

Abstract
Background
Over half of children with rare genetic diseases remain undiagnosed despite maximal clinical evaluation and DNA‐based genetic testing. As part of an Undiagnosed Diseases Program applying transcriptome (RNA) sequencing to identify the causes of these unsolved cases, we studied a child with severe infantile osteopetrosis leading to cranial nerve palsies, bone deformities, and bone marrow failure, for whom whole‐genome sequencing was nondiagnostic.
Methods
We performed transcriptome (RNA) sequencing of whole blood followed by analysis of aberrant transcript isoforms and osteoclast functional studies.
Results
We identified a pathogenic deep intronic variant in CLCN7 creating an unexpected, frameshifting pseudoexon causing complete loss of function. Functional studies, including osteoclastogenesis and bone resorption assays, confirmed normal osteoclast differentiation but loss of osteoclast function.
Conclusion
This is the first report of a pathogenic deep intronic variant in CLCN7, and our approach provides a model for systematic identification of noncoding variants causing osteopetrosis—a disease for which molecular‐genetic diagnosis can be pivotal for potentially curative hematopoietic stem cell transplantation. Our work illustrates that cryptic splice variants may elude DNA‐only sequencing and supports broad first‐line use of transcriptome sequencing for children with undiagnosed diseases.
Keywords: CLCN7, osteopetrosis, RNA‐sequencing, transcriptomics, undiagnosed diseases
Over half of children with rare genetic diseases remain undiagnosed despite maximal DNA‐based genetic testing. Using transcriptome (RNA) sequencing, we identified a deep intronic variant in CLCN7 as the cause of a child's previously undiagnosed severe osteopetrosis syndrome. This work illustrates that cryptic splice variants may elude DNA‐only sequencing and supports first‐line use of transcriptome sequencing for undiagnosed diseases.

1. INTRODUCTION
Despite remarkable progress in genetic diagnostics over the past decade and the identification of many new Mendelian gene‐disease associations, nearly all current clinical and research diagnoses are coding variants or readily interpretable canonical splice site variants (Gloss & Dinger, 2018; MacArthur et al., 2014). At the same time, it is estimated that a remarkable 50%–70% of rare Mendelian disorders still fail to reach a genetic diagnosis, leading to immense challenges and sometimes missed treatment opportunities for patients, their families, and the healthcare system (Boycott et al., 2017; Dragojlovic et al., 2018; Retterer et al., 2015; Splinter et al., 2018). A leading hypothesis is that noncoding variants affecting transcription and splicing may explain a significant subset of the remaining undiagnosed cases, in addition to oligogenic, complex structural, and somatic variants that are not well assessed by current analytic pipelines. However, in contrast to coding variants, noncoding variants are challenging to interpret from DNA sequence alone (Chong et al., 2015; Zhang & Lupski, 2015). Recently, we and others have shown that transcriptome (RNA) sequencing may be used as a tool for unbiased discovery of pathogenic noncoding variants across several disease categories such as congenital malformations, myopathies, metabolic disorders, and other conditions (Cummings et al., 2017; Evrony et al., 2017; Gonorazky et al., 2019; Kremer, Wortmann, & Prokisch, 2018). Indeed, transcriptome sequencing presents an opportunity to rigorously evaluate the “noncoding variant hypothesis” of undiagnosed diseases by virtue of its genome‐wide interrogation of the in vivo transcriptional consequences of noncoding variants.
As part of our Undiagnosed Diseases Program, we are implementing systematic transcriptome sequencing to determine its utility across additional disease categories. Here, we describe the successful application of transcriptome sequencing to reveal a pathogenic deep intronic variant in a girl with infantile malignant osteopetrosis, a severe and often lethal metabolic bone disorder. This work has important implications for other unsolved cases of osteopetrosis, a condition for which a specific genetic diagnosis can have significant clinical impact. It also strongly supports implementation of transcriptome sequencing earlier and more broadly for undiagnosed genetic diseases.
Osteopetrosis is a rare, heterogenous genetic disorder characterized by an uncontrolled increase in bone mass (osteosclerosis) (Stark & Savarirayan, 2009). Its pathogenesis is decreased bone resorption due to either abnormal differentiation or function of osteoclasts, the cell type dedicated to breaking down bone (Sobacchi, Schulz, Coxon, Villa, & Helfrich, 2013). There are several forms of the disease ranging in age of onset, severity, and involvement of other body systems. Many forms are severe, manifesting in infancy or childhood with cranial and skeletal deformities; bone marrow failure leading to infections, bleeding, and anemia; and neurologic complications due to compression of vital nerves and structures by bone overgrowth (Stark & Savarirayan, 2009). Without treatment, the severe forms of the disease are invariably lethal (Del Fattore, Cappariello, & Teti, 2008).
Osteopetrosis and related osteosclerotic disorders are associated with variants in at least 16 genes that map to all stages of osteoclast differentiation and function, with a final common pathway of disrupted bone resorption (Palagano, Menale, Sobacchi, & Villa, 2018; Penna, Capo, Palagano, Sobacchi, & Villa, 2019; Sobacchi et al., 2013). However, 10%–20% of osteopetrosis cases fail to reach a molecular‐genetic diagnosis (Palagano et al., 2018; Sobacchi et al., 2013). Moreover, there are only a few reports of noncanonical pathogenic variants (e.g., splice‐altering coding variants or noncoding variants more than a few base pairs from the exon‐intron junction) (Palagano et al., 2015, 2017), and to the best of our knowledge, RNA sequencing has not been previously reported as an approach for its diagnosis.
In addition to enabling more informed prognoses and reproductive options for families, identifying the specific genetic etiology of a patient's osteopetrosis can be important for clinical management of the disease (Wu et al., 2017). One reason is that some genetic forms of the disease are amenable to curative hematopoietic stem cell transplantation (HSCT), while others are not (Schulz, Steward, & Sobacchi., 2015; Wu et al., 2017). Osteopetrosis cases due to mutations in TNFSF11/RANKL, which encodes a cytokine essential for differentiation of hematopoietic progenitors to osteoclasts, are not candidates for HSCT because this cytokine is mainly expressed by nonhematopoietic cells in bone. Additionally, some forms of osteopetrosis, for example those caused by mutations in OSTM1, have a primary neurodegenerative phenotype that does not improve with HSCT, precluding this high‐risk procedure. Notably, CLCN7‐autosomal recessive osteopetrosis (OMIM #611490), which accounts for 20% of diagnosed infantile osteopetrosis cases, can manifest with either a neurodegenerative or non‐neurodegenerative phenotype (Penna et al., 2019). In the former, patients are developmentally delayed and develop seizures, hypotonia, spasticity, cerebral atrophy, and white matter abnormalities (Pangrazio et al., 2010). While these forms of non‐HSCT responsive osteopetrosis can often be distinguished clinically from forms that are amenable to HSCT, this can sometimes be uncertain. As described below, this was initially the situation for our proband. A timely genetic diagnosis can therefore be an important adjunct for deciding whether to perform HSCT, a high‐risk procedure that should be conducted early to avoid the disease's irreversible damage and to reduce the complications and mortality of HSCT that increase with age (Schulz et al., 2015).
2. CASE PRESENTATION
Our proband is a 3‐year‐old girl with generalized osteosclerosis, developmental delay, multiple progressive cranial neve palsies including vision loss, and progressive hematopoietic dysfunction. She was born to consanguineous (first‐cousin) parents of Egyptian‐Arab descent after an unremarkable pregnancy. Shortly after birth, her family relocated to Egypt where she was initially evaluated at 4 months of age due to abnormal rotary eye movements and left facial weakness. Clinical evaluation revealed left cranial nerve VII palsy, bilateral ptosis, and bilateral nystagmus. Electroretinography and visual evoked potential showed possible optic nerve dysfunction. Electroencephalography (EEG) exhibited left focal sharp waves with occasional propagation to the right hemisphere, but without clinical seizures. Brain MRI showed mild prominence of sulci and gyri concerning for possible minimal brain atrophy.
At 18 months of age, the family relocated to the United States where renewed clinical evaluation identified global developmental delays with delayed speech and motor skills, hypotonia, macrocephaly (97th percentile) that was not present at birth, left cranial nerve VI and VII palsies, hyperopia and astigmatism, bilateral optic disc pallor and optic nerve hypoplasia, nasal congestion, nasolacrimal duct obstruction, and feeding difficulties. Her physical exam was also notable for frontal bossing, prominent occipital protuberance, micrognathia, a high arched palate, delayed eruption of primary teeth, narrow auditory canals, mild pectus carinatum, genu valgus, anterior bowing of the upper limbs, and an unstable gait. Additional family history was noncontributory.
Laboratory testing (Table S1) was notable for mild leukocytosis, mild anemia, vitamin D deficiency with normal calcium levels and elevated parathyroid hormone (secondary hyperparathyroidism), elevated lactate dehydrogenase, elevated alkaline phosphatase, elevated aspartate aminotransferase (AST), normal alanine aminotransferase (ALT), and elevated creatine kinase (CK) and CK‐BB. A repeat EEG was normal, and a repeat brain MRI showed a thickened calvarium but was otherwise unremarkable. Importantly, a skeletal X‐ray survey (Figure 1A) demonstrated diffuse sclerosing bony dysplasia, intramedullary sclerosis, a striated and under‐trabeculated appearance of long bones, bowing deformity of the tibias and femurs, sclerosis of ribs and clavicles some of which had a bone within bone appearance, and a thickened skull with a “hair‐on‐end” appearance.
Figure 1.
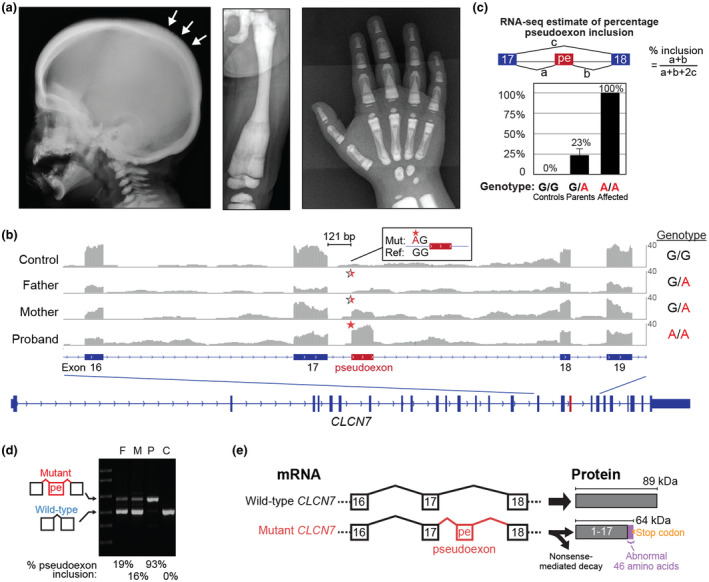
Clinical features of the proband and identification of a pathogenic pseudoexon in CLCN7. (a) Representative images taken at 2 years 9 months of age from a full skeletal X‐ray survey showing bone changes consistent with osteopetrosis. Left panel: lateral view of the skull showing widening and increased sclerosis of the diploic space and “hair on end” appearance (arrows). Middle panel: anterior‐posterior view of the left femur showing a bowing deformity and increased intramedullary sclerosis with a striated appearance. Right panel: anterior‐posterior view of the right hand showing increased bone density, predominantly in the medullary cavities, with a “bone within bone” appearance. Additional X‐rays not shown exhibit similar findings of diffuse sclerosing dysplasia of all bones of the hands, feet, ribs, spine, clavicles, and long bones. (b) RNA sequencing identifies a novel pseudoexon in intron 17 of CLCN7, 121 bp downstream of exon 17. Y‐axis is scaled from 0 to 40 reads. The control sample is representative of three unrelated controls. The genotypes on the right of the coverage tracks (G>A) are relative to the transcript strand. Half and full red stars mark the site of the heterozygous and homozygous variant, respectively. As illustrated in the inset, the variant is 2 bp upstream of the pseudoexon splice acceptor junction (Mut, mutant sequence; Ref, human genome reference sequence). (c) Quantification of percentage pseudoexon inclusion in the proband, parents, and unrelated controls (n = 3) using RNA‐seq splice junction‐spanning reads. pe, pseudoexon. Error bars: std. error. (d) RT‐PCR validation of the pseudoexon using primers spanning from exon 15 to exon 19. Lanes: Ladder, F (father), M (mother), P (proband), C (unrelated control). (e) Schematic of the predicted effects of the pathogenic pseudoexon. Western immunoblots to detect the degree of truncated protein production were unsuccessful with two different commercial antibodies and one custom‐made CLCN7 antibody due to antibody nonspecificity (data not shown)
Altogether, the proband's osteosclerosis, cranial nerve deficits, and hematologic abnormalities were consistent with a clinical diagnosis of infantile malignant osteopetrosis.
Over the next two years, she required multidisciplinary care for both new and progressive disease manifestations, including bilateral optic atrophy and vision loss, nystagmus, bilateral cranial nerve VI and VII palsies, bilateral conductive hearing loss, nasolacrimal duct stenosis, chronic aspiration, obstructive sleep apnea treated by tonsillectomy and adenoidectomy, progressive anemia and thrombocytopenia, long bone deformities, and persistent failure to achieve developmental speech and motor milestones.
Extensive clinical genetic testing was nondiagnostic, including: (a) peripheral blood karyotype, (b) chromosomal microarray, (c) high bone mass genetic panel, and, (d) trio whole‐exome sequencing. The microarray did reveal multiple regions of homozygosity consistent with consanguinity, one of which included CLCN7 (chloride voltage‐gated channel 7), a known osteopetrosis gene coding for a chloride/proton antiporter that maintains acidification of the osteoclast resorption lacunae. However, clinical targeted CLCN7 sequencing, including coding regions and deletion/duplication analysis, did not reveal any variants. During the above clinical and genetic evaluations, the decision about HSCT as a potentially curative treatment was complicated by the disease burden at presentation, the lack of a confirmed molecular‐genetic diagnosis, and the uncertainty whether her osteopetrosis was of an osteoclast‐poor or neurodegenerative form, neither of which would be appropriate for HSCT (Wu et al., 2017).
3. RESULTS
At the age of 3 years, the patient was enrolled in our Undiagnosed Diseases Program at New York University Grossman School of Medicine. We performed whole‐genome sequencing of the proband, including careful analysis of all known osteopetrosis genes, but this did not reveal any pathogenic variants. We hypothesized that a cryptic noncoding variant may be the cause of the proband's disease, and that this could be detected by transcriptome sequencing of the proband's peripheral blood. We were motivated by the fact that 13 of 16 osteopetrosis and osteopetrosis‐related disorder genes (all except for TNFSF11, TNFRSF11A, and LRP5) are expressed at detectable levels in blood (Transcripts Per Million >1) according to the Genotype‐Tissue Expression (GTEx) database (Melé et al., 2015). We therefore proceeded to conduct high‐depth trio transcriptome (RNA) sequencing of parental and proband blood samples with an average of 358 million reads per sample. The high‐depth of sequencing was chosen in order to increase power of detection for lower‐expressed genes.
Analysis of RNA sequencing data for transcriptional abnormalities (gene expression and splicing) across all known osteopetrosis genes revealed an aberrant novel 109 bp exon (chr16:1,450,267‐1,450,376; hg38) inside intron 17 of CLCN7 (Figure 1B). Quantification of the abnormal CLCN7 pseudoexon isoform using splice junction‐spanning reads indicated it was the only isoform present in the proband, and it was present at low level in the father and moderate level in the mother along with the normal isoform (Figure 1C). Moreover, the pseudoexon was absent from three unrelated in‐house blood controls (Figure 1B,C) and the GTEx project that profiled a large number of healthy individuals across a wide range of tissues (Melé et al., 2015).
In the CLCN7 transcript, the pseudoexon was located 121 bp downstream from the exon 17‐intron17 splice junction and 2 bp downstream from a homozygous deep intronic G>A variant that created a novel “AG” splice acceptor site (Figure 1B). The variant (c.1617+119G>A, NM_001287.5 transcript reference; chr16:1,450,378 C>T, hg38 reference) is heterozygous in the parents and absent from the gnomAD population control database. Importantly, the variant locus is not well conserved across mammals (Figure S1) and has a low CADD (Combined Annotation Dependent Depletion (Rentzsch, Witten, Cooper, Shendure & Kircher, 2018 #540)) score (<1). Therefore, standard computational prediction methods based on conservation could not identify this as a candidate variant without the functional transcriptome data. Normalized CLCN7 gene expression was 1.6, 2.6 ± 0.7, and 3.8 ± 0.5 FPKM (fragments per kilobase per million mapped reads; mean ± SD) in the proband, parents, and controls, respectively.
RT‐PCR validation confirmed the pseudoexon coordinates observed by transcriptome sequencing (Figure 1D). Quantification of RT‐PCR products using band intensity (normalized for the size of the amplicon) as a rough estimate of relative isoform levels estimated that 93% of transcripts in the proband and 16‐19% of transcripts in the parents contain the pseudoexon (Figure 1D). The predicted impact of the pseudoexon on the protein is to produce a truncated protein (64 kDa mutant protein versus 89 kDa wild‐type protein) ending in 46 abnormal amino acids due to the pseudoexon and frameshifting of the subsequent exon 18 (p.Ile540AlafsTer47) (Figure 1E). The lower expression levels of CLCN7 in the affected family versus the controls (p = .04, two‐tailed t test) suggests that the abnormal CLCN7 transcript may be undergoing nonsense‐mediated decay (NMD) and provides further evidence of the pathogenicity of the pseudoexon. To the best of our knowledge, this is the first case of osteopetrosis attributed to an aberrant exon in CLCN7 and the first case attributed to a deep intronic variant, illustrating the potential of transcriptome sequencing for identification of cryptic splicing abnormalities.
Next, we studied the functional impact of the proband's genetic defect. Two possible processes may be dysfunctional in osteopetrosis, depending on the affected gene: osteoclast differentiation (TNFSF11 and TNFRSF11A genes) or osteoclast function (CLCN7 and other osteopetrosis genes). We therefore evaluated both processes using in vitro osteoclast assays of patient‐derived samples (Figure 2A).
Figure 2.
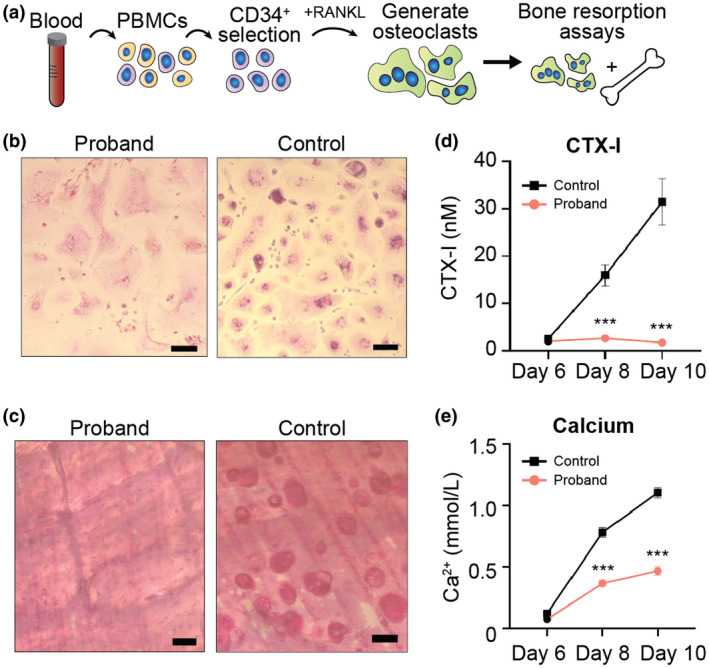
Osteoclastogenesis and bone resorption assays. (a) Schematic of osteoclast generation (osteoclastogenesis) and bone resorption assays. PBMCs, peripheral blood mononuclear cells. (b) Tartrate resistant acid phosphatase (TRAP)‐staining of osteoclasts generated from the proband and a control. Osteoclasts are successfully generated from proband samples, but are larger than control osteoclasts. Note, small cells in proband and control images are cells that have not yet fully differentiated into osteoclasts. Scale bars, 100 μm. (c) Microscopy of bone resorption pits in the bone resorption assay illustrates that proband osteoclasts are unable to break down bone (absent pits) versus the control sample that harbors numerous pits. Scale bars, 50 μm. (d) CTX‐I levels in culture media during the bone resorption assay. Data are means ± SEM (n = 8 replicates). ***Indicates p < .001 (unpaired two‐tailed Student's t test). (e) Calcium levels in culture media during the bone resorption assay. Data are means ± SEM (n = 8 replicates). ***Indicates p < .001 (unpaired two‐tailed Student's t test).
CD34+ hematopoietic progenitor cells were purified from proband peripheral blood mononuclear cells and an umbilical cord blood control sample, followed by expansion in culture (Figure 2A and Figure S2). Osteoclast formation was then stimulated with RANKL. As expected for CLCN7‐mediated osteopetrosis (Coudert, de Vernejoul, Muraca, & Del Fattore, 2015; Villa, Guerrini, Cassani, Pangrazio, & Sobacchi, 2009), multi‐nucleated and tartrate‐resistant acid phosphatase (TRAP)‐positive osteoclasts were successfully formed in the proband sample, although they were larger than control osteoclasts (Figure 2B and Figure S3), a finding seen in other cases of osteopetrosis and suggesting they are functionally abnormal (Chen, Zhang, Hock, Wang, & Yu, 2016). Successful differentiation of proband osteoclasts was further confirmed by readily detectable TRAP activity in media conditioned by proband osteoclasts, albeit at reduced levels compared to control osteoclasts, consistent with a functional abnormality (Figure S4). Additionally, phalloidin staining of actin showed actin rings (specialized structures formed by osteoclasts) in both proband and control osteoclasts, again confirming that osteoclast differentiation is intact in proband cells (Figure S3).
To evaluate the function of proband‐derived osteoclasts, we performed bone resorption assays in which the osteoclasts are incubated with bone samples. Bone pits, a marker of bone resorption were completely absent from proband samples but prevalent in controls (Figure 2C). The bone resorption in this assay was then quantified using both carboxy‐terminal collagen I crosslinks (CTX‐I) and calcium levels. CTX‐I levels were negligible in the proband sample (Figure 2D) as has been demonstrated in a mouse model of Clcn7 osteopetrosis (Neutzsky‐Wulff, Karsdal, & Henriksen, 2008), indicating a significant, if not complete disruption, of the ability of proband osteoclasts to break down bone. Calcium levels in the culture media during this assay increased over time in both the proband and control samples (Figure 2E), however, the increase in the proband was more than twofold less than in the control, again confirming a defect in bone resorption. The observed increase in calcium in the proband sample versus the more striking absence of bone pits and low‐levels of CTX‐I indicates that the observed increase in calcium in the proband is likely a result of cellular calcium metabolism rather than bone resorption.
Altogether, the normal osteoclastogenesis, but abnormal pit formation, CTX‐I levels, and calcium levels in bone resorption assays are consistent with the genetic defect in CLCN7 identified by transcriptome sequencing. Although osteoclast formation is preserved in the proband, the loss of CLCN7 function suggested by our studies causes a significant disruption of the ability of osteoclasts to break down bone and the observed clinical phenotype.
4. DISCUSSION
Our study utilized RNA sequencing to identify a deep intronic noncoding variant as the cause of a severe osteopetrosis syndrome. To the best of our knowledge, this is the first report of a pathogenic deep intronic variant in CLCN7. Deep intronic variants have been reported previously as a cause of osteopetrosis in TCIRG1 (Palagano et al., 2015). Interestingly, some of these variants clustered in one deep intronic location, suggesting that some introns may be susceptible to disruption and appear as hotspots for pathogenic variants. Systematic application of transcriptome sequencing in undiagnosed osteopetroses will further delineate the architecture of noncoding variants important for proper function of the genes associated with the syndrome.
Despite our diagnostic success in this case, it is important to highlight an important limitation of RNA‐sequencing: the disease gene may not be expressed in an accessible tissue such as blood. Although 13 of 16 osteopetrosis and osteopetrosis‐related disorder genes are expressed in blood, the two genes most often associated with osteoclast‐poor osteopetrosis, TNFSF11 and TNFRSF11A (coding for RANKL and RANK, respectively), are not well expressed in blood. This suggests that transcriptome sequencing of blood may be particularly suitable for osteoclast‐rich, but not osteoclast‐poor osteopetrosis. Therefore, when genetic testing is nondiagnostic and bone biopsy is performed and consistent with osteoclast‐rich osteopetrosis, transcriptome sequencing should be considered. In the future, for diseases in which blood is not a good transcriptome surrogate for affected tissue, including osteoclast‐poor osteopetrosis, a possible approach may be to derive pluripotent stem cells from the patient followed by differentiation into the affected cell types.
Noncoding variants, in particular deep intronic variants, are difficult to interpret from DNA sequence alone (Chong et al., 2015; Vaz‐Drago, Custódio, & Carmo‐Fonseca, 2017; Zhang & Lupski, 2015). Although in this case we had a high suspicion for CLCN7 prior to transcriptome sequencing given the clinical phenotype and its presence in a region of homozygosity, DNA sequence analysis did not identify the pathogenic variant due to its low conservation and location deep inside intron 17. Notably, computational splicing prediction with SpliceAI, a recently‐developed machine‐learning approach (Jaganathan et al., 2019), performed concurrently with our transcriptome sequencing assigned this variant a score of 0.67—a moderate prediction for creating a novel splice acceptor site. This suggests that computational prediction of splice‐altering variants should be integrated with whole‐genome and transcriptome sequencing in studies of undiagnosed genetic diseases.
We emphasize, however, that despite the potential of computational prediction tools, transcriptome sequencing should remain part of a first‐line approach for undiagnosed diseases, because: (a) computational splicing prediction tools are unable to predict the full range of outcomes of predicted splice variants, for example, in our case the downstream pseudoexon splice donor site, or intron retention events (Jian, Boerwinkle, & Liu, 2014; Rowlands, Baralle, & Ellingford, 2019); (b) targeted validation of the numerous computational predictions would be uneconomical versus transcriptome sequencing; and, (c) transcriptome sequencing can reveal the impact of other types of rare noncoding pathogenic variants for which we lack good computational predictions (e.g., UTR, promoter, and enhancer variants), as well as complex structural variants (Cummings et al., 2017; Gonorazky et al., 2019). Transcriptome sequencing may also contain orthogonal evidence for a pathogenic variant, such as allelic imbalance, gene expression changes secondary to NMD, and dysregulated expression of genes in downstream pathways. Transcriptome sequencing is therefore a versatile adjunct to whole‐genome sequencing in facilitating the identification and interpretation of pathogenic noncoding variants.
An additional abnormality detected by RNA sequencing in our affected family was lower overall expression of CLCN7, which may be due to NMD. Bulk transcriptome sequencing, however, does not account for possible differences in the proportions of cell types in each sample. For example, osteopetrosis patients often have higher numbers of leukocytes (Sobacchi, Villa, Schulz, & Kornak, 2016). Future transcriptomic studies of undiagnosed diseases may be able to utilize single‐cell RNA‐sequencing in addition to bulk RNA sequencing to not only assess for differences in cell type proportions, but also provide data for normalizing bulk expression levels to reveal changes that would otherwise be masked by these differences.
An accurate genetic diagnosis can be especially important for the most severe forms of osteopetrosis, such as CLCN7 autosomal recessive osteopetrosis. HSCT was not performed for our proband initially due to her early onset of irreversible disease complications, systemic involvement including respiratory infections and chronic aspiration that would put her at risk for procedure‐related morbidity and mortality, her developmental delay that exceeded the expected effects of her vision and hearing loss, and the lack of a molecular‐genetic diagnosis. Despite our subsequent identification of the molecular‐genetic diagnosis, her severe disease course and uncertainty regarding possible primary neurodegeneration currently precludes HSCT. This points to the importance of future research to better delineate genotype‐phenotype correlations for CLCN7‐recessive osteopetrosis, especially in terms of identifying neurodegenerative forms that usually cannot be predicted from the genotype (Schulz et al., 2015; Sobacchi et al., 2016). Further clinical benefit of genetic diagnosis would also be aided by more rapid results. The total time from enrollment of the proband to reporting the genetic diagnosis was 5 months. In the future, it will be feasible to shorten this to as little as 1 month by use of rapid sequencing and by performing whole‐genome and transcriptome sequencing concurrently rather than sequentially, especially in cases in which molecular diagnosis may impact clinical management.
Beyond HSCT as a potential treatment for osteopetrosis, pathogenic pseudoexons are especially amenable to treatment by anti‐sense oligonucleotides (ASOs), including a recent notable case where a custom ASO treatment was developed for a patient with a neurodegenerative syndrome in less than one year (Kim et al., 2019). However, so far, clinical development of ASOs has been focused on central nervous system, kidney, and liver diseases due to challenges in delivering ASOs to other cell types (Geary, Norris, Yu, & Bennett, 2015; Juliano, 2016). Delivery to hematopoietic cells, the progenitors of osteoclasts, has been challenging and in vivo delivery to osteoclasts has not been demonstrated, although one in vitro study has successfully targeted osteoclast precursors (Canalis, Grossman, Carrer, Schilling, & Yu, 2020). The potential ASO side effect of thrombocytopenia would also be a concern in a patient with impaired hematopoiesis. Continued improvements in targeting ASOs to other cell types will expand their potential as curative treatments for other individuals with pathogenic pseudoexons, which pose a unique opportunity for ASO therapy. Achieving delivery of ASOs to osteoclasts with acceptable toxicity would enable life‐saving treatments for osteopetroses that are caused by splicing defects similar to those seen in our proband when HSCT is not an option.
This work adds to the growing body of evidence supporting transcriptomics as an important tool for diagnosing rare genetic diseases that were previously undiagnosed despite maximal utilization of DNA sequencing. We conclude that a reasonable path forward for rigorously testing the “noncoding variant hypothesis of undiagnosed diseases” is to systematically apply transcriptome sequencing to these cases. Our Undiagnosed Diseases Program, along with other similar programs (Splinter et al., 2018; Wright et al., 2015), have therefore set implementation of transcriptome sequencing as a key mission with the goal of working towards finding a diagnosis for every person with a rare genetic disease.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
N.Y. identified the proband and performed the clinical evaluation. O.C. and G.E. performed the genomic analysis, identified and characterized the pathogenic variant, and wrote the manuscript. K.M., I.M., and K.H. performed the osteoclastogenesis and osteoclast function assays. J.P. and G.E. managed and supervised the Undiagnosed Diseases Program.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the family for participating in this study, members of the NYU Undiagnosed Diseases Program (Dr. Aravinda Chakravarti, Dr. Heather Lau, Ellen Moran, and Kara Anstett), Dr. Oded Regev (NYU) and Dr. Ansgar Schulz (University Medical Center, Ulm, Germany) for helpful discussions, and the NYU Center for Biospecimen Research & Development. K.M. and K.H.’s work was supported by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No. 814244.
Chorin O, Yachelevich N, Mohamed K, et al. Transcriptome sequencing identifies a noncoding, deep intronic variant in CLCN7 causing autosomal recessive osteopetrosis. Mol Genet Genomic Med. 2020;8:e1405 10.1002/mgg3.1405
Odelia Chorin and Naomi Yachelevich contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Boycott, K. M. , Rath, A. , Chong, J. X. , Hartley, T. , Alkuraya, F. S. , Baynam, G. , … Lochmüller, H. (2017). International cooperation to enable the diagnosis of all rare genetic diseases. The American Journal of Human Genetics, 100(5), 695–705. 10.1016/j.ajhg.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis, E. , Grossman, T. R. , Carrer, M. , Schilling, L. , & Yu, J. (2020). Antisense oligonucleotides targeting Notch2 ameliorate the osteopenic phenotype in a mouse model of Hajdu Cheney syndrome. Journal of Biological Chemistry, 295, 3952–3964. 10.1074/jbc.RA119.011440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Zhang, K. , Hock, J. , Wang, C. , & Yu, X. (2016). Enhanced but hypofunctional osteoclastogenesis in an autosomal dominant osteopetrosis type II case carrying a c.1856C>T mutation in CLCN7. Bone Research, 4, 16035 10.1038/boneres.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, J. X. , Buckingham, K. J. , Jhangiani, S. N. , Boehm, C. , Sobreira, N. , Smith, J. D. , … Bamshad, M. J. (2015). The genetic basis of Mendelian phenotypes: Discoveries, challenges, and opportunities. The American Journal of Human Genetics, 97(2), 199–215. 10.1016/j.ajhg.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudert, A. E. , de Vernejoul, M.‐C. , Muraca, M. , & Del Fattore, A. (2015). Osteopetrosis and its relevance for the discovery of new functions associated with the skeleton. International Journal of Endocrinology, 2015, 372156 10.1155/2015/372156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, B. B. , Marshall, J. L. , Tukiainen, T. , Lek, M. , Donkervoort, S. , Foley, A. R. , … MacArthur, D. G. (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Science Translational Medicine, 9(386), eaal5209 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Fattore, A. , Cappariello, A. , & Teti, A. (2008). Genetics, pathogenesis and complications of osteopetrosis. Bone, 42(1), 19–29. 10.1016/j.bone.2007.08.029 [DOI] [PubMed] [Google Scholar]
- Dragojlovic, N. , Elliott, A. M. , Adam, S. , van Karnebeek, C. , Lehman, A. , Mwenifumbo, J. C. , … Lynd, L. D. (2018). The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: A benchmarking study. Genetics in Medicine, 20(9), 1013–1021. 10.1038/gim.2017.226 [DOI] [PubMed] [Google Scholar]
- Evrony, G. D. , Cordero, D. R. , Shen, J. , Partlow, J. N. , Yu, T. W. , Rodin, R. E. , … Walsh, C. A. (2017). Integrated genome and transcriptome sequencing identifies a noncoding mutation in the genome replication factor DONSON as the cause of microcephaly‐micromelia syndrome. Genome Research, 27, 1323–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary, R. S. , Norris, D. , Yu, R. , & Bennett, C. F. (2015). Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Advanced Drug Delivery Reviews, 87, 46–51. 10.1016/j.addr.2015.01.008 [DOI] [PubMed] [Google Scholar]
- Gloss, B. S. , & Dinger, M. E. (2018). Realizing the significance of noncoding functionality in clinical genomics. Experimental & Molecular Medicine, 50(8), 97 10.1038/s12276-018-0087-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonorazky, H. D. , Naumenko, S. , Ramani, A. K. , Nelakuditi, V. , Mashouri, P. , Wang, P. , … Dowling, J. J. (2019). Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. The American Journal of Human Genetics, 104(3), 466–483. 10.1016/j.ajhg.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan, K. , Kyriazopoulou Panagiotopoulou, S. , McRae, J. F. , Darbandi, S. F. , Knowles, D. , Li, Y. I. , … Farh, K.‐H. (2019). Predicting splicing from primary sequence with deep learning. Cell, 176(3), 535–548.e524. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Jian, X. , Boerwinkle, E. , & Liu, X. (2014). In silico tools for splicing defect prediction: A survey from the viewpoint of end users. Genetics in Medicine, 16(7), 497–503. 10.1038/gim.2013.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano, R. L. (2016). The delivery of therapeutic oligonucleotides. Nucleic Acids Research, 44(14), 6518–6548. 10.1093/nar/gkw236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Hu, C. , Moufawad El Achkar, C. , Black, L. E. , Douville, J. , Larson, A. , … Yu, T. W. (2019). Patient‐customized oligonucleotide therapy for a rare genetic disease. New England Journal of Medicine, 381(17), 1644–1652. 10.1056/NEJMoa1813279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer, L. S. , Wortmann, S. B. , & Prokisch, H. (2018). “Transcriptomics”: Molecular diagnosis of inborn errors of metabolism via RNA‐sequencing. Journal of Inherited Metabolic Disease, 41(3), 525–532. 10.1007/s10545-017-0133-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur, D. G. , Manolio, T. A. , Dimmock, D. P. , Rehm, H. L. , Shendure, J. , Abecasis, G. R. , … Gunter, C. (2014). Guidelines for investigating causality of sequence variants in human disease. Nature, 508(7497), 469–476. 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele, M. , Ferreira, P. G. , Reverter, F. , DeLuca, D. S. , Monlong, J. , Sammeth, M. , … Guigo, R. (2015). The human transcriptome across tissues and individuals. Science, 348(6235), 660 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzsky‐Wulff, A. V. , Karsdal, M. A. , & Henriksen, K. (2008). Characterization of the bone phenotype in ClC‐7‐deficient mice. Calcified Tissue International, 83(6), 425 10.1007/s00223-008-9185-7 [DOI] [PubMed] [Google Scholar]
- Palagano, E. , Blair, H. C. , Pangrazio, A. , Tourkova, I. , Strina, D. , Angius, A. , … Sobacchi, C. (2015). Buried in the middle but guilty: Intronic mutations in the TCIRG1 gene cause human autosomal recessive osteopetrosis. Journal of Bone and Mineral Research, 30(10), 1814–1821. 10.1002/jbmr.2517 [DOI] [PubMed] [Google Scholar]
- Palagano, E. , Menale, C. , Sobacchi, C. , & Villa, A. (2018). Genetics of osteopetrosis. Current Osteoporosis Reports, 16(1), 13–25. 10.1007/s11914-018-0415-2 [DOI] [PubMed] [Google Scholar]
- Palagano, E. , Susani, L. , Menale, C. , Ramenghi, U. , Berger, M. , Uva, P. , … Sobacchi, C. (2017). Synonymous mutations add a layer of complexity in the diagnosis of human osteopetrosis. Journal of Bone and Mineral Research, 32(1), 99–105. 10.1002/jbmr.2929 [DOI] [PubMed] [Google Scholar]
- Pangrazio, A. , Pusch, M. , Caldana, E. , Frattini, A. , Lanino, E. , Tamhankar, P. M. , … Sobacchi, C. (2010). Molecular and clinical heterogeneity in CLCN7‐dependent osteopetrosis: Report of 20 novel mutations. Human Mutation, 31(1), E1071–E1080. 10.1002/humu.21167 [DOI] [PubMed] [Google Scholar]
- Penna, S. , Capo, V. , Palagano, E. , Sobacchi, C. , & Villa, A. (2019). One disease, many genes: Implications for the treatment of osteopetroses. Frontiers in Endocrinology, 10, 85 10.3389/fendo.2019.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2018). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47, D886–D894. 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer, K. , Juusola, J. , Cho, M. T. , Vitazka, P. , Millan, F. , Gibellini, F. , … Bale, S. (2015). Clinical application of whole‐exome sequencing across clinical indications. Genetics in Medicine, 18, 696 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Rowlands, F. C. , Baralle, D. , & Ellingford, M. J. (2019). Machine learning approaches for the prioritization of genomic variants impacting pre‐mRNA splicing. Cells, 8(12), 1513 10.3390/cells8121513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz, M. , Steward, C. G. , Villa, A. , & Sobacchi, C. (2015).Osteopetrosis: Consensus guidelines for diagnosis, therapy and follow‐up. European Society for Immunodeficiencies. Retrieved from https://esid.org/content/download/14303/398344/file/00_OP_Guidelines_V3.pdf
- Sobacchi, C. , Schulz, A. , Coxon, F. P. , Villa, A. , & Helfrich, M. H. (2013). Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nature Reviews Endocrinology, 9(9), 522–536. 10.1038/nrendo.2013.137 [DOI] [PubMed] [Google Scholar]
- Sobacchi, C. , Villa, A. , Schulz, A. , & Kornak, U. (2016). CLCN7‐related osteopetrosis In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., & Amemiya A. (Eds.), GeneReviews. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Splinter, K. , Adams, D. R. , Bacino, C. A. , Bellen, H. J. , Bernstein, J. A. , Cheatle‐Jarvela, A. M. , … Ashley, E. A. (2018). Effect of genetic diagnosis on patients with previously undiagnosed disease. New England Journal of Medicine, 379, 2131–2139. 10.1056/NEJMoa1714458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, Z. , & Savarirayan, R. (2009). Osteopetrosis. Orphanet Journal of Rare Diseases, 4(1), 5 10.1186/1750-1172-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz‐Drago, R. , Custódio, N. , & Carmo‐Fonseca, M. (2017). Deep intronic mutations and human disease. Human Genetics, 136(9), 1093–1111. 10.1007/s00439-017-1809-4 [DOI] [PubMed] [Google Scholar]
- Villa, A. , Guerrini, M. M. , Cassani, B. , Pangrazio, A. , & Sobacchi, C. (2009). Infantile malignant, autosomal recessive osteopetrosis: The rich and the poor. Calcified Tissue International, 84(1), 1–12. 10.1007/s00223-008-9196-4 [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , Fitzgerald, T. W. , Jones, W. D. , Clayton, S. , McRae, J. F. , van Kogelenberg, M. , … Firth, H. V. (2015). Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome‐wide research data. The Lancet, 385(9975), 1305–1314. 10.1016/S0140-6736(14)61705-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. C. , Econs, M. J. , DiMeglio, L. A. , Insogna, K. L. , Levine, M. A. , Orchard, P. J. , … Polgreen, L. E. (2017). Diagnosis and management of osteopetrosis: Consensus guidelines from the osteopetrosis working group. Journal of Clinical Endocrinology and Metabolism, 102(9), 3111–3123. 10.1210/jc.2017-01127 [DOI] [PubMed] [Google Scholar]
- Zhang, F. , & Lupski, J. R. (2015). Non‐coding genetic variants in human disease. Human Molecular Genetics, 24(R1), R102–R110. 10.1093/hmg/ddv259 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.