

Abstract
Background
In GM1 gangliosidosis the lack of function of β‐galactosidase results in an accumulation of GM1 ganglioside and related glycoconjugates in visceral organs, and particularly in the central nervous system, leading to severe disability and premature death. In the type 2 form of the disease, early intervention would be important to avoid precocious complications. To date, there are no effective therapeutic options in preventing progressive neurological deterioration. Substrate reduction therapy with Miglustat, a N‐alkylated sugar that inhibits the enzyme glucosylceramide synthase, has been proposed for the treatment of several lysosomal storage disorders such as Gaucher type 1 and Niemann Pick Type C diseases. However, data on Miglustat therapy in patients with GM1 gangliosidosis are still scarce.
Methods
We report here the results of Miglustat administration in four Italian children (average age: 55 months, range 20–125) affected by GM1 gangliosidosis type 2 treated in three different Italian pediatric hospitals specialized in metabolic diseases.
Conclusion
This treatment was safe and relatively well tolerated by all patients, with stabilization and/or slowing down of the neurological progression in three subjects.
Keywords: GM1 gangliosidosis, Miglustat, pediatric
In GM1 gangliosidosis the lack of function of β‐galactosidase results in an accumulation of GM1 gangliosides and related glycoconjugates in visceral organs, especially the central nervous system, leading to severe disability and premature death. In the type 2 form of the disease, early intervention would be important to avoid precocious complications. We report here the results of Miglustat administration in four Italian children (average age: 55 months, range 20–125) affected by GM1 gangliosidosis type 2. This treatment was safe and relatively well tolerated by all patients, with stabilization and/or slowing down of the neurological progression in three subjects.
1. INTRODUCTION
GM1 gangliosidosis is an autosomal recessive disorder caused by the deficiency of β‐galactosidase (GLB1), a lysosomal hydrolase that removes the terminal β‐galactosyl residues from GM1 gangliosides, glycoproteins, and glycosaminoglycans. Lack of β‐galactosidase function results in an accumulation of GM1 ganglioside and related glycoconjugates in visceral organs, and particularly in the central nervous system (CNS) (Sandhoff & Harzer, 2013). The incidence of GM1 gangliosidosis is estimated to be 1 in 100.000–200.000 live births, although prevalence widely differs in the world (Brunetti‐Pierri & Scaglia, 2008).
β‐galactosidase is encoded by GLB1, a gene located on chromosome 3p21.33 (Yamamoto et al., 1990). To date, more than 200 mutations have been identified (HGMD professional database 2015.3; https://portal.biobase‐international.com/cgibin/portal/login.cgi), differing in the residual enzyme activity that inversely correlates with the clinical severity (Suzuki, Nakamura, & Fukuoka, 1978). Despite the large genetic variability, currently, three main clinical forms have been identified: type 1 (infantile), type 2 (late infantile and juvenile), and type 3 (adult). GM1 gangliosidosis type 1 is characterized by early psychomotor deterioration, visceromegaly, macular cherry‐red spot, skeletal deformities, and death within the first 2 years of life (Suzuki & Oshima, 1993). Type 2 form presents with slowly progressive neurological signs as early locomotor problems, strabismus, muscle weakness, seizures, recurrent bronchopneumonia, and less severe dysmorphisms and skeletal involvement than in type 1. GM1 gangliosidosis type 2 can be divided into a late infantile and a juvenile form. In the late infantile form death occurs within adolescence while patients with the juvenile form can survive longer, with great individual clinical variability (Regier & Tifft, 2013). The adult form (type 3) is characterized by a less severe course and late onset, usually in juvenile age (Cox, 2015; Jarnes Utz et al., 2017; Regier, Proia, D'Azzo, & Tifft, 2016). Data on neuroimaging findings in the different forms of GM1 gangliosidosis are scarce; the reported patterns are nonspecific, including diffuse atrophy and white matter abnormalities (Chen et al., 1998; De Grandis, Di Rocco, Pessagno, Veneselli, & Rossi, 2009; Di Rocco et al., 2005; Gururaj et al., 2005). Type 1 may also present with delayed myelination and hyposignal on T2‐weighted images in the thalami, while type 2 may be associated with T2 hypointensity in the globi pallidi, and type 3 shows T2 hyperintensity within the putamina (Chen et al., 1998; De Grandis et al., 2009; Di Rocco et al., 2005; Erol, Alehan, Pourbagher, Canan, & Vefa, 2006; Gururaj et al., 2005).
To date, there are no effective drugs for the management of GM1 gangliosidosis that is mainly based on palliative treatments. Especially in the infantile and juvenile forms of GM1 gangliosidosis, early intervention would be important to avoid severe disability and premature death. However, the failure to access glycolipid storage products in CNS cells during critical developmental periods and the absence of clinical markers to evaluate treatment efficacy have hindered progress in developing effective therapies (Jarnes Utz et al., 2017).
Recently, the application of a substrate reduction therapy (SRT) has been evaluated in several lysosomal storage disorders (Hollak & Wijburg, 2014; Parenti, Andria, & Valenzano, 2015). The rationale of SRT is based on the use of synthesis inhibitors of substrates accumulated in lysosomes using specific iminosugars (Jeyakumar, Butters, Dwek, & Platt, 2002). NB‐DNJ (Miglustat, Zavesca®) is a N‐alkylated sugar that is a reversible competitive inhibitor of the glucosylceramide synthase, an enzyme catalyzing the first committed step in the synthesis of lacto‐ and globo‐series glycolipids (Platt, Neises, Dwek, & Butters, 1994). The ability of Miglustat to cross the blood–brain barrier blocking glycolipid storage synthesis has opened new interesting options for the treatment of multiple lysosomal storage disorders, especially those with CNS involvement, such as Gaucher and Niemann Pick type C diseases (Cox et al., 2000; Ficicioglu, 2008; Patterson, Vecchio, Prady, Abel, & Wraith, 2007). Miglustat has also been proposed for treatment of other sphingolipidoses, such as GM1 gangliosidosis. Indeed, GM1 gangliosidosis mouse models demonstrated Miglustat effectiveness in reducing brain GM1 ganglioside content (Kasperzyk et al., 2004; Treiber, Morand, & Clozel, 2007). Data on Miglustat efficacy in patients with GM1 gangliosidosis are scarce but promising, as reported in a few pediatric and adult patients affected by type 1 and 2 forms (Deodato et al., 2017; Garzone et al., 2013; Tifft, Adams, & Morgan, 2007).
Here, we describe the clinical course and adverse effects of Miglustat therapy in four pediatric patients with GM1 gangliosidosis type 2.
2. MATERIALS AND METHODS
After obtaining informed consent and approval from the local ethics committees, we reviewed the clinical and neuroradiological data of four patients with GM1 gangliosidosis treated in three different Italian pediatric hospitals specialized in metabolic diseases. The decision to initiate treatment was based on: (a) good safety profile of Miglustat, (b) good clinical results in other lysosomal storage diseases, and (c) promising preclinical data on GM1 mouse models. Miglustat was dosed according to guidelines for pediatric patients (younger than 12 years) affected by Niemann Pick type C disease. The drug was melted in water to facilitate the assumption and the dosage was progressively increased according to body surface area. Data on drug tolerance and compliance were collected from the mothers with follow‐up calls or during clinical assessment every 3 months. All patients were followed up at regular intervals by a multidisciplinary team and the development of side effects was monitored. Symptoms and signs and neurological evaluation of patients before and after Miglustat therapy are summarized in Table 1.
Table 1.
Symptoms and signs and neurological evaluation before and after Miglustat therapy
| Patient | Age at onset of symptoms | Age at Miglustat initiation | Age at last follow‐up during Miglustat therapy | Symptoms and signs | Neurological evaluation | ||
|---|---|---|---|---|---|---|---|
| Before Miglustat therapy | After Miglustat therapy | Before Miglustat therapy | After Miglustat therapy | ||||
| 1 | 11 months | 20 months | 5 years |
|
|
|
WPPSI‐III: mild worsening in fine motor skills and language |
| 2 | 5 years | 10 years and 5 months | 16 years |
|
Stable |
|
Stable motor and cognitive impairment |
| 3 | 5 years | 3 years and 8 months | 10 years and 5 months | Asymptomatic |
|
Normal |
|
| 4 | 18 months | 2 years and 9 months | 6 years and 7 months |
|
|
|
|
2.1. Patient 1
Patient #1 was born at term after an uneventful pregnancy by spontaneous delivery. He was the first child of a healthy nonconsanguineous couple, with no contributory family history. Normal growth and neurodevelopmental milestones were referred until the age of 11 months. At this time, he was referred to the orthopedist for thoracolumbar kyphosis that was treated with a corset. The first clinical evaluation did not reveal any dysmorphological signs. Spine X‐Ray showed anterior vertebral beaking from D11 to L4, more evident in L1 and L2, associated with marked thoracic kyphosis. Electrocardiogram, echocardiogram, Boel test, fundus oculi and slit lamp examination, neurological assessment, abdomen echography, and hand X‐ray were normal. Routine blood tests, aminoacidemia/uria, acylcarnitines, urinary glycosaminoglycans screening test, and organic acids were normal. At 18 months of age, the patient was operated of an abdominal cystic lymphangioma.
Increased levels of urinary oligosaccharides were found using thin‐layer chromatographic (TLC) method, while fluorimetric enzyme assay in leucocytes revealed that β‐galactosidase (GLB1) enzyme activity was below normal range (6.9 nM mg−1 h−1; normal values 100–600 nM mg−1 h−1). GLB1 gene analysis was performed, revealing compound heterozygous mutations in c.602G>A p.(Arg201His) (inherited from the mother) and c.841C>T p.(His 281Tyr) (inherited from the father).
Brain MRI and MR spectroscopy performed at 21 months of age were normal (Figure 2A,B). Cognitive assessment was performed with "Wechsler Preschool And Primary Scale Of Intelligence" (WPPSI‐III) resulting normal in both verbal and performance IQ scores.
Figure 2.
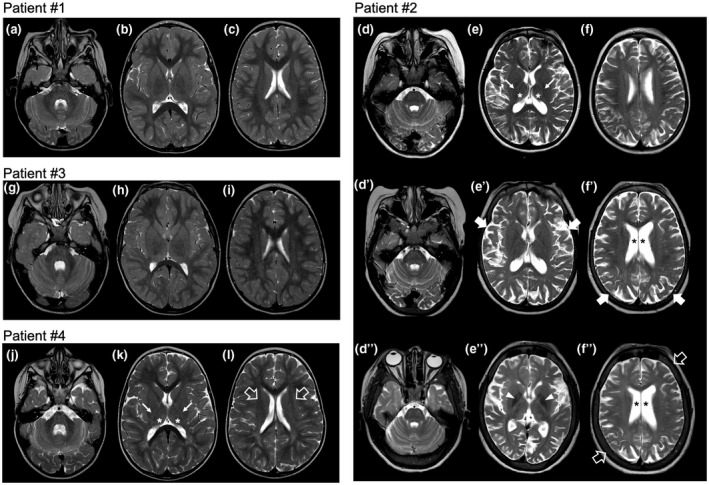
Brain MRI features of the patients. (A–C) Axial T2‐weighted images of patient #1 performed at 21 months of age reveal normal brain anatomy and signal intensity. (D–F) Axial T2‐weighted images of patient #2 performed at 8 years of age show diffuse white matter hyperintensity, more evident at the level of posterior limbs of internal capsules (arrows), associated with small thalami (asterisks) and mild enlargement of the cerebral and cerebellar subarachnoid spaces. (D′–F′) Corresponding axial T2‐weighted images of the same patient performed at 10 years and 5 months reveal progressive enlargement of the cerebral and cerebellar subarachnoid spaces (thick arrows) and enlargement of the lateral ventricles (asterisks). (D″–F″) Follow‐up axial T2‐weighted images at 16 years reveal further progression of the cerebral atrophy, with thickening of the skull bones (empty arrows) and hypointensity of the globipallidi (arrowheads). (G–I) Axial T2‐weighted images of patient #3 performed at 6 years and 2 months of age reveal normal brain anatomy and signal intensity. (J–L) Axial T2‐weighted images of patient #4 performed at 2 years of age demonstrate mild hyperintensity of the white matter of the centri semiovali (empty arrows) and posterior limbs of internal capsules (arrows). There is also mild enlargement of the cerebral and cerebellar subarachnoid spaces. Note the small thalami (asterisks)
At 20 months of age, after 2 months of preliminary dietary treatment (lactose‐free aliments), Miglustat therapy was started at a dose of 100 mg/day (subdivided into two doses) and progressively increased to 240 mg/day. At 4 years of age, the total drug amount was fractioned in three doses a day, while at 5 years, in four doses a day. Daily assumption of a probiotic product (Saccharomyces Boulardii) was also started. In the following years, height, weight, and head circumference growth curves were stable, between the 25th and 50th centiles. Orthopedic evaluations revealed a significant improvement of kyphosis with a nonstop use of the corset until the age of 4 years. Spine X‐Rays confirmed a significant improvement of the thoracic hyperkyphosis and a stable appearance of the vertebral anomalies. Follow‐up brain MRI scans at 4 and 5 years of age did not reveal any abnormality. Cognitive assessment using WPPSI‐III performed at 5 years of age showed mild worsening in fine motor skills and language. Routine blood tests were normal, except for mild and transient increase in alanine amino transpherase (ALT) levels.
2.2. Patient 2
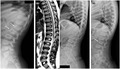
Patient #2 is the first child of healthy nonconsanguineous parents with negative family history. She was born at term after an uneventful pregnancy. She showed a normal psychomotor development until the age of 5 years when she presented with developmental regression associated with attention deficit, dysarthria, and extrapyramidal symptoms. She came to our attention at the age of 9 years when she developed atypical absences with generalized sharp waves (3 Hz) followed by slow waves lasting 20‐12′ at EEG, and she started antiepileptic therapy with lamotrigine. Neurological examination showed severe dysarthria, mild hypotonia, brisk reflex with mild bilateral clonus, awkwardness, bradykinesia with freezing, and gait ataxia; she could not walk without assistance. Neuropsychological evaluation showed severe cognitive impairment (IQ of 34 at WISC‐III Scale) and EDSS score of 7.5. Brain MRI performed at 8 years of age revealed mild cerebral and cerebellar atrophy, white matter signal alterations consistent with lack of myelin, dysmyelination, and reduced thalamic volume (Figure 2D–F). Brain MR spectroscopy was normal. Spinal MRI and X‐ray showed diffuse platyspondyly with mild anterior wedging of the lumbar vertebrae (Figure 1b,c). Routine blood tests were normal, as well as genetic exams including cytogenetic studies, Array – Comparative Genomic Hybridization (Array – CGH), and analysis of subtelomeric regions and MECP2 gene.
Figure 1.
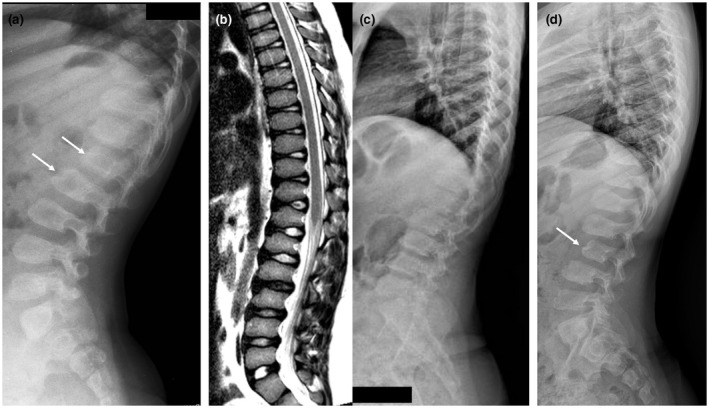
Spinal X‐ray and MRI features of the patients. (a) Spinal X‐ray, lateral view, of patient #1 performed at 6 years showing platyspondyly with anterior vertebral beaking from D11 to L4, more evident in L1 and L2 (arrows). (b) Spinal MRI, T2‐weighted sagittal image, and (c) Spinal X‐ray, lateral view, of patient #2 performed at 15 years of age demonstrates diffuse platyspondyly with mild anterior wedging of the lumbar vertebrae. (d) Spinal X‐ray, lateral view, of patient #3 performed at 9 years of age reveals diffuse platyspondyly with mild anterior wedging of the lumbar vertebrae, more evident in L3 (arrow)
Metabolic exams, including blood chromatography of the amino acids, organic acidurias, mucopolysaccharidosis (MPS) urinary screening, plasma chitotriosidase, and fucosidase, mannosidase, were normal while the activity of β‐galactosidase was reduced in lymphocytes (16.2 nM mg−1 h−1; normal values 100–600 nM mg−1 h−1) as well as in cultured skin fibroblasts, leading to the diagnosis of GM1 gangliosidosis. The genetic analysis revealed compound heterozygous mutations in GLB1 in c.602G>A p.(Arg201His) and the novel c.247dup1 p.(Tyr83Leu fsx8).
At the age of 10 years and 5 months, the clinical picture and neurological examination were stable. Brain MRI showed mild progression of cerebral atrophy with enlargement of the lateral ventricles (Figure 2D′–F′). Therapy with Miglustat was started (age 10 years and 5 months) at the dosage of 100 mg three times a day, gradually increased to 200 mg three times a day. She initially presented occasional diarrhea and mild abdominal pain, spontaneously improving in the following weeks. In the first 4 months of therapy, her body weight decreased from 42.6 to 38.9 kg (BMI from 24.8 to 21.9). Subsequently, in the following 6 years, the BMI was quite stable between 21.1 and 22.3. She has now been on a stable dosage of 600 mg/day for 1 year with good compliance and without relevant adverse events. At last follow‐up, at the age of 16 years, neurological examination showed stable motor and cognitive impairment. Brain MRI revealed further progression of mild cortical atrophy and T2 hypointensity at the level of the globi pallidi, substantia nigra, red nuclei, and cerebellar dentate nuclei, associated with thickening of the skull bones (Figure 2D″–F″).
2.3. Patient 3
Patient #3 is the younger brother of Patient #2. He was born at term after an uneventful pregnancy. He was asymptomatic when GM1 gangliosidosis were diagnosed at the age of 3 years, and he harbored the same compound mutations of his sister. Treatment with Miglustat was started at the age of 3 years and 8 months but was stopped after a week for the appearance of severe hyporexia. Four months later, the treatment was reinitiated at the dosage of 70 mg/twice daily and gradually increased up to 300 mg/day (subdivided into 3 doses). Up to date, he has been on a stable dosage for 3 years with good compliance and insignificant side effects such as occasional abdominal pain with diarrhea. At the age of 5 years, he started to develop dysarthria and attention deficit.
Brain MRI with MR spectroscopy performed at 6 years and 2 months was normal (Figure 2G–I). Spinal X‐ray revealed diffuse platyspondyly with anterior wedging of the lumbar vertebrae, especially of L3 (Figure 1d). At 10 years of age, he presented epileptic alterations at EEG during sleep, without evident clinical seizures and was treated with therapy with topiramate. Neurological examination revealed moderate intellectual disability, clumsiness, dysarthria, significative hyperkinesia, attention deficit, behavioral problems with heteroaggressivity, and psychomotor agitation outbursts. His EDSS score was 5. At last follow‐up, performed at 10 years and 5 months of age, neurological examination revealed a stable clinical course.
2.4. Patient 4
Patient #4 was born at term after an uneventful pregnancy by spontaneous delivery. At the age of 18 months, he presented with hypotonia, delayed psychomotor development, and a steppage gait characterized by widened base of support and retropulsion of the pelvis. He had slightly coarse facies with downturned corners of the mouth, convergent strabismus of the left eye, and open bite with sialorrhea. Abdominal ultrasound and metabolic screening tests were normal. Brain MRI performed at 2 years of age revealed white matter signal alterations consistent with lack of myelin and dysmyelination in the posterior limbs of internal capsules and pons, mild subarachnoid spaces enlargement, and reduced thalamic volume (Figure 2J–L). At the age of 2 years and 4 months, GM1 gangliosidosis was diagnosed by the β‐galactosidase enzymatic assay and GLB1 gene molecular analysis, which highlighted the presence in heterozygosis of the c.572G>A (p. Ser191Asn) and c.1445G>A (p. Arg482Hys) mutations. The boy started therapy with Miglustat at the age of 2 years and 9 months, at the dosage of 200 mg (subdivided into two doses).
At the age of 3 years and 2 months, the patient was again hospitalized for night cry, asthenia, and pain. The abdomen ultrasound was normal, while a barium X‐ray examination showed gastroesophageal reflux and therapy was initiated with esomeprazole. The neurological exam showed total regression of language, associated with pyramidal and extrapyramidal signs.
At the age of 3 years and 7 months, frequent daily seizures appeared, lasting few seconds and characterized by pathological eyes rolling and staring. An EEG showed high‐voltage slow waves and spike waves complexes on the right derivations. He began therapy with levetiracetam at the dosage of 400 mg/die with clinical improvement.
When he was 6 years old, he was hospitalized because of significant weight loss, dehydration, sickness, and vomiting. Oral feeding was strongly compromised. A percutaneous gastrostomy was placed. Seven months later, therapy with Miglustat was also suspended. Last clinical follow‐up at 7 years of age revealed severe intellectual disability and total gross and fine motor regression. He depended on his parents for daily survival.
3. DISCUSSION
We present our experience of Miglustat administration in four children affected by GM1 gangliosidosis type 2. In GM1 gangliosidosis type 2, the accumulation of GM1 ganglioside in the CNS begins in the early stages of development. Therefore, it has been hypothesized that the progression of the disease could only be avoided with early diagnosis and intervention (Del Vecchio et al., 2008; Jarnes Utz et al., 2017). Some therapeutic strategies have been implemented in animal models, such as bone marrow transplantation (Sano, Tessitore, Ingrassia, & d'Azzo, 2005), intracranial transplant of neuronal stem cells (Sawada et al., 2009), SRT (Elliot‐Smith et al., 2008; Kasperzyk et al., 2004; Suzuki et al., 2007), gene therapies (Baek et al., 2010), and enzyme replacement therapy (Condori et al., 2016). Additionally, a recent study demonstrated that the intracerebroventricular administration of recombinant human β‐galactosidase in a mouse model of GM1 gangliosidosis reduced brain levels of ganglioside and oligosaccharide substrates (Chen et al., 2019).
In human patients, several interventions have been undertaken to mitigate the relentless progression of disease (Regier et al., 2016). However, the failure to access glycolipid storage products in CNS during critical developmental periods and the absence of clinical markers to evaluate treatment efficacy have hindered progress in developing effective therapies (Jarnes Utz et al., 2017).
Substrate reduction therapy with oral administration of the iminosugar NB‐DNJ has been demonstrated to be effective in crossing the blood–brain barrier and blocking glycolipid storage synthesis (Kasperzyk et al., 2004; Treiber et al., 2007), thus revealing a potential therapeutic role in lysosomal storage disorders with neurological involvement (Platt et al., 1994). In particular, Miglustat has been approved in Europe and the United States for the treatment of patients with type 1 Gaucher disease with mild‐to‐moderate clinical manifestations for whom intravenous enzyme replacement therapy is not an option (Cox et al., 2000; Ficicioglu, 2008). Subsequently, Miglustat has also been approved for treating progressive neurologic manifestations in children and adults with Niemann Pick disease type C (Patterson et al., 2007).
The mechanism of Miglustat action has not been completely clarified yet. The efficacy of NB‐DNJ is related to its ability to cross the blood–brain barrier, inhibit the synthesis of storage products, and stabilize the enzyme with an enhancement of the catalytic residual activity (Alfonso et al., 2005; Kuter et al., 2013). Furthermore, several studies have proved the potential role of Miglustat administration in the regulation of the abnormal CNS inflammatory response observed in GM1 gangliosidosis (Jeyakumar et al., 2003), reducing levels of proinflammatory cytokines such as tumor necrosis factor‐α (TNF‐α) and interleukin‐1 beta (IL1‐β) (Jeyakumar et al., 2001; Masciullo et al., 2010; Santoro et al., 2013).
Despite the demonstrated effectiveness of Miglustat in other storage disorders, its use in GM 1 gangliosidosis type 2 has been reported only in a few patients. In 2007, Tifft et al. (2007) reported that Miglustat administration improved neurological functions in two patients with juvenile GM1 gangliosidosis. Recently, Deodato et al. (2017) described similar neurological improvement in the juvenile form of the disease. In our patients, we observed stabilization and/or slowing down of the neurological progression in three of four patients. Of note, the administration of Miglustat was started few months after the genetic confirmation. In particular, patients #1 and #3 received an early diagnosis of GM1 gangliosidosis type 2 and the treatment was started within the fourth year of age, before the onset of neurological symptoms. In patient #1, early treatment limited the disease progression until 5 years of age, when minor motor problems were noticed. Moreover, the improvement of the kyphosis without other novel skeletal manifestations and the absence of neuroimaging abnormalities were indicative of substantial stabilization of the clinical course due to the treatment. In patient #3, who was asymptomatic at the beginning of therapy, we noted a very slow progression of symptomatology, appeared at the age of 5 years and evident since the age of 8 years, thus suggesting a possible role of Miglustat in slowing down the neurological deterioration.
Patient #2 started the therapy rather late in the course of the disease, at the age of 10 years, when neurological symptoms and MRI alterations were already present. Surprisingly, in this case, we observed a stable motor and cognitive impairment for the following 5 years, while brain MRI showed only a minimal progression of the cerebral atrophy. On the other hand, Miglustat therapy was insufficient to avoid worsening of the clinical course in patient #4, who presented with a severe neurological phenotype since the first months of life.
Consensus guidelines for clinical management of children with Niemann Pick type C recommend close clinical monitoring after the initiation of Miglustat therapy (NP‐C Guidelines Working Group, 2009). However, several studies have proved that Miglustat is safe and generally well tolerated, with minor and often transient adverse effects, such as tremor and gastrointestinal disturbances (Belmatoug et al., 2011). In particular, gastrointestinal problems, including diarrhea, abdominal pain, nausea, and weight loss, are mainly dose‐related and limited to the initial phase of the treatment. The mechanism is most likely the inhibition of intestinal disaccharidases leading to reduced absorption of dietary disaccharides and increase in undigested intraluminal oligosaccharides creating an osmotic gradient with the influx of water and electrolytes in the gastrointestinal tract. Subsequent colonic bacterial fermentation reduces caloric supply and triggers flatulence, abdominal distension, pain, and nausea. Of note, these symptoms generally disappear or decrease with dose escalation or discontinued administrations in association with a low‐carbohydrate/lactose diet and probiotic administration (Cox, 2015; Patterson et al., 2007). In this series, two patients presented minor gastrointestinal problems. In particular, patient #2 developed transient diarrhea and abdominal pain, while patient #3 presented severe hyporexia that subsided after brief treatment suspension and did not reappear after Miglustat reintroduction at lower and fractionated doses while occasional abdominal pain with diarrhea persisted. Interestingly, in patient #1, Miglustat therapy was started after 2 months of a preliminary diet based on lactose‐free products and daily assumption of probiotic Saccharomyces Boulardii, thus avoiding gastroenteric symptoms (carbohydrates were preserved in order to maintain an adequate nutrition).
In conclusion, our findings suggest that Miglustat is relatively well tolerated in children with GM1 type 2 gangliosidosis and seems more effective if used to slow down, rather than treat, both visceral and neurological manifestation, as in Niemann Pick type C pediatric patients (Di Rocco, Dardis, Madeo, Barone, & Fiumara, 2012). The rarity of GM1 gangliosidosis type 2 disease, fast clinical progression, and short lifespan make studies on clinical surveillance and therapeutic efficacy tough, as effective interventions are only possible before irreversible damage occurs. Since definitive conclusions cannot be drawn from small case series, further studies on larger number of patients and with longer follow‐up duration are needed to evaluate of the long‐term therapeutic effects of Miglustat in GM1 type II gangliosidosis.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
ACKNOWLEDGMENTS
All authors contributed equally to the study design and performance.
Fischetto R, Palladino V, Mancardi MM, et al. Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol Genet Genomic Med. 2020;8:e1371 10.1002/mgg3.1371
DATA AVAILABILITY STATEMENT
The data supporting the findings of the article can be requested to the authors at the following.
REFERENCES
- Alfonso, P. , Pampín, S. , Estrada, J. , Rodríguez‐Rey, J. C. , Giraldo, P. , Sancho, J. , & Pocoví, M. (2005). Miglustat (NB‐DNJ) works as a chaperone for mutated acid beta‐glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells, Molecules & Diseases, 35, 268–276. 10.1016/j.bcmd.2005.05.007 [DOI] [PubMed] [Google Scholar]
- Baek, R. C. , Broekman, M. L. , Leroy, S. G. , Tierney, L. A. , Sandberg, M. A. , d'Azzo, A. , … Sena‐Esteves, M. (2010). AAV‐mediated gene delivery in adult GM1‐gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS One, 5, e13468 10.1371/journal.pone.0013468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmatoug, N. , Burlina, A. , Giraldo, P. , Hendriksz, C. J. , Kuter, D. J. , Mengel, E. , & Pastores, G. M. (2011). Gastrointestinal disturbances and their management in miglustat‐treated patients. Journal of Inherited Metabolic Disease, 34, 991–1001. 10.1007/s10545-011-9368-7 [DOI] [PubMed] [Google Scholar]
- Brunetti‐Pierri, N. , & Scaglia, F. (2008). GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Molecular Genetics and Metabolism, 94, 391–396. 10.1016/j.ymgme.2008.04.012 [DOI] [PubMed] [Google Scholar]
- Chen, C. Y. , Zimmerman, R. A. , Lee, C. C. , Chen, F. H. , Yuh, Y. S. , & Hsiao, H. S. (1998). Neuroimaging findings in late infantile GM1 gangliosidosis. AJNR. American Journal of Neuroradiology, 19, 1628–1630. [PMC free article] [PubMed] [Google Scholar]
- Chen, J. C. , Luu, A. R. , Wise, N. , Angelis, R. , Agrawal, V. , Mangini, L. , … Yogalingam, G. (2019). Intracerebroventricular enzyme replacement therapy with Beta‐Galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. The Journal of Biological Chemistry. 10.1074/jbc.RA119.009811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condori, J. , Acosta, W. , Ayala, J. , Katta, V. , Flory, A. , Martin, R. , … Radin, D. N. (2016). Enzyme replacement for GM1‐gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β‐galactosidase: RTB lectin fusion. Molecular Genetics and Metabolism, 117, 199–209. 10.1016/j.ymgme.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, T. M. (2015). Innovative treatments for lysosomal diseases. Best Practice & Research Clinical Endocrinology & Metabolism, 29, 275–311. 10.1016/j.beem.2015.01.001 [DOI] [PubMed] [Google Scholar]
- Cox, T. , Lachmann, R. , Hollak, C. , Aerts, J. , van Weely, S. , Hrebícek, M. , … Zimran, A. (2000). Novel oral treatment of Gaucher’s disease with Nbutyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet, 355, 1481–1485. 10.1016/S0140-6736(00)02161-9 [DOI] [PubMed] [Google Scholar]
- De Grandis, E. , Di Rocco, M. , Pessagno, A. , Veneselli, E. , & Rossi, A. (2009). MR imaging findings in 2 cases of late infantile GM1 gangliosidosis. AJNR. American Journal of Neuroradiology, 30, 1325–1327. 10.3174/ajnr.A1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio G. C., De Santis A., Giordano P., Amendola G., Baronci C., Del Principe D., … De Mattia D.; AIEOP ITP Study Group . (2008). Management of acute childhood idiopathic thrombocytopenic purpura according to AIEOP consensus guidelines: Assessment of Italian experience. Acta Haematologica, 119, 1–7. 10.1159/000112837 [DOI] [PubMed] [Google Scholar]
- Deodato, F. , Procopio, E. , Rampazzo, A. , Taurisano, R. , Donati, M. A. , Dionisi‐Vici, C. , … Scarpa, M. (2017). The treatment of juvenile/adult GM1‐gangliosidosis with Miglustat may reverse disease progression. Metabolic Brain Disease, 32, 1529–1536. 10.1007/s11011-017-0044-y [DOI] [PubMed] [Google Scholar]
- Di Rocco, M. , Dardis, A. , Madeo, A. , Barone, R. , & Fiumara, A. (2012). Early Miglustat therapy in infantile Niemann Pick Type C. Pediatric Neurology, 47, 40–43. 10.1016/j.pediatrneurol.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Di Rocco, M. , Rossi, A. , Parenti, G. , Allegri, A. E. , Filocamo, M. , Pessagno, A. , … Biancheri, R. (2005). Different molecular mechanisms leading to white matter hypomyelination in infantile onset lysosomal disorders. Journal of Pediatric Neurobiology, Neurology and Neurosurgery, 36, 265–269. 10.1055/s-2005-865863 [DOI] [PubMed] [Google Scholar]
- Elliot‐Smith, E. , Speak, A. O. , Lloyd‐Evans, E. , Smith, D. A. , van der Spoel, A. C. , Jeyakumar, M. , … Platt, F. M. (2008). Beneficial effect of substrate reduction therapy in a mouse model of GM1‐Gangliosidosis. Molecular Genetics and Metabolism, 94, 204–211. 10.1016/j.ymgme.2008.02.005 [DOI] [PubMed] [Google Scholar]
- Erol, I. , Alehan, F. , Pourbagher, M. A. , Canan, O. , & Vefa, Y. S. (2006). Neuroimaging findings in infantile GM1 gangliosidosis. European Journal of Paediatric Neurology, 10, 245–248. 10.1016/j.ejpn.2006.08.005 [DOI] [PubMed] [Google Scholar]
- Ficicioglu, C. (2008). Review of miglustat for clinical management in Gaucher disease type 1. Therapeutics and Clinical Risk Management, 4, 425–431. 10.2147/tcrm.s6865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzone, A. , Pompilio, A. , Ciuccarelli, F. , Andresciani, E. , Ficcadenti, A. , De Meo, M. , … Moretti, V. (2013). Miglustat off‐label in a paediatric formulation for a rare metabolic disease: Early infantile GM1 gangliosidosis. European Journal of Hospital Pharmacy, 20, A1–A238. 10.1136/ejhpharm-2013-000276.543 [DOI] [Google Scholar]
- Gururaj, A. , Sztriha, L. , Hertecant, J. , Johansen, J. G. , Georgiou, T. , Campos, Y. , … d'Azzo, A. (2005). Magnetic resonance imaging findings and novel mutations in GM1 gangliosidosis. Journal of Child Neurology, 20, 57–60. 0.1177/08830738050200010901 [DOI] [PubMed] [Google Scholar]
- Hollak, C. E. , & Wijburg, F. A. (2014). Treatment of lysosomal storage disorders: Successes and challenges. Journal of Inherited Metabolic Disease, 37, 587–598. 10.1007/s10545-014-9718-3 [DOI] [PubMed] [Google Scholar]
- Jarnes Utz, J. R. , Kim, S. , King, K. , Ziegler, R. , Schema, L. , Redtree, E. S. , & Whitley, C. B. (2017). Infantile Gangliosidoses: Mapping a Timeline of Clinical Changes. Molecular Genetics and Metabolism, 121, 170–179. 10.1016/j.ymgme.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyakumar, M. , Butters, T. D. , Dwek, R. A. , & Platt, F. M. (2002). Glycosphingolipid lysosomal storage diseases: Therapy and pathogenesis. Neuropathology and Applied Neurobiology, 28, 343–357. 10.1046/j.1365-2990.2002.00422.x [DOI] [PubMed] [Google Scholar]
- Jeyakumar, M. , Norflus, F. , Tifft, C. J. , Cortina‐Borja, M. , Butters, T. D. , Proia, R. L. , … Platt, F. M. (2001). Enhanced survival in Sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood, 97, 327–329. 10.1182/blood.v97.1.327 [DOI] [PubMed] [Google Scholar]
- Jeyakumar, M. , Thomas, R. , Elliot‐Smith, E. , Smith, D. A. , van der Spoel, A. C. , d'Azzo, A. , … Platt, F. M. (2003). Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain, 126, 974–987. 10.1093/brain/awg089 [DOI] [PubMed] [Google Scholar]
- Kasperzyk, J. L. , El‐Abbadi, M. M. , Hauser, E. C. , D'Azzo, A. , Platt, F. M. , & Seyfried, T. N. (2004). N‐butyldeoxygalactonojirimycin reduces neonatal brain ganglioside content in a mouse model of GM1 gangliosidosis. Journal of Neurochemistry, 89, 645–653. 10.1046/j.1471-4159.2004.02381.x [DOI] [PubMed] [Google Scholar]
- Kuter, D. J. , Mehta, A. , Hollak, C. E. , Giraldo, P. , Hughes, D. , Belmatoug, N. , … Zimran, A. (2013). Miglustat therapy in type1 Gaucher disease: Clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells, Molecules & Diseases, 51, 116–124. 10.1016/j.bcmd.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Masciullo, M. , Santoro, M. , Modoni, A. , Ricci, E. , Guitton, J. , Tonali, P. , & Silvestri, G. (2010). Substrate reduction therapy with miglustat in chronic GM2 gangliosidosis type Sandhoff: Results of a 3 year follow‐up. Journal of Inherited Metabolic Disease, 33, S355–S361. 10.1007/s10545-010-9186-3 [DOI] [PubMed] [Google Scholar]
- NP‐C Guidelines Working Group , Wraith J. E., Baumgartner M. R., Bembi B., Covanis A., Levade T., Mengel E., … Patterson M. C. (2009). Recommendations on the diagnosis and management of Niemann‐Pick disease type C. Molecular Genetics and Metabolism, 98, 152–165. 10.1016/j.ymgme.2012.03.012 [DOI] [PubMed] [Google Scholar]
- Parenti, G. , Andria, G. , & Valenzano, K. J. (2015). Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Molecular Therapy, 23, 1138–1148. 10.1038/mt.2015.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, M. C. , Vecchio, D. , Prady, H. , Abel, L. , & Wraith, J. E. (2007). Miglustat for treatment of Niemann‐Pick C disease: A randomised controlled study. The Lancet Neurology, 6, 765–772. 10.1016/S1474-4422(07)70194-1 [DOI] [PubMed] [Google Scholar]
- Platt, F. M. , Neises, G. R. , Dwek, R. A. , & Butters, T. D. (1994). N‐butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. The Journal of Biological Chemistry, 269, 8362–8365. [PubMed] [Google Scholar]
- Regier, D. S. , Proia, R. L. , D'Azzo, A. , & Tifft, C. J. (2016). The GM1 and GM2 Gangliosidoses: Natural history and progress toward therapy. Pediatric Endocrinology Review: PER, 13, 663–673. [PMC free article] [PubMed] [Google Scholar]
- Regier, D. S. , & Tifft, C. J. (2013). GLB1‐related disorders In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A., (Eds.), GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2015. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK164500/ [PubMed] [Google Scholar]
- Sandhoff, K. , & Harzer, K. (2013). Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. The Journal of Neuroscience, 33, 10195–10208. 10.1523/JNEUROSCI.0822-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano, R. , Tessitore, A. , Ingrassia, A. , & d'Azzo, A. (2005). Chemokine‐induced recruitment of genetically modified bone marrow cells into the CNS of GM1‐gangliosidosis mice corrects neuronal pathology. Blood, 106, 2259–2268. 10.1182/blood-2005-03-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, N. , Colombini, A. , Silvestri, D. , Grassi, M. , Giordano, P. , Parasole, R. , … Rizzari, C. (2013). Screening for coagulopathy and identification of children with acute lymphoblastic leukemia at a higher risk of symptomatic venous thrombosis: An AIEOP experience. Journal of Pediatric hematology/oncology, 35, 348–355. 10.1097/MPH.0b013e31828dc614 [DOI] [PubMed] [Google Scholar]
- Sawada, T. , Tanaka, A. , Higaki, K. , Takamura, A. , Nanba, E. , Seto, T. , … Yamano, T. (2009). Intracerebral cell transplantation therapy for murine GM1 gangliosidosis. Brain & Development, 31, 717–724. 10.1016/j.braindev.2008.11.004 [DOI] [PubMed] [Google Scholar]
- Suzuki, Y. , Ichinomiya, S. , Kurosawa, M. , Ohkubo, M. , Watanabe, H. , Iwasaki, H. , … Brady, R. O. (2007). Chemical chaperone therapy: Clinical effect in murine G(M1)‐gangliosidosis. Annals of Neurology, 62, 671–675. 10.1002/ana.21284 [DOI] [PubMed] [Google Scholar]
- Suzuki, Y. , Nakamura, N. , & Fukuoka, K. (1978). GM1‐gangliosidosis: Accumulation of ganglioside GM1 in cultured skin fibroblasts and correlation with clinical types. Human Genetics, 43, 127–131. 10.1007/bf00293589 [DOI] [PubMed] [Google Scholar]
- Suzuki, Y. , & Oshima, A. (1993). A beta‐galactosidase gene mutation identified in both Morquio B disease and infantile GM1 gangliosidosis. Human Genetics, 91, 407 10.1007/bf00217370 [DOI] [PubMed] [Google Scholar]
- Tifft, C. , Adams, T. R. , & Morgan, C. P. (2007). Miglustat improves function in patients with juvenile GM1 gangliosidosis. Molecular Genetics and Metabolism, 92(4), 24. [Google Scholar]
- Treiber, A. , Morand, O. , & Clozel, M. (2007). The pharmacokinetics and tissue distribution of the glucosylceramide synthase inhibitor miglustat in the rat. Xenobiotica, 37, 298–314. 10.1080/00498250601094543 [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y. , Hake, C. A. , Martin, B. M. , Kretz, K. A. , Ahern‐Rindell, A. J. , Naylor, S. L. , … O’Brien, J. S. (1990). Isolation, characterization, and mapping of a human acid beta‐galactosidase cDNA. DNA and Cell Biology, 9, 119–127. 10.1089/dna.1990.9.119 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of the article can be requested to the authors at the following.