

ABSTRACT
Yes-associated protein 1 (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) (YAP/TAZ) are transcriptional coactivators that regulate genes involved in proliferation and transformation by interacting with DNA-binding transcription factors. Remarkably, YAP/TAZ are essential for cancer initiation or growth of most solid tumors. Their activation induces cancer stem cell attributes, proliferation, and metastasis. The oncogenic activity of YAP/TAZ is inhibited by the Hippo cascade, an evolutionarily conserved pathway that is governed by two kinases, mammalian Ste20-like kinases 1/2 (MST1/2) and Large tumor suppressor kinase 1/2 (LATS1/2), corresponding to Drosophila's Hippo (Hpo) and Warts (Wts), respectively. One of the most influential aspects of YAP/TAZ biology is that these factors are transducers of cell structural features, including polarity, shape, and cytoskeletal organization. In turn, these features are intimately related to the cell's ability to attach to other cells and to the surrounding extracellular matrix (ECM), and are also influenced by the cell's microenvironment. Thus, YAP/TAZ respond to changes that occur at the level of whole tissues. Notably, small GTPases act as master organizers of the actin cytoskeleton. Recent studies provided convincing genetic evidence that small GTPase signaling pathways activate YAP/TAZ, while the Hippo pathway inhibits them. Biochemical studies showed that small GTPases facilitate the YAP-Tea domain transcription factor (TEAD) interaction by inhibiting YAP phosphorylation in response to serum stimulation, while the Hippo pathway facilitates the YAP-RUNX3 interaction by increasing YAP phosphorylation. Therefore, small GTPase pathways activate YAP/TAZ by switching its DNA-binding transcription factors. In this review, we summarize the relationship between the Hippo pathway and small GTPase pathways in the regulation of YAP/TAZ.
KEYWORDS: YAP, TAZ, Hippo, Rho, Rac, Cdc42, Ras
Inhibition of YAP/TAZ by the Hippo pathway
YAP contains two consecutive WW domains and was originally identified as a Yes-associated protein [1]. TAZ is a YAP paralog in mammals and displays a similar domain organization, but has only one WW domain. The schematic depiction of YAP, TAZ, and their Drosophila homolog Yorkie (Yki) is illustrated in Figure 1. The biological function of YAP as a transcriptional coactivator was first demonstrated by the observation that YAP interacts with Runt related transcription factor 2 (RUNX2)/PEBP2αA and strongly enhances RUNX2-mediated transactivation activity [2]. TAZ was also shown to function as a transcriptional coactivator of RUNX2 [3]. The important biological function of YAP was highlighted 10 years later when Yki was identified as an endpoint effector of the Hippo pathway in flies. The Hippo pathway includes Hpo and Wts kinases, which inhibit cell proliferation and activate apoptosis. Interestingly, Yki, a homolog of YAP, was identified as a Wts-interacting protein by yeast two-hybrid screening [4]. Functionally, the overexpression of Yki results in massive tissue overgrowth (increase in cell number), a phenocopy of the loss of function of Hpo or Wts. Mechanistically, Wts phosphorylates and inactivates Yki by sequestering the protein in the cytoplasm. Thus, the Hippo core components can negatively regulate Yki by phosphorylation. Importantly, the components of the Hippo pathway are structurally conserved in mammals [5]. MST1/2 and LATS1/2 are mammalian homologs of fly Hpo and Wts, respectively (Figure 2). Therefore, when Drosophila Yki was identified in 2005 as the downstream target of the Hippo tumor suppressor [4], studies on YAP shed new light on the context of the potentially conserved Hippo pathway in mammalian cells and its cellular and physiological role in cell proliferation and apoptosis. Soon after the identification of Yki, several landmark studies established the paradigm of the conserved Hippo pathway as the major signaling cascade regulating organ size in vivo and cell contact inhibition of proliferation in vitro [6]. It was concluded that the cell-to-cell interactions upon confluence trigger the Hippo pathway, resulting in the phosphorylation of Yki/YAP, and leading to cytoplasmic sequestration.
Figure 1.
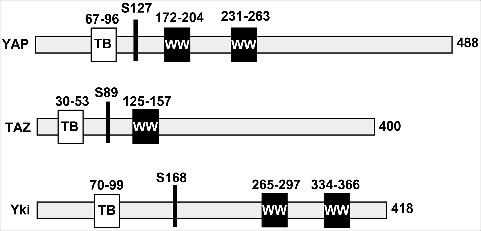
The schematic drawing of YAP, TAZ, and Yki. The key features of YAP, TAZ, and Yki are indicated (e.g., WW domain (WW) and TEAD-binding region (TB)). The major Hippo target sites where phosphorylation creates a binding site for 14–3-3 proteins are also highlighted (S127 of YAP, S89 of TAZ, and S168 of Yki).
Figure 2.
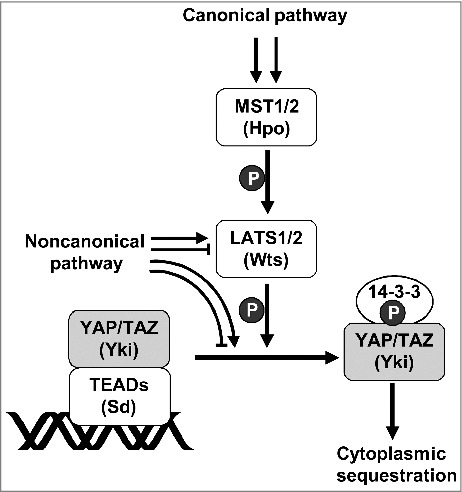
Hippo signaling pathway. MST1/2 (Hpo in Drosophila) phosphorylates LATS1/2 (Wts in Drosophila) and activates the kinase activity of LATS1/2. The activated LATS1/2 phosphorylates YAP/TAZ (Yki in Drosophila). The phosphorylated YAP/TAZ are released from their DNA-binding partners, TEADs (Sd in Drosophila), and exported to the cytoplasm through the interaction with 14–3-3. Thereby, the Hippo pathway inactivates the oncogenic activity of YAP/TAZ. MST1/2 can be activated by multiple pathways. The MST1/2 -> LATS1/2 -> YAP/TAZ pathway is known as the canonical Hippo signaling pathway. LATS1/2 can be regulated by a MST1/2-independent mechanism. YAY/TAZ can be phosphorylated by various kinases other than LATS1/2. The pathways regulating YAP/TAZ in a MST1/2-independent manner are known as non-canonical pathways.
However, the role of RUNX proteins (Lozenge (Lz) in Drosophila) as DNA-binding partners of YAP in the Hippo pathway was unclear. Instead, the TEAD family DNA-binding transcription factors were shown to interact with YAP [7]. Several recent studies demonstrated that TEADs in mammals and Scalloped (Sd) in the fly are major transcriptional factors mediating the biological outcome of YAP and TAZ (YAP/TAZ) and Yki, respectively, that are governed by the Hippo pathway. For example, TEADs are essential in mediating YAP-dependent gene expression and functional outcomes, including cell growth, oncogenic transformation, and epithelial-to-mesenchymal transition (EMT) [8]. Similarly, a Yki mutant that is not capable of interacting with Sd was unable to induce tissue overgrowth in vivo [9]. These results suggested that the oncogenic activities of YAP/TAZ are dependent on their interaction with TEADs and that their activities are inhibited by the Hippo pathway (Figure 2). A recent integrative analysis, using epigenomic and transcriptomic approaches, revealed that TEAD4 is the major transcription factor through which YAP drives cell proliferation and EMT [8].
Mechanisms for the activation of YAP/TAZ
For years, LATS1/2-mediated phosphorylation was the only known mechanism that regulates YAP/TAZ. This led to the assumption that inactivation of the Hippo pathway may be a major mechanism for the activation of YAP/TAZ in cancer [10, 11]. However, Hippo pathway mutations are extremely rare in human tumors [12]. In fact, inactivation of LATS1/2 is inconsequential, or only partially relevant for YAP/TAZ regulation. In addition, inactivation of Hippo pathway components in mice ultimately leads to tumor development but requires a longer latency than in mice overexpressing YAP [13]. These results suggest that inactivation of the Hippo pathway is not sufficient to induce tumorigenesis without additional YAP activity. Thus, it appears that the Hippo pathway is not the only mechanism that regulates YAP/TAZ. These regulatory elements include cellular mechano-transduction, RHO-GTPase signaling, inflammation, metabolism, and, to a certain extent, contact inhibition and G protein-coupled receptor (GPCR) signaling.
Activation of YAP/TAZ by RHO family small GTPases
The RHO family of GTPases, a subfamily of the RAS superfamily, consists of small (∼21 kDa) G proteins. RAS homolog family member A (RHOA), RAS-related C3 botulinum toxin substrate 1 (RAC1), and Cell division cycle 42 (CDC42) are the most well-characterized and are established master regulators of cytoskeletal dynamics and morphology [14, 15]. RHO-GTPases are overexpressed in numerous cancers, including breast, lung, colon, head and neck, bladder, and gastric tumors [16]. A prominent feature of the Hippo pathway is that stress fibers (F-actin) are increased at low cell densities and inhibit the Hippo pathway, thereby reducing YAP phosphorylation and promoting nuclear YAP accumulation [17]. Therefore, small GTPase signaling governs actin cytoskeleton dynamics and affects the Hippo pathway, suggesting cross-talk between small GTPase signaling and the Hippo pathway.
Ridley and Hall reported that activation of RHOA by GTP binding induces the formation of F-actin stress fibers [18]. It is now well known that RHO/RAC stimulate RHO-associated protein kinase (ROCK) and p21-activated kinase (PAK), which induce LIM kinase-1 (LIMK) activity and inactivate cofilin, resulting in F-actin accumulation [19]. F-actin sequesters angiomotin (AMOT), an inhibitor of YAP, and thereby promotes YAP activation and nuclear translocation [20]. Attachment of cells to ECM induces YAP activation and nuclear translocation through RHOA-mediated F-actin accumulation [21]. Activation of YAP/TAZ by RHO-mediated F-actin dynamics is required for survival of human embryonic stem cells (hESC) [22]. Gαq also promotes the YAP-dependent growth of uveal melanoma cells through RHO/RAC-mediated F-actin accumulation [23]. RHOA signaling is also involved in activation of Sphingosine-1-phosphatate (S1P)-mediated YAP and myocardin-related transcription factor A (MRTF-A). MRTF and YAP/TAZ are major mechanosensitive transcriptional co-activators that link cytoskeleton organization to gene expression [24]. Activation of MRTF-A and YAP regulates RHOA-mediated CCN1 (also known as CYR61) expression [25]. Serum starvation of cells induces cytoplasmic localization of MRTF-A and YAP, and S1P treatment leads to increased nuclear accumulation of MRTF-A and YAP [25].
Cell mechanics and the status of the cytoskeleton represent a central mechanism in the control of YAP/TAZ activity. A rigid ECM maintains active YAP/TAZ in the nucleus, while more elastic matrices lead to YAP/TAZ inactivation. This regulation requires RHO GTPase activity, but appears to be independent of the Hippo/LATS pathway, because depletion of LATS1/2 does not block YAP and TAZ regulation in cells cultured in soft gels [26]. However, two studies reported that RHO-mediated F-actin accumulation induces YAP activity through inhibition of LATS kinase activity [17, 21]. Lysophosphatidic acid (LPA) and S1P induce G12/13-mediated RHO activation, and then activate YAP/TAZ through inhibition of LATS1/2 kinase activity [27]. Also, cyclic stretch induces YAP activity through JNK-mediated binding of LIM domains containing 1 [LIMD1 (LATS inhibitor)] to LATS1 [28]. These reports show that RHO signaling regulates YAP activity in a LATS-dependent manner. Therefore, RHO signaling regulates YAP activity via LATS-dependent and -independent mechanisms.
A large body of data implicates RHO-GTPases in the pathogenesis of kidney disease. Kidney podocytes are highly specialized, terminally differentiated cells that form the final barrier to urinary protein loss. Scott et al. determined that the deletion of Cdc42, but not of Rac1 or RhoA, in mouse podocytes results in congenital nephrotic syndrome and glomerulosclerosis [29]. Blattner et al. also showed that Cdc42 is necessary and Rac1 is dispensable for the maintenance of podocyte structure and function [30]. Notably, the deletion of Yap or Cdc42 in mouse podocytes results in very similar defects [31]. Histologically, both mutant mice exhibited features characteristic of focal segmental glomerulosclerosis (FSGS), which is the most common primary glomerular disease [31]. In human primary FSGS, Cdc42 expression [32] and Yap expression [31] are markedly lower than in normal glomerular podocytes. These results suggest that Cdc42 and Yap are involved in nephrogenesis and that Cdc42 may be an upstream regulator of Yap. This possibility was further investigated by Reginensi et al. They found that the deletion of either Cdc42 or Yap in cap mesenchyme cells, the precursors of podocytes, causes essentially the same defects as those found in nephrogenesis [33]. Mechanistically, the deletion of Cdc42 reduces the nuclear localization of Yap, leading to lower levels of Yap-dependent gene expression. These results provide strong evidence that Yap is a downstream target of Cdc42 signaling in nephron progenitor cells.
These findings prompt the question of how Cdc42 regulates the nuclear localization of Yap. When podocytes are injured, Cdc42 and its downstream effector, Neural Wiskott-Aldrich syndrome protein (Nwasp), are downregulated. Subsequently, the loss of stress fibers, caused by Cdc42/Nwasp deficiency, results in lower Yap levels and podocyte apoptosis [32]. Importantly, the level of stress fibers is regarded as a regulator of Yap [26, 33]. Therefore, the Cdc42-Nwasp-stress fiber signal pathway may play a central role in regulating Yap activity and podocyte apoptosis.
These studies indicated that YAP/TAZ are critical mediators of fibrosis in podocytes. Fibrosis, which is defined as the excessive accumulation of extracellular matrix, is a common pathological process in soft tissues and organs, including not only kidney but also lung, liver, and skin [34–39]. The activation of YAP and TAZ was recently found to be involved in the induction of fibrosis in lung and liver. In idiopathic pulmonary fibrosis, both YAP and TAZ levels are elevated, and display a predominantly nuclear localization [36]. Moreover, YAP and TAZ knockdown in mouse lung and liver fibroblasts reduces the levels of proteins associated with myofibroblast differentiation [36]. In addition, mice heterozygous for TAZ show a remarkable resilience to bleomycin-induced pulmonary fibrosis [40].
Activation of YAP/TAZ by GPCRs
GPCRs recognize numerous extracellular signals and transduce them to heterotrimeric G proteins that further transmit these intracellular signals to the appropriate downstream effectors, and thereby play a main role in various signaling pathways [41]. An important discovery came about with the demonstration that soluble ligands signaling through GPCR and RHO can promote YAP/TAZ nuclear localization and transcriptional activities [27]. Mechanistically, LPA and S1P trigger Gα12/13- and Gαq/11-coupled GPCRs to activate RHO-GTPases. Activation of RHO-GTPases serves as a key mediator in the activation of YAP/TAZ from upstream GPCRs. The role of GPCR-RHO as an upstream regulator of YAP/TAZ was further confirmed by the observations that YAP mediates the oncogenic activity of mutant Gαq/11 in the development of uveal melanoma and that the YAP inhibitor verteporfin blocks the growth of uveal melanoma tumor cells harboring Gαq/11 mutations [42]. In addition, the genes encoding Gαq and Gα11 are frequently mutated in uveal melanoma [42]. Stimulation of GPCRs by LPA and S1P treatment induces the expression of the YAP target genes CTGF, CYR61, and ankyrin repeat domain 1 (ANKRD1) [42].
Recent studies show that the overexpression of oncogenic Gαq also promotes nuclear localization of YAP in cell culture and in human tumors. The mechanism by which oncogenic Gαq activates YAP is unclear. One study purported that Gαq ultimately affects LATS-mediated phosphorylation of YAP [27], while another suggested that Gαq promotes changes in AMOT in the cytoskeleton independently of LATS [23]. Therefore, GPCR-activated Gαq-, Gα12/13-, and Gαi/o-coupled receptors typically stimulate cell proliferation, similar to the function of activated YAP/TAZ [43]. However, GPCR signaling does not always activate YAP/TAZ. While Gαq/11-, Gα12/13-, and Gαi/o-coupled receptors can inhibit LATS1/2 kinase activity, Gαs-coupled receptors can activate LATS1/2 kinase ([Fig 3).27] Gαs signaling can induce YAP/TAZ phosphorylation mainly via cyclic adenosine phosphate (cAMP) and protein kinase A (PKA).
Figure 3.
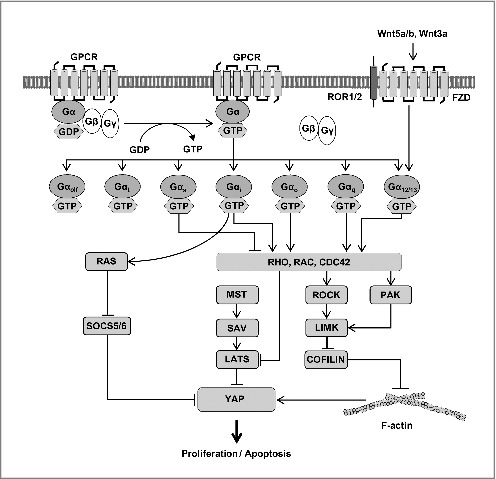
Regulation of YAP activity by GPCR and WNT-RHO-GTPase signaling. Involvement of small GTPases in the regulation of YAP/TAZ is summarized. When GPCR is activated, GTP-bound Gα proteins activate YAP through RHO-GTPase and RAS-GTPase and promote several cellular responses. Gαi, Gαo, Gαq, and Gα12/13 activate YAP by inhibiting LATS1/2 kinase activity and stabilizing F-actin. On the other hand, Gαs inhibits YAP activity. RHO-GTPases can stabilize F-actin through the regulation of the ROCK-LMK-COFILIN pathway and PAK. RAS activates YAP by inhibiting SOCS5/6. The WNT-FZD-ROR1/2 pathway can activate YAP through Gα12/13.
Connection between the RAS pathway and the Hippo pathway
Mammals possess three RAS genes (H-RAS, N-RAS, and K-RAS) that encode proteins that cycle between the inactive GDP-bound and the active GTP-bound forms [44, 45]. RAS mutations are found in more than 30% of human tumors, with K-RAS mutations accounting for ∼85% of all RAS mutations in human cancers [46, 47]. Although active RAS mutants are able to transform nearly all immortalized cell lines, they are usually unable to transform normal primary cells, except in the presence of a cooperating oncogene or in association with the loss of certain tumor suppressor genes [48]. Introduction of oncogenic Ras into primary mouse cells generally results in cell cycle arrest mediated by higher levels of a variety of cyclin-dependent kinase inhibitors, or in apoptosis [49]. Subsequent studies revealed that oncogenic Ras strongly stimulates the activation of p53 through the induction of p19Arf (p14ARF in humans), which represses the p53 inhibitor Mdm2 [50, 51]. The existence of additional p53-independent pathways in Ras-induced apoptosis was also indicated by the ability of RasG12V to promote apoptosis when the Ras-induced increase in NFκb activity is suppressed in p53−/− mouse embryo fibroblasts [52]. Thus, multiple pathways exist for Ras-induced apoptosis as a defense mechanism against transformation.
Khokhlatchev et al. identified an additional p53-independent pathway, whereby active K-RAS initiates apoptosis through the direct recruitment of its putative effector NORE, which is stably associated with the proapoptotic MST1 [53]. Subsequent studies revealed that oncogenic K-RAS signaling is linked to the Hippo pathway in two distinct ways. First, oncogenic RAS signaling promotes the association of MST2 with RASSF1A (heterodimerizing with NORE) and LATS1. This induces the phosphorylation of YAP1, allowing its translocation to the nucleus and binding to p73, resulting in transcription of the proapoptotic target gene p53 upregulated modulator of apoptosis (PUMA) [54]. These results reveal a new pathway (oncogenic K-RAS → RASSF1A → MST1/2 → LATS1/2 → YAP-p73 → PUMA) and demonstrate an additional connection between the RAS and the Hippo pathways. The surprising finding that LATS-dependent YAP phosphorylation can promote YAP translocation to the nucleus (and binding to p73) and thereby cause activation of gene expression, is opposite to the classic role of LATS-dependent phosphorylation, which keeps YAP out of the nucleus. Several studies support the observation that phosphorylated YAP is localized to the nucleus under some conditions [7, 17, 55]. Second, LATS1 binds to and sequesters the ubiquitin ligase MDM2, leading to the stabilization of the tumor suppressor p53 and the induction of apoptosis [56]. Similarly, Aylon et al. reported that LATS2 is required for the p53-dependent oncogenic stress checkpoint [57]. These findings indicate the involvement of additional distinct pathways (oncogenic K-RAS → RASSF1A → MST1/2 → LATS1/2 → MDM2 → p53 pathway). Altogether, these results suggest connections between the RAS pathway and the Hippo pathway.
Recently, another relationship between oncogenic K-RAS and YAP was identified. In almost all cases of oncogene-induced tumors, resistance to cancer therapy and tumor relapse occur. Since somatic K-RAS mutations are found in the most common activating lesions in humans, including pancreatic, lung, and colon cancer [47], the identification of potential resistance mechanisms is of great therapeutic relevance. Kapoor et al. and Shao et al. examined the long-term effects of the shutdown of oncogenic K-Ras in established pancreatic cancer and lung cancer in mice [58, 59]. All tumors initially regressed but began to relapse after about 2 weeks. Notably, Yap was induced in relapsed mouse tumors and enabled their maintenance after the suppression of oncogenic K-Ras. These studies identified Yap as a central driver of compensation for the loss of K-Ras signaling in K-Ras-dependent cancer.
Activation of YAP/TAZ by WNT signaling
Wnt5a and Wnt3a signals are transduced through the Frizzled (FZD)/ receptor tyrosine kinase like orphan receptor 1 (ROR) pathway [60]. The FZD family receptors and the ROR1/2 co-receptors promote the activation of several alternative (non-canonical) Wnt signaling pathways, while the LRP5/6 co-receptors are linked to the activation of the canonical Wnt signaling pathway. Konsavage et al. demonstrated that Wnt signaling promotes cancer cell proliferation through the upregulation of YAP [61, 62]. Subsequent studies revealed that WNT-FZD/ROR signaling inhibits LATS1/2 kinase activity via Gα12/13-RHO and then induces YAP/TAZ activation ([Fig 3).62, 63]
Interestingly, the activation of YAP/TAZ inhibits canonical WNT-β-catenin signaling. For example, WNT pathway inhibitors [Dickkopf 1 (DKK1), Bone morphogenetic protein 4 (BMP4), and Insulin like growth factor binding protein 4 (IGFBP4)] are the major target genes of YAP/TAZ-TEAD [63]. Previously, Li and Iyengar suggested that the Gαq pathway represses canonical WNT-β-catenin signaling [64]. Taken altogether, these results suggest that YAP/TAZ activated by the alternative WNT pathway, induces WNT inhibitors, thereby inhibiting canonical WNT-β-catenin signaling. YAP/TAZ would mediate the biological functions of alternative WNT signaling, including gene expression, osteogenic differentiation, and cell migration [64].
Reciprocal regulation of YAP by RAC signaling and the Hippo pathway
The RUNX family of transcription factors plays a pivotal role in normal development and neoplasia [65]. RUNX1 is essential for hematopoiesis and is often mutated in leukemia. Likewise, RUNX2 is required for osteogenesis, and mutations in this gene are associated with bone disease [65]. RUNX3 is a well-documented tumor suppressor that is frequently inactivated by DNA hyper-methylation in various kinds of cancers [65]. The identification of YAP/TAZ as transcriptional coactivators was based on the observation that YAP/TAZ dramatically enhances RUNX2-mediated transcription [2, 3]. However, the YAP/TAZ-RUNX interaction was not further investigated for a long time, most likely due to the fixed concept that YAP/TAZ stimulates proliferation while RUNX family members stimulate differentiation. Recent studies highlight that RUNX3 is another important DNA-binding partner of YAP-TAZ. Qiao et al. and Jang et al. uncovered a tripartite signaling complex of RUNX3, TEAD, and YAP, in which a direct interaction between RUNX3 and TEAD reduces the affinity of TEAD for DNA and thereby inhibits the proliferative effects of the TEAD-YAP complex [7, 66]. For example, introduction of RUNX3 resulted in lower expression of CTGF and CYR61, which are known as targets of TEAD-YAP complex associated with cell growth and transformation [66].
In a separate study, Jang et al. performed a large-scale functional genetic screening to elucidate the genetic modifiers of RUNX expression using Lz, a Drosophila homolog of mammalian RUNX family members [7]. The screen revealed genetic interactions between the Lz, Rac, and Hippo pathways. An analysis of the gene interactions revealed that the defective phenotype resulting from oncogenic Yki was markedly suppressed by Lz and enhanced by Rac-Triple functional domain protein (Trio) [ 7]. Molecular analysis using mammalian homologs revealed that, in serum-deprived or high cell density conditions, LATS1/2-mediated YAP phosphorylation facilitated the dissociation of the YAP-TEAD4 complex and the formation of the YAP-RUNX3 complex. When cells were stimulated to proliferate by serum deprivation followed by serum treatment, activated RAC-TRIO signaling inhibited LATS1/2-mediated YAP phosphorylation. Consequently, YAP dissociated from RUNX3 and associated with TEAD, thereby replacing the YAP-RUNX3 complex with YAP-TEAD. RUNX3 contributed to both the formation and dissociation of the YAP-TEAD complex, via the formation of a YAP-TEAD-RUNX3 ternary complex. Therefore, RAC and Hippo signaling regulate YAP activity by exchanging the YAP-binding partners TEAD4 and RUNX3 [7]. These results identified a novel regulatory mechanism mediated by the Hippo and RAC-TRIO pathways that switches the binding partner of YAP (Figure 4).
Figure 4.
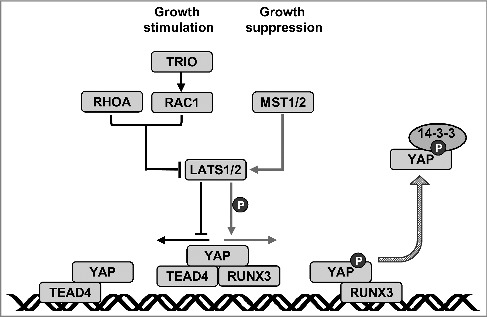
Reciprocal regulation of YAP activity by RHO signaling and the Hippo pathway. YAP, TEAD, and RUNX3 form a ternary complex that does not interact with the TEAD- binding sites. When RAC-TRIO signaling is activated, LATS1/2 is inhibited and YAP phosphorylation is decreased. Then, RUNX3 dissociates from the ternary complex, resulting in a YAP-TEAD complex which binds to TEAD-binding sites. When the Hippo pathway is activated, LATS1/2 is activated and YAP phosphorylation is increased. Then, TEAD dissociates from the ternary complex to form a YAP-RUNX3 complex. Therefore, Hippo pathway-mediated YAP phosphorylation not only inhibits YAP-TEAD complex formation but also facilitates YAP-RUNX3 complex formation.
RUNX3 is downregulated in gastric cancers by hyper-activated YAP-TEAD, and the ectopic expression of RUNX3 suppresses the growth of these tumors [7, 66]. Thus, a failure of YAP-RUNX3 complex formation is expected to be associated with gastric tumorigenesis. Since the Hippo pathway effectively inactivates YAP, the role of the YAP-RUNX3 complex in tumorigenesis is likely not limited by YAP inactivation. If phosphorylated YAP is simply an inactive form of YAP, the interaction between inactivated YAP and RUNX3 would be dispensable. However, ectopic expression of RUNX3 in gastric cancer cells reduces tumorigenicity, and the tumor-suppressive activity of RUNX3 is dependent on its ability to interact with YAP in the nucleus [7, 66]. In addition, the phospho-YAP/RUNX3 complex is detected only in the nucleus [7]. Therefore, phosphorylated YAP may serve another function by exchanging TEAD for RUNX3 before it is exported to the cytoplasm. For example, when proliferating cells stop growing, they must turn on genes that are required for cell cycle arrest and turn off growth-related genes. The switch between the growth-promoting YAP-TEAD4 complex and the growth-suppressing YAP-RUNX3 complex may alter the pattern of gene expression. This interpretation is supported by the observation that YAP activates RUNX-mediated transcription of Osteocalcin [2] and Itch [67]. Additional studies are required to elucidate the role of the YAP-RUNX3 complex in cell cycle regulation.
Prospects
The Hippo kinase cascade has long been considered a major regulator of YAP/TAZ via its ability to phosphorylate YAP/TAZ and inhibit their activities. This idea was based on the assumption that the default activity of YAP/TAZ is oncogenic. However, recent studies have identified novel upstream regulatory elements that activate YAP/TAZ, including RHO, RAC, CDC42, and RAS signaling. Therefore, YAP/TAZ activities can be activated or inactivated by multiple pathways. Furthermore, the assumption that the default role of YAP/TAZ is oncogenic should also be reconsidered. Although YAP/TAZ interacts with various DNA-binding transcription factors, as well as with TEADs, only TEADs are considered to be the major partners of YAP/TAZ. This is mainly due to the assumption that YAP/TAZ most often plays an oncogenic role. Recent studies report that RUNX3 interacts with phosphorylated YAP in the nucleus and that the tumor suppressor activity of RUNX3 is enhanced by this interaction. These findings challenge the paradigm that YAP/TAZ coactivators are oncogenes and that TEADs are the major partners of these coactivators. YAP/TAZ were originally identified as coactivators of RUNX2. As the identification of other factors (small GTPases) that activate YAP/TAZ challenged the idea that the Hippo pathway is the major regulator of YAP/TAZ, future studies focusing on the activities of coactivators with various DNA-binding partners will further contribute to knowledge in this field.
Acknowledgments
Suk-Chul Bae is supported by a Creative Research Grant (2014R1A3A2030690) through the National Research Foundation (NRF) of Korea. Ju-Won Jang is supported by the Basic Science Research Program (2014R1A1A2009492) through the NRF funded by the Ministry of Education of Korea. Min-Kyu Kim is supported by the Basic Science Research Program (2017R1A6A3A11028050) through the NRF funded by the Ministry of Education of Korea.
Funding Statement
This work was supported by the National Research Foundation of South Korea (NRF-2014R1A3A2030690), National Research Foundation of South Korea (NRF-2017R1A6A3A11028050), National Research Foundation of South Korea (NRF-2018R1C1B6009179).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Sudol M. Yes-associated protein (yap65) is a proline-rich phosphoprotein that binds to the sh3 domain of the yes proto-oncogene product. Oncogene. 1994;9(8):2145–2152. PMID:8035999. [PubMed] [Google Scholar]
- [2].Yagi R, Chen LF, Shigesada K, et al. A ww domain-containing yes-associated protein (yap) is a novel transcriptional co-activator. EMBO J. 1999;18(9):2551–2562. doi: 10.1093/emboj/18.9.2551. PMID:10228168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hong JH, Hwang ES, McManus MT, et al. Taz, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309(5737):1074–1078. doi: 10.1126/science.1110955. PMID:16099986. [DOI] [PubMed] [Google Scholar]
- [4].Huang J, Wu S, Barrera J, et al. The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating yorkie, the drosophila homolog of yap. Cell. 2005;122(3):421–434. doi: 10.1016/j.cell.2005.06.007. PMID:16096061. [DOI] [PubMed] [Google Scholar]
- [5].Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell. 2007;130(6):1120–1133. doi: 10.1016/j.cell.2007.07.019. PMID:17889654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13(3):188–192. doi: 10.1016/j.ccr.2008.02.011. PMID:18328423. [DOI] [PubMed] [Google Scholar]
- [7].Jang JW, Kim MK, Lee YS, et al. Rac-lats1/2 signaling regulates yap activity by switching between the yap-binding partners tead4 and runx3. Oncogene. 2017;36(7):999–1011. doi: 10.1038/onc.2016.266. PMID:27425596. [DOI] [PubMed] [Google Scholar]
- [8].Zhao B, Ye X, Yu J, et al. Tead mediates yap-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. doi: 10.1101/gad.1664408. PMID:18579750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang J, Smolen GA, Haber DA. Negative regulation of yap by lats1 underscores evolutionary conservation of the drosophila hippo pathway. Cancer Res. 2008;68(8):2789–2794. doi: 10.1158/0008-5472.CAN-07-6205. PMID:18413746. [DOI] [PubMed] [Google Scholar]
- [10].Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. PMID:20951342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meng Z, Moroishi T, Guan KL. Mechanisms of hippo pathway regulation. Genes Dev. 2016;30(1):1–17. doi: 10.1101/gad.274027.115. PMID:26728553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Harvey KF, Zhang X, Thomas DM. The hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–257. doi: 10.1038/nrc3458. PMID:23467301. [DOI] [PubMed] [Google Scholar]
- [13].Zanconato F, Cordenonsi M, Piccolo S. Yap/taz at the roots of cancer. Cancer Cell. 2016;29(6):783–803. doi: 10.1016/j.ccell.2016.05.005. PMID:27300434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Etienne-Manneville S, Hall A. Rho gtpases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. PMID:12478284. [DOI] [PubMed] [Google Scholar]
- [15].Alan JK, Lundquist EA. Mutationally activated rho gtpases in cancer. Small GTPases. 2013;4(3):159–163. doi: 10.4161/sgtp.26530. PMID:24088985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Porter AP, Papaioannou A, Malliri A. Deregulation of rho gtpases in cancer. Small GTPases. 2016;7(3):123–138. doi: 10.1080/21541248.2016.1173767. PMID:27104658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wada K, Itoga K, Okano T, et al. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138(18):3907–3914. doi: 10.1242/dev.070987. PMID:21831922. [DOI] [PubMed] [Google Scholar]
- [18].Ridley AJ, Hall A. The small gtp-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–399. doi: 10.1016/0092-8674(92)90163-7. PMID:1643657. [DOI] [PubMed] [Google Scholar]
- [19].Edwards DC, Sanders LC, Bokoch GM, et al. Activation of lim-kinase by pak1 couples rac/cdc42 gtpase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1(5):253–259. doi: 10.1038/12963. PMID:10559936. [DOI] [PubMed] [Google Scholar]
- [20].Mana-Capelli S, Paramasivam M, Dutta S, et al. Angiomotins link f-actin architecture to hippo pathway signaling. Mol Biol Cell. 2014;25(10):1676–1685. doi: 10.1091/mbc.E13-11-0701. PMID:24648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhao B, Li L, Wang L, et al. Cell detachment activates the hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26(1):54–68. doi: 10.1101/gad.173435.111. PMID:22215811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ohgushi M, Minaguchi M, Sasai Y. Rho-signaling-directed yap/taz activity underlies the long-term survival and expansion of human embryonic stem cells. Cell Stem Cell. 2015;17(4):448–461. doi: 10.1016/j.stem.2015.07.009. PMID:26321201. [DOI] [PubMed] [Google Scholar]
- [23].Feng X, Degese MS, Iglesias-Bartolome R, et al. Hippo-independent activation of yap by the gnaq uveal melanoma oncogene through a trio-regulated rho gtpase signaling circuitry. Cancer Cell. 2014;25(6):831–845. doi: 10.1016/j.ccr.2014.04.016. PMID:24882515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Speight P, Kofler M, Szaszi K, et al. Context-dependent switch in chemo/ mechanotransduction via multilevel crosstalk among cytoskeleton-regulated mrtf and taz and tgfbeta-regulated smad3. Nat Commun. 2016;7:11642. doi: 10.1038/ncomms11642. PMID:27189435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yu OM, Miyamoto S, Brown JH. Myocardin-related transcription factor a and yes-associated protein exert dual control in g protein-coupled receptor- and rhoa-mediated transcriptional regulation and cell proliferation. Mol Cell Biol. 2016;36(1):39–49. PMID:26459764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dupont S, Morsut L, Aragona M, et al. Role of yap/taz in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. PMID:21654799. [DOI] [PubMed] [Google Scholar]
- [27].Yu FX, Zhao B, Panupinthu N, et al. Regulation of the hippo-yap pathway by g-protein-coupled receptor signaling. Cell. 2012;150(4):780–791. doi: 10.1016/j.cell.2012.06.037. PMID:22863277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Codelia VA, Sun G, Irvine KD. Regulation of yap by mechanical strain through jnk and hippo signaling. Curr Biol. 2014;24(17):2012–2017. doi: 10.1016/j.cub.2014.07.034. PMID:25127217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Scott RP, Hawley SP, Ruston J, et al. Podocyte-specific loss of cdc42 leads to congenital nephropathy. J Am Soc Nephrol. 2012;23(7):1149–1154. doi: 10.1681/ASN.2011121206. PMID:22518006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Blattner SM, Hodgin JB, Nishio M, et al. Divergent functions of the rho gtpases rac1 and cdc42 in podocyte injury. Kidney Int. 2013;84(5):920–930. doi: 10.1038/ki.2013.175. PMID:23677246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schwartzman M, Reginensi A, Wong JS, et al. Podocyte-specific deletion of yes-associated protein causes fsgs and progressive renal failure. J Am Soc Nephrol. 2016;27(1):216–226. doi: 10.1681/ASN.2014090916. PMID:26015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang Z, Zhang L, Chen Y, et al. Cdc42 deficiency induces podocyte apoptosis by inhibiting the nwasp/stress fibers/yap pathway. Cell Death Dis. 2016:7(e2142). doi: 10.1038/cddis.2016.51. PMID:26986510.23555292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reginensi A, Scott RP, Gregorieff A, et al. Yap- and cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013;9(3):e1003380. doi: 10.1371/journal.pgen.1003380. PMID:23555292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Piersma B, de Rond S, Werker PM, et al. Yap1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. 2015;185(12):3326–3337. doi: 10.1016/j.ajpath.2015.08.011. PMID:26458763. [DOI] [PubMed] [Google Scholar]
- [35].Miranda MZ, Bialik JF, Speight P, et al. Tgf-beta1 regulates the expression and transcriptional activity of taz protein via a smad3-independent, myocardin-related transcription factor-mediated mechanism. J Biol Chem. 2017;292(36):14902–14920. doi: 10.1074/jbc.M117.780502. PMID:28739802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu F, Lagares D, Choi KM, et al. Mechanosignaling through yap and taz drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L344–357. doi: 10.1152/ajplung.00300.2014. PMID:25502501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liang M, Yu M, Xia R, et al. Yap/taz deletion in gli(+) cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol. 2017;28(11):3278–3290. doi: 10.1681/ASN.2015121354. PMID:28768710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Szeto SG, Narimatsu M, Lu M, et al. Yap/taz are mechanoregulators of tgf-beta-smad signaling and renal fibrogenesis. J Am Soc Nephrol. 2016;27(10):3117–3128. doi: 10.1681/ASN.2015050499. PMID:26961347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mannaerts I, Leite SB, Verhulst S, et al. The hippo pathway effector yap controls mouse hepatic stellate cell activation. J Hepatol. 2015;63(3):679–688. doi: 10.1016/j.jhep.2015.04.011. PMID:25908270. [DOI] [PubMed] [Google Scholar]
- [40].Mitani A, Nagase T, Fukuchi K, et al. Transcriptional coactivator with pdz-binding motif is essential for normal alveolarization in mice. Am J Respir Crit Care Med. 2009;180(4):326–338. doi: 10.1164/rccm.200812-1827OC. PMID:19498055. [DOI] [PubMed] [Google Scholar]
- [41].Lappano R, Maggiolini M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10(1):47–60. doi: 10.1038/nrd3320. PMID:21193867. [DOI] [PubMed] [Google Scholar]
- [42].Yu FX, Luo J, Mo JS, et al. Mutant gq/11 promote uveal melanoma tumorigenesis by activating yap. Cancer Cell. 2014;25(6):822–830. doi: 10.1016/j.ccr.2014.04.017. PMID:24882516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94. doi: 10.1038/nrc2069. PMID:17251915. [DOI] [PubMed] [Google Scholar]
- [44].Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. PMID:3304147. [DOI] [PubMed] [Google Scholar]
- [45].Malumbres M, Pellicer A. Ras pathways to cell cycle control and cell transformation. Front Biosci. 1998;3:d887–912. doi: 10.2741/A331. PMID:9696882. [DOI] [PubMed] [Google Scholar]
- [46].Downward J. Targeting ras signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- [47].Karnoub AE, Weinberg RA. Ras oncogenes: Split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. doi: 10.1038/nrm2438. PMID:18568040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by ej c-ha-ras oncogene. Nature. 1983;304(5927):648–651. doi: 10.1038/304648a0. PMID:6877385. [DOI] [PubMed] [Google Scholar]
- [49].Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell. 1997;88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9. PMID:9054499. [DOI] [PubMed] [Google Scholar]
- [50].Palmero I, Pantoja C, Serrano M. P19arf links the tumour suppressor p53 to ras. Nature. 1998;395(6698):125–126. doi: 10.1038/25870. PMID:9744268. [DOI] [PubMed] [Google Scholar]
- [51].Bates S, Phillips AC, Clark PA, et al. P14arf links the tumour suppressors rb and p53. Nature. 1998;395(6698):124–125. doi: 10.1038/25867. PMID:9744267. [DOI] [PubMed] [Google Scholar]
- [52].Mayo MW, Wang CY, Cogswell PC, et al. Requirement of nf-kappab activation to suppress p53-independent apoptosis induced by oncogenic ras. Science. 1997;278(5344):1812–1815. doi: 10.1126/science.278.5344.1812. PMID:9388187. [DOI] [PubMed] [Google Scholar]
- [53].Khokhlatchev A, Rabizadeh S, Xavier R, et al. Identification of a novel ras-regulated proapoptotic pathway. Curr Biol. 2002;12(4):253–265. doi: 10.1016/S0960-9822(02)00683-8. PMID:11864565. [DOI] [PubMed] [Google Scholar]
- [54].Matallanas D, Romano D, Yee K, et al. Rassf1a elicits apoptosis through an mst2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell. 2007;27(6):962–975. doi: 10.1016/j.molcel.2007.08.008. PMID:17889669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hong AW, Meng Z, Yuan HX, et al. Osmotic stress-induced phosphorylation by nlk at ser128 activates yap. EMBO Rep. 2017;18(1):72–86. doi: 10.15252/embr.201642681. PMID:27979971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Matallanas D, Romano D, Al-Mulla F, et al. Mutant k-ras activation of the proapoptotic mst2 pathway is antagonized by wild-type k-ras. Mol Cell. 2011;44(6):893–906. doi: 10.1016/j.molcel.2011.10.016. PMID:22195963. [DOI] [PubMed] [Google Scholar]
- [57].Aylon Y, Yabuta N, Besserglick H, et al. Silencing of the lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant h-ras-driven cell transformation. Oncogene. 2009;28(50):4469–4479. doi: 10.1038/onc.2009.270. PMID:19855428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kapoor A, Yao W, Ying H, et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell. 2014;158(1):185–197. doi: 10.1016/j.cell.2014.06.003. PMID:24954535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shao T, Zheng Y, Zhao B, et al. Recombinant expression of different mutant k-ras gene in pancreatic cancer bxpc-3 cells and its effects on chemotherapy sensitivity. Sci China Life Sci. 2014;57(10):1011–1017. doi: 10.1007/s11427-014-4724-0. PMID:25216706. [DOI] [PubMed] [Google Scholar]
- [60].Asem MS, Buechler S, Wates RB, et al. Wnt5a signaling in cancer. Cancers (Basel). 2016;8(9):E79. doi: 10.3390/cancers8090079. PMID:27571105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Konsavage WM, Jr., Kyler SL, Rennoll SA, et al. Wnt/beta-catenin signaling regulates yes-associated protein (yap) gene expression in colorectal carcinoma cells. J Biol Chem. 2012;287(15):11730–11739. doi: 10.1074/jbc.M111.327767. PMID:22337891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Seo WI, Park S, Gwak J, et al. Wnt signaling promotes androgen-independent prostate cancer cell proliferation through up-regulation of the hippo pathway effector yap. Biochem Biophys Res Commun. 2017;486(4):1034–1039. doi: 10.1016/j.bbrc.2017.03.158. PMID:28366633. [DOI] [PubMed] [Google Scholar]
- [63].Park HW, Kim YC, Yu B, et al. Alternative wnt signaling activates yap/taz. Cell. 2015;162(4):780–794. doi: 10.1016/j.cell.2015.07.013. PMID:26276632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li G, Iyengar R. Calpain as an effector of the gq signaling pathway for inhibition of wnt/beta -catenin-regulated cell proliferation. Proc Natl Acad Sci U S A. 2002;99(20):13254–13259. doi: 10.1073/pnas.202355799. PMID:12239346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ito Y, Bae SC, Chuang LS. The runx family: Developmental regulators in cancer. Nat Rev Cancer. 2015;15(2):81–95. doi: 10.1038/nrc3877. PMID:25592647. [DOI] [PubMed] [Google Scholar]
- [66].Qiao Y, Lin SJ, Chen Y, et al. Runx3 is a novel negative regulator of oncogenic tead-yap complex in gastric cancer. Oncogene. 2016;35(20):2664–2674. doi: 10.1038/onc.2015.338. PMID:26364597. [DOI] [PubMed] [Google Scholar]
- [67].Levy D, Reuven N, Shaul Y. A regulatory circuit controlling itch-mediated p73 degradation by runx. J Biol Chem. 2008;283(41):27462–27468. doi: 10.1074/jbc.M803941200. PMID:18701449. [DOI] [PubMed] [Google Scholar]