg11 and si:ch1073‐82l19.1 arose from the whole genome duplication described in teleosts (Figure 3a). So, two independent events are noticed in Human and zebrafish genomes probably diluting the heterologous rescue capacity.
4. DISCUSSION
ZYG11B protein is characterized as a member of an E3 ubiquitin ligase complex and is involved in substrate recognition for subsequent proteasome degradation. It is a multi‐domain protein including a Von Hippel–Lindau‐box, three leucine‐rich repeat motifs and an armadillo helical domain (ARM). ZYG11B is demonstrated to be associated with Cullin‐2, an E3 ubiquitin ligase, in several models as C. elegans or animal and human cell lines (Balachandran et al., 2016; Liu, Vasudevan, & Kipreos, 2004; Yamaki, Kagawa, Hatta, Natsume, & Kawahara, 2016). In addition to the anaphase‐promoting complex/cyclosome (APC/C), CUL2/ZYG11 complex is necessary for meiosis completion at metaphase to anaphase transition at meiosis II (Liu et al., 2004; Sonneville & Gönczy, 2004). Moreover, ZYG11B through the CUL2‐based complex is also demonstrated to be involved in cell differentiation through the Patched1 receptor (Ptcd1), a twelve transmembrane domain receptor of Shh (Yamaki et al., 2016). Thus far, there is no evidence of ZYG11B involvement in developmental disorders. Identification of a de novo nonsense variant in ZYG11B in the exome of an OAVS patient was then interesting. Unfortunately, we do not find any additional variant in 143 OAVS patients. This finding is not surprising as genetic heterogeneity is highly suspected in OAVS: we only identified few variants in the first OAVS gene MYT1 (Berenguer et al., 2017; Lopez et al., 2016).
The ZYG11B variant produces in vitro a truncated protein whose intracellular localization is altered, as demonstrated by overexpression experiments. Potential pathogenicity of the variant could then be linked to these data. Moreover, knockdown of ZYG11B expression induces down‐regulation of cellular proliferation, maybe linked to cell cycle control, a complementary role of ZYG11A and ZYG11B previously demonstrated (Balachandran et al., 2016).
RA is a toxic environmental agent causing OAVS features (Coberly et al., 1996; Lammer et al., 1985) and as the MYT1‐RA relationship was previously hypothesized (Berenguer et al., 2017; Lopez et al., 2016), investigation of the relationship with ZYG11B was performed. ZYG11B expression appears down‐regulated by cellular RA exposure.
To support the involvement of ZYG11B in craniofacial development, we then produced a zebrafish model of zyg11 knockdown. Interestingly, embryos are strongly impacted at the craniofacial level, based on observation of the well‐described meckel‐palatoquadrate‐basihyal‐ceratohyal‐hyosymplectic architecture. Interestingly, a wavy notochord phenotype is also observed. Wavy notochord has already been observed in genetic or toxic zebrafish models of copper deficiency (Mendelsohn et al., 2006; Raldúa & Babin, 2007). Indeed, in zebrafish atp7a mutant calamity or atp7a knockdown models present with a wavy notochord rescuable by exogenous copper. Moreover, environmental copper chelating agents are identified based on observation of wavy notochord as BLT‐1 (Raldúa & Babin, 2007; Sandoval et al., 2013) or kalihinol F (Sandoval et al., 2013). This could be an additional clue for a synergic role of environmental and genetic factors leading to the same phenotype.
In summary, we describe a de novo nonsense variant in ZYG11B, associated with OAVS phenotype. In vitro studies show that the resulting truncated protein presents a subcellular mislocalization. Moreover, ZYG11B belongs to RA regulated transcriptome, such as the first OAVS gene MYT1 (Lopez et al., 2016). Furthermore, in zebrafish model, zyg11 is involved in craniofacial cartilage and notochord development. Taken together, and considering the molecular heterogeneity described in this spectrum, these findings support ZYG11B as a new causative gene for OAVS.
The only reported genes associated with OAVS are MYT1 (Berenguer et al., 2017; Lopez et al., 2016), AMIGO2 (Rengasamy Venugopalan et al., 2019) and ZYG11B (in the present study). All these genes are poorly characterized and their developmental functions are not fully understood. Moreover, their known functions are different. Molecular hypotheses for OAVS physiopathology are still difficult to highlight. More data and candidate genes are needed to isolate a common biological pathway involved in OAVS.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
AT‐S, AT and CR conceived and designed the experiments. AT‐S, EL, MB, SG, and MB performed the experiments. AT‐S, AT, SM, EL, and CR analyzed the data. AT‐S, AT, CR wrote the paper. SM, BA, DL, and CR edited, revised and approved the final version of the manuscript.
Supporting information
Table S1‐Fig S1
ACKNOWLEDGEMENTS
The authors warmly thank the clinical geneticists from the FECLAD (Fédération des Centres Labellisés pour les Anomalies du Développement) for providing patients. They thank Mr Marie Yannick from the ICM in La Pitié Salpêtrière Hospital for his technical support in sequencing by GS Junior technology and Integragen society for exome sequencing facilities. They thank the patients and their families.
Tingaud‐Sequeira A, Trimouille A, Marlin S, et al. Functional and genetic analyses of ZYG11B provide evidences for its involvement in OAVS. Mol Genet Genomic Med. 2020;8:e1375 10.1002/mgg3.1375
DATA AVAILABILITY STATEMENT
The identified variant (NM_024646, c.1609G>T) was submitted to ClinVar database (submission identification: SUB6959693). All data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- Balachandran, R. S. , Heighington, C. S. , Starostina, N. G. , Anderson, J. W. , Owen, D. L. , Vasudevan, S. , & Kipreos, E. T. (2016). The ubiquitin ligase CRL2ZYG11targets cyclin B1 for degradation in a conserved pathway that facilitates mitotic slippage. Journal of Cell Biology, 215(2), 151–166. 10.1083/jcb.201601083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber, J. C. K. (2018). Reassignment of HMX1 indicates copy number variation within 4p16.1 may be an alternative cause of oculoauricular phenotypes. American Journal of Medical Genetics Part A, 176(9), 2034–2036. 10.1002/ajmg.a.40385 [DOI] [PubMed] [Google Scholar]
- Barisic, I. , Odak, L. , Loane, M. , Garne, E. , Wellesley, D. , Calzolari, E. , … Tucker, D. (2014). Prevalence, prenatal diagnosis and clinical features of oculo‐auriculo‐vertebral spectrum: A registry‐based study in Europe. European Journal of Human Genetics, 22(8), 1026–1033. 10.1038/ejhg.2013.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beleza‐Meireles, A. , Hart, R. , Clayton‐Smith, J. , Oliveira, R. , Reis, C. F. , Venâncio, M. , … Tassabehji, M. (2015). Oculo‐auriculo‐vertebral spectrum: Clinical and molecular analysis of 51 patients. European Journal of Medical Genetics, 58(9), 455–465. 10.1016/j.ejmg.2015.07.003 [DOI] [PubMed] [Google Scholar]
- Berenguer, M. , Tingaud‐Sequeira, A. , Colovati, M. , Melaragno, M. I. , Bragagnolo, S. , Perez, A. B. A. , … Rooryck, C. (2017). A novel de novo mutation in MYT1, the unique OAVS gene identified so far. European Journal of Human Genetics, 25(9), 1083–1086. 10.1038/ejhg.2017.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragagnolo, S. , Colovati, M. E. S. , Souza, M. Z. , Dantas, A. G. , F de Soares, M. F. , Melaragno, M. I. , & Perez, A. B. (2018). Clinical and cytogenomic findings in OAV spectrum. American Journal of Medical Genetics, Part A, 176(3), 638–648. 10.1002/ajmg.a.38576 [DOI] [PubMed] [Google Scholar]
- Coberly, S. , Lammer, E. , & Alashari, M. (1996). Retinoic acid embryopathy: Case report and review of literature. Pediatric Pathology & Laboratory Medicine, 16(5), 823–836. 10.1080/15513819609169308 [DOI] [PubMed] [Google Scholar]
- Derbent, M. , Y?lmaz, Z. , Baltac?, V. , Sayg?l?, A. , Varan, B. , & Tokel, K. (2003). Chromosome 22q11.2 deletion and phenotypic features in 30 patients with conotruncal heart defects. American Journal of Medical Genetics, 116A(2), 129–135. 10.1002/ajmg.a.10832 [DOI] [PubMed] [Google Scholar]
- Dos Santos, P. A. C. , de Oliveira, S. F. , Freitas, E. L. , Safatle, H. P. N. , Rosenberg, C. , Ferrari, I. , & Mazzeu, J. F. (2014). Non‐overlapping 22q11.2 microdeletions in patients with oculo‐auriculo‐vertebral spectrum. American Journal of Medical Genetics, Part A, 164(2), 551–553. 10.1002/ajmg.a.36231 [DOI] [PubMed] [Google Scholar]
- Glineur, R. , Louryan, S. , Lemaître, A. , Evrard, L. , Rooze, M. , & De Vos, L. (1999). Cranio‐facial dysmorphism: Experimental study in the mouse, clinical applications. Surgical and Radiologic Anatomy, 21(1), 41–47. 10.1007/BF01635051 [DOI] [PubMed] [Google Scholar]
- Goldenhar, M. (1952). Associations malformatives de l’oeil et de l’oreille, en particulier le syndrome dermoïde epibulbaire‐appendices auriculaires‐fistula auris congenita et ses relations avec la dysostose mandibulo‐faciale. Journal De Génétique Humaine, 243–282. [Google Scholar]
- Gorlin, R. J. , Cohen, M. , & Hennekam, R. C. M. (2001). Branchial arch and oro‐acral disorders In Syndromes of the Head and Neck (pp. 790–798). USA: Oxford University Press. [Google Scholar]
- Gorlin, R. J. , Jue, K. L. , Jacobsen, U. , & Goldschmidt, E. (1963). Oculoauriculovertebral dysplasia. The Journal of Pediatrics, 63(5), 991–999. 10.1016/S0022-3476(63)80233-4 [DOI] [PubMed] [Google Scholar]
- Lammer, E. J. , Chen, D. T. , Hoar, R. M. , Agnish, N. D. , Benke, P. J. , Braun, J. T. , … Sun, S. C. (1985). Retinoic acid embryopathy. The New England Journal of Medicine, 313(14), 837–841. 10.1056/NEJM198510033131401 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Vasudevan, S. , & Kipreos, E. T. (2004). CUL‐2 and ZYG‐11 promote meiotic anaphase II and the proper placement of the anterior‐posterior axis in C. elegans. Development, 131(15), 3513–3525. 10.1242/dev.01245 [DOI] [PubMed] [Google Scholar]
- Lopez, E. , Berenguer, M. , Tingaud‐Sequeira, A. , Marlin, S. , Toutain, A. , Denoyelle, F. , … Rooryck, C. (2016). Mutations in MYT1, encoding the myelin transcription factor 1, are a rare cause of OAVS. Journal of Medical Genetics, 53(11), 752–760. 10.1136/jmedgenet-2016-103774 [DOI] [PubMed] [Google Scholar]
- Mendelsohn, B. A. , Yin, C. , Johnson, S. L. , Wilm, T. P. , Solnica‐Krezel, L. , & Gitlin, J. D. (2006). Atp7a determines a hierarchy of copper metabolism essential for notochord development. Cell Metabolism, 4(2), 155–162. 10.1016/j.cmet.2006.05.001 [DOI] [PubMed] [Google Scholar]
- Mondal, D. , Shenoy, R. S. , & Mishra, S. (2017). Retinoic acid embryopathy. International Journal of Applied and Basic Medical Research, 7(4), S72–S77. 10.4103/ijabmr.IJABMR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poswillo, D. (1975). Causal mechanisms of craniofacial deformity. British Medical Bulletin, 31(2), 101–106. 10.1093/oxfordjournals.bmb.a071260 [DOI] [PubMed] [Google Scholar]
- Raldúa, D. , & Babin, P. J. (2007). BLT‐1, a specific inhibitor of the HDL receptor SR‐BI, induces a copper‐dependent phenotype during zebrafish development. Toxicology Letters, 175(1–3), 1–7. 10.1016/j.toxlet.2007.08.007 [DOI] [PubMed] [Google Scholar]
- Rengasamy Venugopalan, S. , Farrow, E. , Sanchez‐Lara, P. A. , Yen, S. , Lypka, M. , Jiang, S. , & Allareddy, V. (2019). A novel nonsense substitution identified in the AMIGO2 gene in an Occulo‐Auriculo‐Vertebral spectrum patient. Orthodontics and Craniofacial Research, 22(S1), 163–167. 10.1111/ocr.12259 [DOI] [PubMed] [Google Scholar]
- Sandoval, I. T. , Manos, E. J. , Van Wagoner, R. M. , Delacruz, R. G. , Edes, K. , Winge, D. R. , … Jones, D. A. (2013). Juxtaposition of chemical and mutation‐ induced developmental defects in zebrafish reveal a novel copper‐chelating activity for kalihinol F. Chemistry & Biology, 20(6), 753–763. 10.1016/j.chembiol.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneville, R. , & Gönczy, P. (2004). zyg‐11 and cul‐2 regulate progression through meiosis II and polarity establishment in C. elegans. Development, 131(15), 3527–3543. 10.1242/dev.01244 [DOI] [PubMed] [Google Scholar]
- Stoll, C. , Viville, B. , Treisser, A. , & Gasser, B. (1998).Oculoauriculovertebral Spectrum, 349(February), 345–349. [DOI] [PubMed] [Google Scholar]
- Vendramini‐Pittoli, S. , & Kokitsu‐Nakata, N. M. (2009). Oculoauriculovertebral spectrum: Report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clinical Dysmorphology, 18(2), 67–77. 10.1097/MCD.0b013e328323a7dd [DOI] [PubMed] [Google Scholar]
- Wang, R. , Martínez‐Frías, M. L. , & Graham, J. M. (2002). Infants of diabetic mothers are at increased risk for the oculo‐auriculo‐vertebral sequence: A case‐based and case‐control approach. Journal of Pediatrics, 141(5), 611–617. 10.1067/mpd.2002.128891 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Fan, Y. S. , & Siu, V. M. (2008). A child with features of Goldenhar syndrome and a novel 1.12 Mb deletion in 22q11.2 by cytogenetics and oligonucleotide array CGH: Is this a candidate region for the syndrome? American Journal of Medical Genetics, Part A, 146(14), 1886–1889. 10.1002/ajmg.a.32359 [DOI] [PubMed] [Google Scholar]
- Yamaki, Y. , Kagawa, H. , Hatta, T. , Natsume, T. , & Kawahara, H. (2016). The C‐terminal cytoplasmic tail of hedgehog receptor Patched1 is a platform for E3 ubiquitin ligase complexes. Molecular and Cellular Biochemistry, 414(1–2), 1–12. 10.1007/s11010-015-2643-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐Fig S1
Data Availability Statement
The identified variant (NM_024646, c.1609G>T) was submitted to ClinVar database (submission identification: SUB6959693). All data that support the findings of this study are available on request from the corresponding author.