

ABSTRACT
The tight gene expression regulation controls the development and pathogenesis of human malaria parasite Plasmodium falciparum throughout the complex life cycle. Recent studies have revealed the pervasive nascent transcripts in the genome of P. falciparum, suggesting the existence of a hidden transcriptome involved in the dynamic gene expression. However, the landscape and related biological functions of nascent non-coding RNAs (ns-ncRNAs) are still poorly explored. Here we profiled the transcription dynamics of nascent RNAs by rRNA-depleted and stranded RNA sequencing over the course of 48-h intraerythrocytic developmental cycle (IDC). We identified the genome-wide sources of a total of 2252 ns-ncRNAs, mostly originating from intergenic and untranslated regions of annotated genes. By integrating the nascent RNA abundances with ATAC-seq and ChIP-seq analysis, we uncovered the euchromatic microenvironment surrounding the ns-ncRNA loci, and revealed a positive correlation between ns-ncRNAs and corresponding mRNA abundances. Finally, by gene knock-down strategy, we showed that the cooperation of RNA exosome catalytic subunit PfDis3 and PfMtr4 cofactor played a major role in ns-ncRNAs degradation. Collectively, this study contributes to understanding of the potential roles of short-lived nascent ncRNAs in regulating gene expression in malaria parasites.
KEYWORDS: Nascent non-coding RNA, Plasmodium falciparum, RNA Exosome
Background
Malaria is one of the most devasting infectious diseases worldwide. The causative pathogen, Plasmodium falciparum, harbours complex life cycles in human host and mosquito vector. During the intraerythrocytic developmental cycle (IDC), the number and abundance of gene expression are highly dynamic with the obvious feature of stage-specific expression. This multi-stage life cycle of parasite requires highly precise regulation, which could be achieved potentially at multiple layers such as transcriptional, post-transcriptional, and translational regulation. However, for the unicellular organism of P. falciparum in which the RNA interference (RNAi) pathway is absent [1], most studies focused on the epigenetic regulation at transcriptional level, particularly for the virulence gene families [2–5]. Emerging evidences showed a group of transcription factors, called ApiAP2 family, also contributed to parasite development in either asexual or sexual stages by controlling the dynamic expression of target genes [6–9].
The conventional tools used to measure the levels of gene transcripts such as quantitative real-time PCR (RT-qPCR), microarray, and RNA-seq only detected the abundance of steady-state RNAs. Previously, it reported that the genome-wide distribution of active RNA Polymerase II (RNPII) was not well correlated with the steady-state RNA abundances [10]. Another clue came from the finding that nascent RNA degradation by a ribonuclease, PfRNase II, was involved in the silencing of a subset of var gene family [11]. At present, there are a variety of high-throughput sequencing technology such as GRO-seq (Global run-on sequencing), PRO-seq (Precision nuclear run-on sequencing), NET-seq (Native elongating transcript sequencing) to detect the nascent transcripts [12–14]. Recently, two studies utilizing nascent mRNA sequencing both revealed numerous events of post-transcriptional regulation (PTR) in specific developmental stages, which points to a vital significance of nascent RNA degradation in the complex mechanisms of tight stage-specific gene expression in P. falciparum [15,16].
Among the various nascent transcripts, the cryptic unstable transcripts (CUTs) were the main class of nascent non-coding RNAs (ns-ncRNAs) which were originally described as a principal class of RNPII-associated transcripts in Saccharomyces cerevisiae [17,18]. CUTs had been shown to originate from the widespread bidirectional promoters in the genome of yeast. Unlike the steady-state unannotated transcripts (SUTs), they were subjected to degradation immediately after synthesis by the action of the core RNA exosome-associated cofactors, Trf4–Air1/Air2–Mtr4 polyadenylation (TRAMP) complex [19,20]. In addition, RNA exosome depletion revealed that many human genes were capable of producing CUTs-like unstable transcripts, i.e., Promoter Upstream Transcripts (PROMPTs) in both orientations at ~0.5 to 2.5 kb upstream of active transcription start sites [21]. This class of ns-ncRNAs represented another common characteristic of RNAPII transcribed genes with unknown functions. In P. falciparum, nascent mRNAs from annotated genes have been well profiled and analysed in depth with respect to their transcription and stabilization recently, however, little is known about the existence and related functions of ns-ncRNAs.
In recent years, long non-coding RNAs (lncRNAs) have emerged as a new critical regulatory factor of eukaryotic gene expression by a variety of mechanisms, e.g., recruitment of specific epigenetic regulators by its binding protein complex to modify the local chromatin environment surrounding the targeting genes, thereby modulating gene expression [22]. In some cases, lncRNAs were able to interfere with the formation of transcription machinery or transcription process by direct interaction with RNA polymerases. In P. falciparum, the most attractive finding is that the intronic antisense lncRNA contributes to the mutually exclusive expression of var gene family [23]. In addition, other lncRNAs such as the subtelomeric lncRNAs, natural antisense transcripts (NATs), intronic sterile transcripts, intergenic lncRNAs, and circular RNAs were identified in succession by high-throughput sequencing techniques, albeit the biological functions and underlying mechanisms are still elusive [24–26].
To this end, here we profiled the hidden lncRNA transcriptome by capturing nascent RNA transcripts using EU-labelling method followed by rRNA-removed strand-specific high-throughput sequencing with steady-state RNAs as control. We reported the main sources in the genome where ns-ncRNAs originated in various forms, and the correlation of the dynamic transcriptomes between ns-ncRNAs and corresponding protein-coding genes. We also defined the local chromatin environment of those ns-ncRNAs-associated gene loci by integrated analysis of chromatin accessibility (ATAC-seq) and histone modification (ChIP-seq). Finally, the RNA exosome factors accounting for the degradation of ns-ncRNAs were investigated by genetic manipulation (knock-down) of Pfrrp6, Pfdis3, and Pfmtr4 genes, respectively. Our results provide an in-depth view of the cryptic lncRNAs in malaria parasites, which may contribute to the understanding of the complex mechanism of gene expression in malaria parasites.
Results
A genome-wide source and transcription dynamics of ns-ncRNAs
In this study, we captured the EU-labelling nascent transcripts including mRNAs and lncRNAs across four stages of IDC (TP10, TP20, TP30, TP40) and prepared stranded RNA-seq libraries by rRNA-depletion method for high-throughput sequencing (Fig. 1A and Additional file 2: Supplemental Table S1). After quality evaluation of these sequencing data, the transcriptomes of ns-ncRNAs were constructed de novo for each time point (see Methods and Additional file 1: Figure S1 and S2). To further examine the data quality of nascent transcriptome analysis in this study, we compared the nascent mRNAs with the real-time in vivo mRNA transcriptomes detected by using rapid 4-thiouracil (4-tU) incorporation and microarray analysis by Painter et al. [16], or nascent mRNAs obtained by non-stranded EU-labelled GRO-seq by Lu et al. [15], respectively. As shown in Additional file 1: Figure S3, the correlation between the datasets of Painter and Lu was relatively lower, but our data is comparable to that of Lu et al., which may reflect the technical difference, e.g., labelling with EU vers4 USD-tU, or high-throughput analysis of microarray versus RNA-seq. In addition, as described by Lu et al., we performed the nuclear run-on reaction according to the standard procedure, i.e., 30 min at 37°C. Nevertheless, transcription may have already reached a plateau at 30 min, and a shorter incubation might allow a more dynamic range of transcription. This possibility may be another reason for the disagreement between our data and that of Painter et al.
Figure 1.
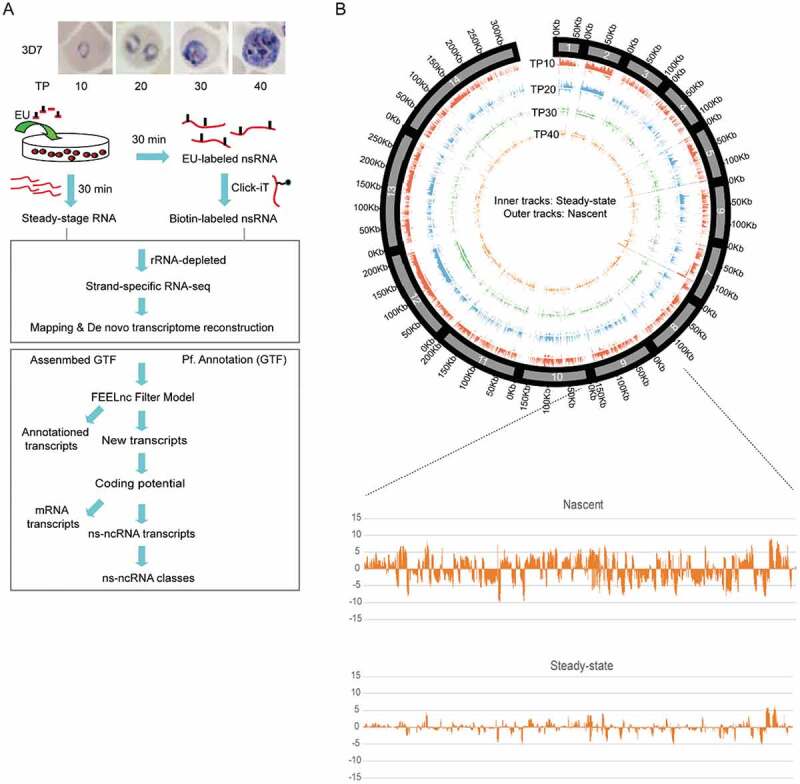
Genome-wide profile of nascent ncRNAs (ns-ncRNAs) during blood-stage development of P. falciparum. (A) Nascent RNA capture and ns-ncRNAs identification pipeline. (B) Upper: Genome-wide abundance distribution of ncRNAs in nascent RNA-seq and steady-state RNA-seq at four stages of IDC was shown in Circus plot. Bottom: an example of ncRNAs signal density (normalized by TPM) of chromosome 9 displaying a whole strand-specific transcripts abundance in nascent RNA-seq and steady-state RNA-seq at TP10 (positive values: ‘+’ strand; negative values: ‘-’ strand). The histogram signals of Circus plot represented normalized reads density by reads per million and then divided by scale factor.
Our data confers us the opportunity to identify those unknown ns-ncRNAs on either chromosomal strands and profile the global expression dynamics throughout the IDC. A genome-wide map of source and expression level of individual ns-ncRNAs uncovered a hidden dynamic non-coding transcriptome over the course of asexual development of P. falciparum (Fig. 1B). Next, we attempted to identify and quantify individual ns-ncRNAs including sense and antisense long non-coding transcripts (lncRNAs) originated from upstream, intron, or downstream untranslated regions (3ʹUTR) of individual gene locus, and intergenic regions [27] (schematic in Fig. 2A, upper). After de novo transcriptome analysis, a total of 2252 unique ns-ncRNAs were identified at the four time points of IDC, i.e., 1878 (63.64%), 701 (23.75%), 361 (12.23%), and 307 (10.40%), respectively, where some ns-ncRNAs reappeared at multiple stages. The main proportions of them were intergenic or antisense ns-ncRNAs (Fig. 2A, bottom, and Additional file 2: Supplemental Table S2), suggesting that the intergenic and 3ʹUTR regions are the main source of ns-ncRNAs production. Importantly, comparative analysis of these ns-ncRNAs with previously identified NATs or lncRNAs [24–26,28] showed that approximately 77% of ns-ncRNAs identified here were novel transcripts (Additional file 1: Figure S4 and Additional file 2: Supplemental Table S2). In addition, for those protein-coding gene locus-associated ns-ncRNAs, i.e., divergent and antisense, the majority of them were produced at the early stage of TP10, and positively correlated with the nascent mRNAs at transcriptional level ((Fig. 2B, C), and Additional file 1: Figure S5). Next, to further investigate the potential link between ns-ncRNAs and protein-coding genes, our analysis focused on the upstream divergent and antisense ns-ncRNAs.
Figure 2.
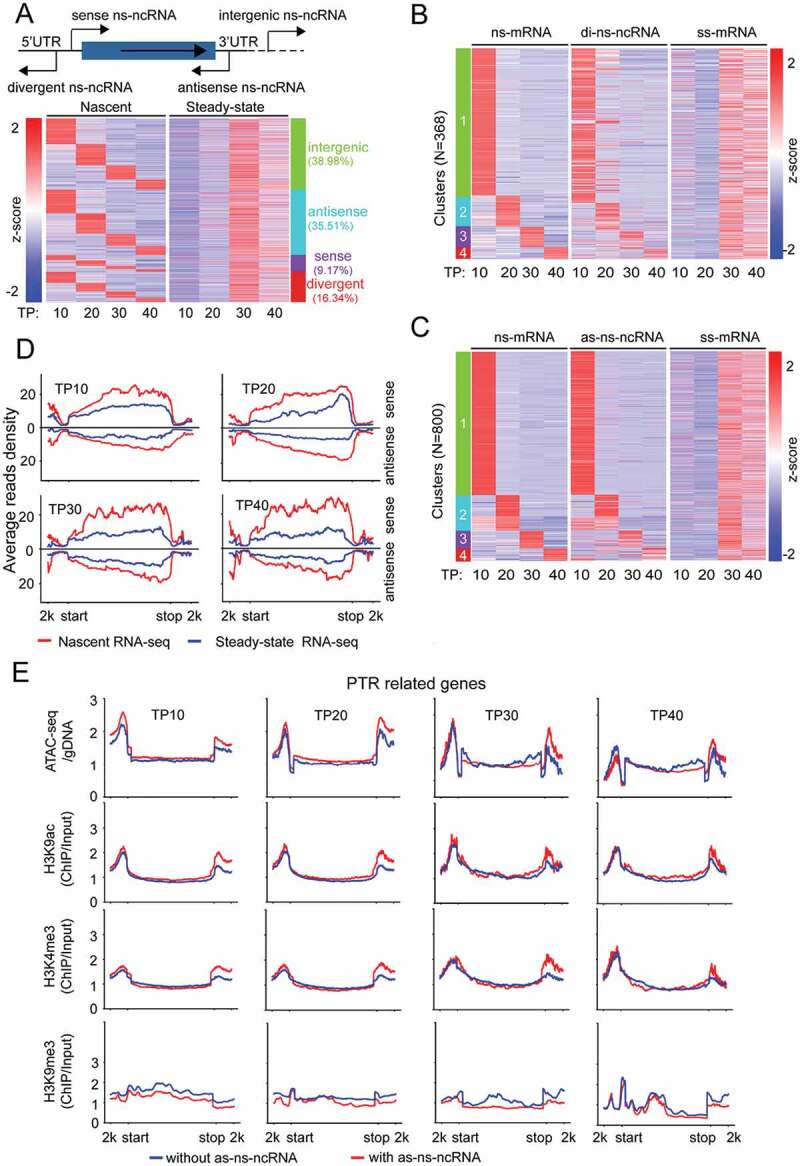
Expression dynamics of nascent ncRNAs (ns-ncRNAs) during blood-stage development of P. falciparum. (A) Upper: schematic diagram of ns-ncRNAs category with regard to the sites they are produced. Bottom: the dynamics of nascent or steady-state ncRNAs measured by nascent RNA-seq (left) or steady-state RNA-seq (right). Profiles of heatmaps were clustered by K-means clustering. (B) and (C), Comparative transcriptional profiles of nascent mRNAs (ns-mRNA), divergent ns-ncRNAs (di-ns-ncRNA), antisense ns-ncRNA (as-ns-ncRNA) and steady-state mRNAs (ss-mRNA) based on normalized TPM values (z-score transformed). Transcripts were ranked in the same order according to the ns-mRNAs. Profiles of ns-mRNAs were clustered by K-means clustering. (D) Average reads coverage profiles for ‘divergent ns-ncRNA – mRNA pairs’ in the whole gene body ranging from – 2kb upstream the gene start site (start) to 2 kb downstream the gene stop site (stop) by nascent RNA-seq (red) and steady-state RNA-seq (blue) throughout the IDC. Reads were normalized using RPKM. (E) Dynamic epigenetic profiles of post-transcriptional regulation-related (PTR-related) gene loci including ATAC-seq and ChIP-seq (H3K9ac, H3K4me3 and H3K9me3) were compared throughout the IDC by post-transcriptional regulation-related genes with as-ncRNAs (red) and without as-ncRNAs (blue).
A positive correlation between ns-ncRNAs and corresponding mRNAs production
Cryptic transcripts surrounding the active RNPII promoters have been recognized as the products of bidirectional promoter activity [19,20]. Although the pattern of such upstream ns-ncRNAs varies among different organisms, virtually two groups have been categorized with respect to the origination site to promoter and length of these transcripts, i.e., TSS-associated RNAs of 20–90 nucleotides in length (TSSa-RNA) and promoter upstream transcripts (PROMPTs) of 0.5–2.5 kb in length [21,29]. To address this issue, we systematically analysed the divergent ns-ncRNAs and mRNAs pairs with a pattern of ‘HTH’ (Head to Head) in P. falciparum (Additional file 1: Figure S6A and Additional file 2: Supplemental Table S2). Most of them could be grouped into the PROMPTs-like ns-ncRNAs with a peak of density at approximately −0.5 ~ −1 kb upstream the gene start site (Additional file 1: Figure S6(B)), whereas no apparent short divergent ns-ncRNAs corresponding to the TSSs-RNAs were detected. Fort the divergent ncRNAs, approximately 91% were detected as ns-ncRNAs at TP10, then descended dramatically after this stage (Additional file 1: Figure S6(C)). Moreover, our GRO-seq revealed positively correlated transcriptional levels between divergent ns-ncRNAs and corresponding mRNAs, which was not observed at the level of steady-state RNA (Fig. 2D and Additional file 1: Figure S5(B)). This may reflect the real promoter activity of the central region between divergent ns-ncRNAs-mRNA pair. Previous studies have suggested that bidirectional promoters within ~1 kb apart were the source of upstream divergent transcripts, and it is likely intrinsic in eukaryotic organisms [12,19]. According to this criteria, about 40% to 60% of divergent ns-ncRNA-mRNA pairs identified here were potentially produced by the bidirectional promoters throughout the IDC (Additional file 1: Figure S6(D)). Meanwhile, for those steady-state mRNA-mRNA pairs, we found a similar transcriptional pattern as that of divergent ns-ncRNA-mRNA (Additional file 1: Figure S6(E)), implying that symmetric transcriptional activities in both orientations may be an intrinsic property of the active RNPII promoters as suggested previously [20].
It has reported that the natural antisense transcripts (NATs) produced from the 3ʹUTR regions of protein-coding genes were pervasive in the genome of P. falciparum [25]. Here a large proportion (35.51%) of cryptic ncRNAs were identified as antisense ns-ncRNAs (Fig. 2A and Additional file 2: Supplemental Table S2). To explore the potential role of antisense ns-ncRNAs in expression regulation of their overlapping protein-coding genes, we evaluated the transcriptional pattern between them. It showed a highly positive correlation of transcriptional abundances between the antisense ns-ncRNAs and nascent mRNAs (Pearson correlation coefficient 0.88, p < 0.01) (Additional file 1: Figure S5(A)), which was not observed for NATs at steady-state RNA level [25]. Interestingly, the antisense ns-ncRNAs likely correlated negatively with the steady-state mRNAs, but the Pearson correlation coefficient is relatively lower (−0.37, p < 0.01).
Nascent ncRNAs were involved in local chromatin structure
The distinct transcriptomes between nascent RNAs and steady-state RNAs may point to a widespread post-transcriptional regulation pathway of gene expression in P.falciparum. With the two datasets in parallel, we are able to categorize the post-transcriptional regulation-related genes from others such as constantly active (CA) or silent (CS) genes (see Materials and Methods) (Additional file 2: Supplemental Table S3). It has been well established that histone modifications are associated with gene transcriptional activity, e.g., H3K9me3 is a heterochromatic marker for transcriptional silencing, while H3K9ac or H3K4me3 are euchromatic makers for transcriptionally active genes. Here we performed systematically comparative analysis by utilizing our data of ATAC-seq and ChIP-seq of H3K9me3 together with H3K9ac and H3K4me3 published previously [30]. As shown in Fig. 2E, the post-transcriptional regulation-related genes generating antisense ns-ncRNAs showed a more ‘open’ state of local chromatin surrounding the 3ʹUTR regions throughout IDC compared with other genes without antisense ns-ncRNAs. Meanwhile, no apparent difference of the chromatin state was observed for those CA or CS genes (Additional file 1: Figure S7A). Interestingly, ATAC levels were relatively higher at the upstream regions of post-transcriptional regulation-related genes with antisense ns-ncRNAs at an early stage, when most divergent ns-ncRNAs were transcribed. A further global Pearson correlation coefficient analysis confirmed that nascent RNA levels had a higher correlation with ATAC-seq levels than histone modification levels (Additional file 1: Figure S8).
The role of ns-ncRNAs in clonally expression of variant genes
The sense ns-ncRNAs at upstream region have been shown to regulate the singular expression of ~ 60 var gene [11], but it was not investigated systematically for these variant gene families including rifin (n = 184), stevor (n = 42), and Pfmc-2tm (n = 13). Here, by normalizing the abundances of nascent RNAs and steady-state RNAs, we found that only var and Pfmc-2tm gene families produced nascent RNAs with expression peaks at TP20 (Fig. 3A, left). In detail, a total of 39 var genes mostly belonged to upsB and upsC subtype, and seven members of Pfmc-2tm genes produced both sense and antisense ns-ncRNAs. These gene members were originally silent at steady-state RNA level (Fig. 3A, right, Additional file 1: Figure S9 and S10, and Additional file 2: Supplemental Table S4). While the sense ns-ncRNAs covered both exon 1 and exon 2 as steady-state mRNAs, the antisense ns-ncRNAs were originated from the introns as the known antisense ncRNAs of var genes [11,23]. Moreover, the transcription profile of the two ns-ncRNAs were consistent throughout the IDC for both var and Pfmc-2tm genes, indicating that the crosstalk of the two promoters plays a crucial role in the mutually exclusive expression mode of var family as suggested previously (Figure 3B and 4A). For the active var gene (PF3D7_0412700) of wild-type 3D7-G7 clone used in this study [11], only the sense ns-ncRNA covering the 3ʹend of exon 1 and the entire exon 2 was observed at early stages. While the antisense ns-ncRNA was absent, a steady-state antisense ncRNA was detected for this active var gene (Fig. 3C). This intronic antisense ncRNA has been shown to activate the upstream promoter activity [23]. Strikingly here we found it was produced at TP20 after the activation of its upstream promoter at TP10. In addition, no antisense ns-ncRNAs from the 3ʹUTR regions were detected. This is consistent with previous finding that almost no NATs were found in variant gene loci, albeit NATs were pervasive for protein-coding genes in P. falciparum [25]. ATAC-seq data also linked the ‘open’ local chromatin state to the production of these cryptic transcripts from either upstream or intronic promoters of these so-called ‘silent’ var genes as described previously [31] (Fig. 3D and Additional file 1: Figure S11(B)). A similar euchromatic state of 5ʹUTR and 3ʹUTR was also observed to be associated with ns-ncRNAs for Pfmc-2tm gene loci (Fig. 4B).
Figure 3.
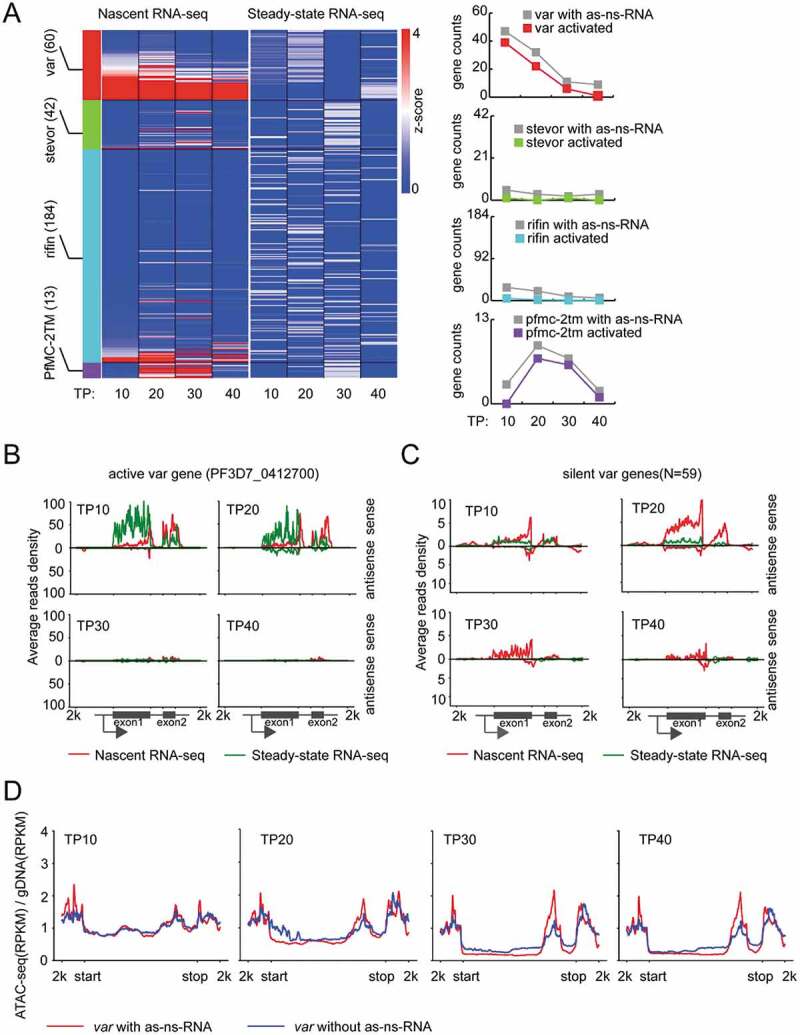
Transcriptional profile of ns-ncRNAs in variant gene families. (A) Left: Expression profiles of each virulence gene families (var, stevor, rifin and Pfmc-2tm) detected by nascent RNA-seq (left heatmap) or steady-state RNA-seq (right heatmap). Right: The counts of all the virulence genes with antisense ns-ncRNAs (grey line) and those genes upregulated by 2-fold in nascent mRNAs compared with steady-state mRNAs at each stage (red, var genes; green, stevor genes; sky blue, rifin genes; purple, Pfmc-2tm genes). The Z-score was calculated by gene expression value, TPM, to evaluate the transcription level with regard to the mean for individual genes. (B) and (C) Average stranded reads coverage profiles of all the silent var genes (N = 59) (B) and the active var gene (PF3D7_0412700) (C) at four time points (TP10, TP20, TP30 and TP40) detected by nascent RNA-seq (red) or steady-state RNA-seq (green), respectively. Reads were normalized using RPKM. The Peaks above the x-axis are sense transcripts, and under the x-axis are antisense transcripts. The schematic of var gene locus was shown at the bottom. (D) ATAC-seq signals for var genes with (red) or without (green) antisense ns-ncRNAs at four time points. y-axis: ATAC-seq (RPKM)/gDNA (RPKM).
Figure 4.
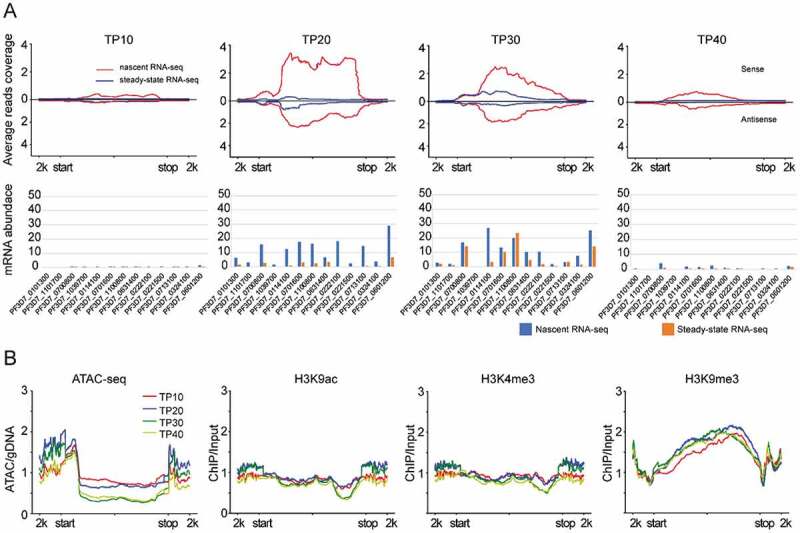
Transcriptional profile and epigenetic state of Pfmc-2tm genes. (A) nascent RNA and steady-state RNA transcription profiles at four time points (line plots) and expression values (bar plots). (B) the chromatin state (ATAC-seq, H3K9ac, H3K4me3 and H3K9me3) of Pfmc-2tm genes at each time points. All reads coverage in line plots were normalized by RPKM. The gene features are −2kb upstream of gene start (start) and +2kb downstream of gene stop (stop).
Nascent RNA abundance reflected the real transcriptional activities of post-transcriptional regulation-related genes
The global transcriptomic profile showed a relatively constant abundance of nascent mRNAs or ncRNAs during the IDC whereas a highly dynamic transcriptional mode was observed for steady-state RNAs (Fig. 5A). In detail, Fig. 5B exhibited the dynamic composition profiles of the three gene groups sorted by differential expression between nascent mRNAs and steady-state mRNAs at various stages of IDC (Additional file 2: Supplemental Table S4). The significantly higher ratio of post-transcriptional regulation-related genes at early developmental stages indicates the tight stage-specific gene expression of these stages parasites likely relies on post-transcriptional regulation by nascent RNA degradation more than later stages in IDC. Further GO analysis of these post-transcriptional regulation-related genes defined different stage-specific physiological processes in which these genes were involved, respectively. For instance, genes associated with metabolism processes were mainly enriched at ring stages (TP10 to TP20), and DNA replication-related genes were enriched in early trophozoites (TP20) when the parasites started to propagate in RBCs (Additional file 1: Figure S12).
Figure 5.
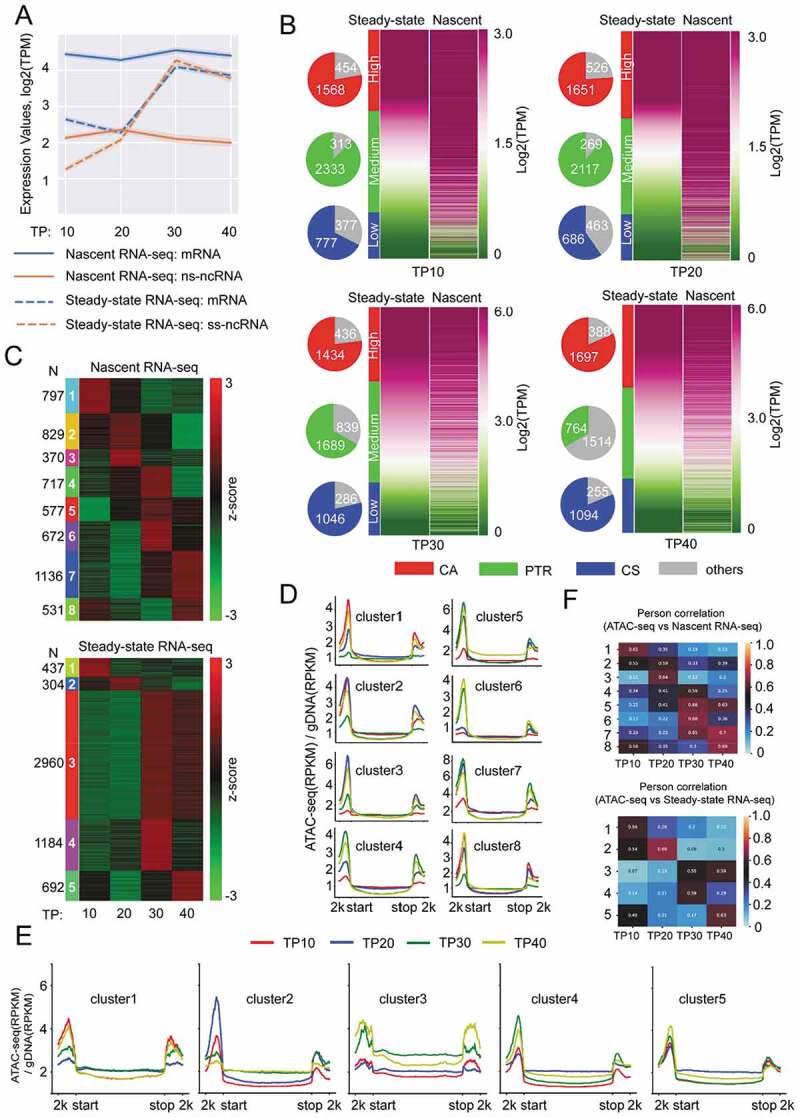
Nascent RNA abundance associated with chromatin accessibility of post-transcriptional regulation-related genes of P. falciparum over the course of IDC. (A) Line plot of average expression TPM (Transcripts Per Million) value (logarithm transformation) for nascent RNA-seq (solid) or steady-state RNA-seq (dashed) among different transcript groups (blue: mRNA; orange, lncRNA) at 4 time points (TP10, TP20, TP30 and TP40). (B) Heatmap depicting three subgroups of genes with high (CA), medium (post-transcriptional regulation), and low (CS) expression levels defined by TPM value (logarithm transformed) (see the detailed definition in Materials and Methods). The pie charts at the left of each heatmap represent gene counts for each level. (C) Heatmap depicting the K-means cluster results for nascent RNA-seq (left) and steady-state RNA-seq (right) based on the TPM values (z-score transformed). The numbers of each gene cluster were indicated on the left (nascent) and right (steady-state). (D) and (E) Average ATAC-seq reads coverage profiles for genes of each cluster (D, nascent RNA-seq; E, steady-state RNA-seq) corresponding to that in Fig. 1C. ATAC-seq signal intensity was calculated as (RPKM)/gDNA (RPKM). RPKM, reads per kilobase per million mapped reads. (F) Pearson correlation coefficient for expression pattern between ATAC-seq and nascent RNA-seq (top panel), or between ATAC-seq and steady-state RNA-seq (bottom panel) in each gene cluster defined in (C).
We subsequently analysed the dynamic variation of total transcript abundances (mRNA + lncRNA) by generating eight clusters for nascent RNAs (N = 5629) and five clusters for steady-state RNAs (N = 5577) respectively, throughout the IDC by using KMeans clustering method (Fig. 5C and Additional file 2: Supplemental Table S5). Obviously, the steady-state transcriptomes exhibited a more dynamic pattern than that of nascent RNAs. Next, to test the correlation between the nascent RNA abundance and local chromatin accessibility, we performed ATAC-seq assay with the wild-type parasite line at the four developmental stages (Additional file 1: Figure S11(A) and Additional file 2: Supplemental Table S6). Recently, two groups have adopted ATAC-seq technique to profile the genome-wide accessible chromatin state in P. falciparum. They both found that the dynamics of the chromatin accessibility pattern matched euchromatic marks (H3K9ac and H3K4me3) and temporal transcription during development; thus, chromatin accessibility measured by ATAC-seq was predictive of mRNA transcripts [31,32]. Here, comparative analysis showed that the Pearson correlation coefficient among the three ATAC-seq datasets was moderate (0.6 ~ 0.7, p < 0.001) at T10 and T20 time points, then declined at later stages, which might be caused by the different genetic backgrounds of parasite lines analysed, or the harvesting time points were not exactly same (Additional file 1: Figure S13). Intriguingly, we observed a positive correlation between transcriptional activity and accessible chromatin environment across the entire IDC for either nascent or steady-state RNAs of those gene clusters (Fig. 5D–F). These data confirm that both the chromatin accessibility extent and nascent RNA abundance reflect the real transcriptional state of post-transcriptional regulation-related genes.
RNA exosome-mediated degradation of ns-ncRNAs
For the degradation pathway of ns-ncRNAs, it has been detailed documented in yeast that TRAMP complex accounts for the metabolism of ns-ncRNAs in nucleus. In P. falciparum, the composition of RNA exosome has been identified recently, i.e., seven core subunits of RNA exosome and two associated catalytic ribonucleases, PfDis3 and PfRrp6 [33]. However, no any other cofactors have been found so far. To investigate the underlying pathways and associated factors involved in ns-ncRNAs degradation, we aimed to down-regulate the expression levels of Pfdis3, Pfrrp6, and mtr4, the essential gene of TRAMP complex, respectively. Because no Mtr4 protein candidate has been identified in P. falciparum so far, here we firstly searched a SKIV2 L family homologue-like Mtr4 gene candidate with DEAD/H domain-contained RNA helicase in the genome of 3D7 line by bioinformatic prediction, which exhibited a distinct evolutional relationship with other organisms (Additional file 1: Figure S14(A)). To further confirm the physical association of this PfMtr4 protein candidate with exosome complex, we generated dual-tagging parasite strain, PfRrp4-HA:PfMtr4-Ty1-Ribo, by using CRISPR-Cas9 as described previously [33] (Additional file 1: Figure S14(B)), and performed IFA and Co-IP assays with this transgenic parasite line. It showed that PfMtr4 located at nuclear periphery with the interaction of PfRrp4 (Additional file 1: Figure S14(C) and (D)), confirming that this Mtr4 homologue is a cofactor of TRAMP complex associated with RNA exosome. Next, we successfully generated inducible gene knock-down lines of Pfdis3, Pfrrp6 and Pfmtr4, respectively, by incorporating the glucosamine (GlcN)-inducible glms ribozyme sequence within the 3ʹUTR region of targeted genes (see the schematic diagram in Fig. 6A, upper). Western blot analysis showed that a significant reduction of target gene expression was achieved by the presence of GlcN drug in culture (Fig. 6A, bottom). These parasite lines provide an opportunity for us to further investigate the regulatory role of individual RNA exosome-associated factors on ns-ncRNAs degradation, and identify potential exosome-independent ns-ncRNAs in P. falciparum.
Figure 6.
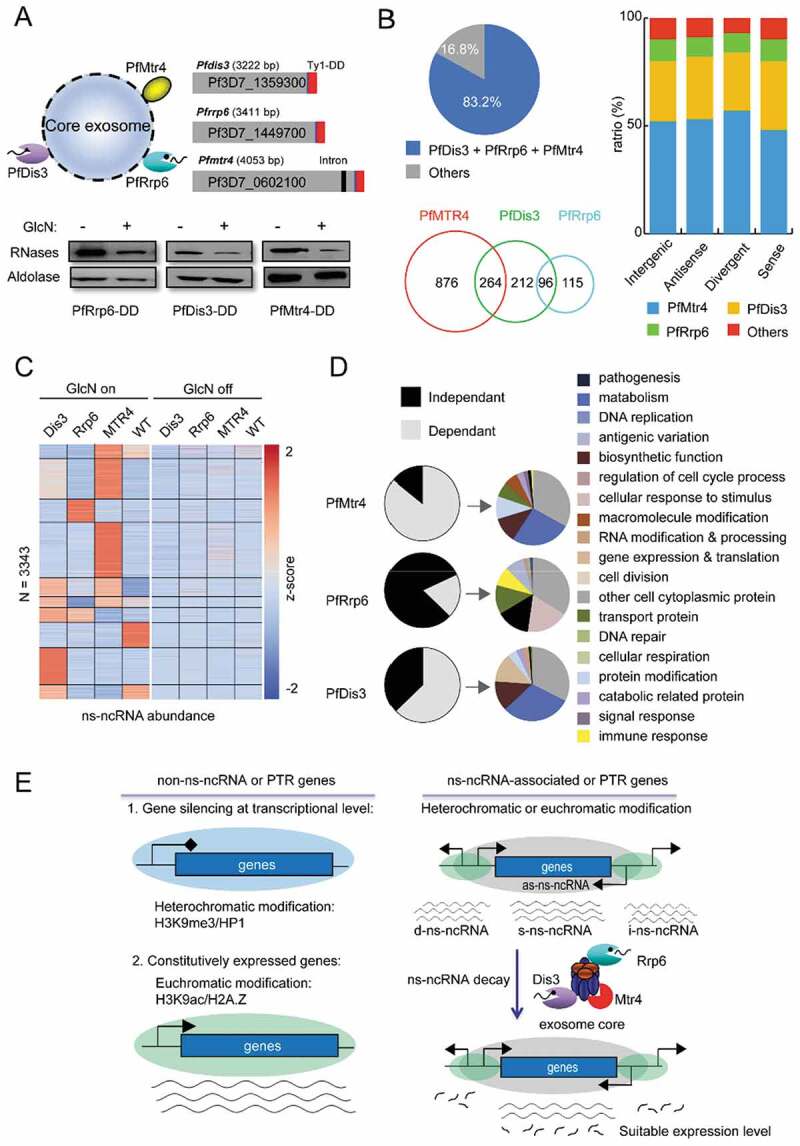
RNA exosome-mediated ns-ncRNAs degradation in P. falciparum. (A) Upper: the schematic representation of RNA exosome composition containing two catalytic subunits and another TRAMP-associated Mtr4 (left) and transgenic constructs of gene knock-down of Pfdis3, Pfrrp6, and Pfmtr4, respectively (right). The C-terminal fusion sequences are of the Ty1 epitope with triple repeats and glms ribozyme in tandem, and western blot analysis of gene knock-down effect for each exosome-associated subunits. (B) The ratio between lncRNAs regulated by exosome-associated subunits (Pfdis3, Pfrrp6 and Pfmtr4) and ns-ncRNAs at ring stage (pie chart); and Venn diagram showing the counts of ns-ncRNAs regulated by individual exosome-related subunits, respectively. The percentages of ns-ncRNAs regulated by different exosome-related genes (Pfdis3, Pfrrp6 and Pfmtr4) at TP10 for different ns-ncRNAs groups as shown in Fig. 2A. (C) Expression profiles of lncRNAs in Pfdis3-DD, Pfrrp6-DD and Pfmtr4-DD strains with drug (left) or without drug (right). (D) The percentages and GO enrichment of genes regulated by ns-ncRNAs dependently (grey) or independently (black) on Pfmtr4, Pfrrp6, and Pfdis3 pathways respectively; (E) Model of the putative regulatory functions of ns-ncRNAs for expression of post-transcriptional regulation-related genes and non-post-transcriptional regulation genes.
Due to the majority of ns-ncRNAs were produced in ring-stage parasites, we carried out stranded RNA-seq assays for Pfdis3, Pfrrp6 and Pfmtr4 knock-down strains with WT 3D7-G7 clone as control (GlcN on versus off) at ring stage (Additional file 2: Supplemental Table S1(B)). After de novo transcriptome analysis, a total of 3343 lncRNAs were obtained from all RNA-seq libraries upon the conditional knock-down of the three genes. Among them, 1563 lncRNAs were transcriptionally upregulated after knock-down of three exosome-associated cofactors. When we compared these differentially expressed lncRNAs with the ns-ncRNAs identified in ring-stage parasites (Fig. 6A), it showed that PfDis3, PfRrp6 and PfMtr4 were involved in the post-transcriptional degradation of 83.2% (1563/1878) ns-ncRNAs (Fig. 6B, left, and Additional file 2: Supplemental Table S7). As described in yeast, the majority of ns-ncRNAs (1140/1878, 60.7%) were associated with the Mtr4-related TRAMP pathway. For the two catalytic cofactors, PfDis3 likely played a more important role in ns-ncRNAs metabolism than PfRrp6 (572 vers211 USD). This finding is consistent with our recent observation that PfDis3 was involved in the degradation of 1046 antisense lncRNAs whereas only 131 sense transcripts were targeted [33]. A further classification of these ns-ncRNAs revealed a similar contribution of the individual RNA exosome-associated cofactors in ns-ncRNAs degradation (Fig. 6B, right). Next, we generated 10 clusters of ns-ncRNAs accumulated in the gene knock-down lines of three RNA exosome-associated cofactors individually as shown in the heatmaps (Fig. 6C and Additional file 2: Supplemental Table S8). Because Mtr4 itself does not harbour ribonuclease activity, its function on ns-ncRNAs degradation is likely partially depending on PfDis3 in P. falciparum. Subsequently, we analysed the genes associated with the ns-ncRNAs regulated by PfDis3, PfRrp6 and PfMtr4 by GO enrichment analysis, respectively (Fig. 6D). While PfDis3-dependent genes were enriched in various metabolic processes, PfRrp6 was likely involved in cell-cell adhesion, pathogenesis, response to stimulus, and antigenic variation. For instance, the ns-ncRNAs of var genes and Pfmc-2tm seem to be degraded by PfRrp6 factor. For the PfMtr4 dependent genes, they were close to that of PfDis3, which was consistent with the ns-ncRNAs analysis in Fig. 6B. Finally, we found higher mRNA levels of the corresponding protein-coding genes when the nascent ncRNAs were upregulated upon PfMTR4 or PfDis3 knockdown, whereas only a small proportion of these mRNAs were upregulated upon PfRrp6 knockdown (Additional file 1: Figure S15). This is consistent with the finding that a much more ns-ncRNAs had been identified by knockdown of PfMTR4 and PfDis3 compared to PfRrp6. We guess the PfRrp6 protein is abundant in the parasites, and the knockdown effect of PfRrp6 is not sufficient to upregulate more target ns-ncRNAs.
Discussion
The discovery of the cryptic unstable RNA population has expanded the reservoir of transcriptome in eukaryotic organisms. However, only a few reports linked the production of ns-ncRNAs with the expression of specific genes [34–36]. Here, by strand-specific nascent RNA-seq analysis, we successfully identified two groups of nascent transcripts in the human malaria parasites at blood stage, i.e., the nascent mRNA linked to post-transcriptional regulation genes, and ns-ncRNAs. For the nascent mRNA, they were likely complementary to the regulatory pathway of histone modification as shown in the model (Fig. 6E). Meanwhile, our data showed that the transcriptional abundances between divergent or antisense ns-ncRNAs and corresponding mRNAs were positively correlated. Particularly, the antisense ns-ncRNAs of those variant genes such as var and Pfmc-2TM genes were usually located in a eukaryotic environment measured by ATAC-seq and ChIP-seq of histone modification. Therefore, the constantly transcribed ns-ncRNAs might contribute to the maintenance of local chromatin structure surrounding the gene locus.
Previous studies have discovered that the antisense lncRNA transcribed from the intron of var gene was linked to the activation of the related var gene [11]. Subsequent research confirmed such correlation by strategies of overexpression or transcription interference of this lncRNA [23]. However, our data showed that both transcriptional peaks of sense mRNAs and antisense ns-ncRNAs were appeared at the early trophozoite stage (TP20), and no apparent antisense ns-ncRNAs were observed in early rings (TP10). This contradictory result raises a question that the positive regulation function of the antisense intronic transcripts is at the transcriptional level or post-transcriptional level. Though the abundance of the steady-state var mRNA was changed consistently after genetic manipulation of the antisense lncRNA, it was not measured at the transcriptional level [23]. Therefore, that work could not exclude the possibility that the antisense lncRNA may contribute to the stabilization of sense mRNA for the active var member, otherwise, the sense nascent RNA would be degraded by ribonucleases as shown in the post-transcriptional regulation-related upsA-subtype var genes [11].
Bidirectional promoter activities have been demonstrated as a common characteristic of eukaryotic protein-coding genes in yeast and mammalians, though the functions of upstream divergent transcripts are not well understood. As for the eukaryotic malaria parasites, bidirectional transcription activities have been demonstrated previously, e.g. the intron of var gene which was involved in the regulation of var expression [9,23,37,38]. Here we observed the existence of divergent nascent ncRNAs with opposite orientation to protein-coding genes, and approximately half of them were likely produced by bidirectional promoters upstream those protein-coding genes. Another interesting observation is that the transcription levels of upstream divergent ns-ncRNAs and mRNAs were comparable, which was not observed in the steady-state RNAs because of the rapid degradation of upstream nascent transcripts.
In eukaryotes, RNA exosome complex is the master cellular machinery for controlling RNA metabolism including structural RNA processing and maturation, mRNA turn over, and RNA quality surveillance [39,40]. In yeast, depletion of the catalytic active exosome cofactor, Rrp6, was sufficient to stabilize those cryptic unstable transcripts (CUTs) [20,41]. However, in human cells, only by depletion of both catalytic cofactors, it was able to achieve comparable effect as the core exosome [21]. In P. falciparum, our data showed that individual gene knock-down of Pfrrp6 or Pfdis3 alone did not stabilize their targeting ns-ncRNAs efficiently as described in human. This may partially interpret that only ~ 44% of ns-ncRNAs identified in this work are regulated by both PfDis3 and PfRrp6. Additionally, here we have identified another RNA exosome-related cofactor candidate, PfMtr4. The reduced expression of this non-catalytic cofactor confirmed that this homologue executes a major role in ns-ncRNAs decay as in model organisms. Intriguingly, in addition to the RNA exosome-dependent ns-ncRNAs, we found that a proportion (16.8%) of detected ns-ncRNAs was not targeted by the traditional decay pathway [12,21,36], which may be linked to a new class of RNA exosome-independent nc-ncRNAs.
Taken together, this work uncovers a genome-wide sources of cryptic transcripts, and the dynamic hidden transcriptomes with the potential of regulatory function on gene expression in falciparum malaria parasites. In addition, we also identified the RNA exosome-associated cofactors accounting for ns-ncRNAs degradation. These findings will help us gain insight into the gene expression world and contribute to our understanding of the complex mechanisms of tight regulated gene expression in the development of this parasitic pathogen.
Materials and methods
Parasite culture and transfection
Parasite strain 3D7-G7 clone was cultivated in vitro with human erythrocytes as described previously [11]. The parasites were synchronized by two rounds treatment of 5% D-sorbitol (Sigma-Aldrich, 240,850) at the ring stage (~ 8 hours post-invasion). To generate dual-labelling parasite strain of Pfrrp4-HA:Pfmtr4-Ty1-Ribo, we used Pfrrp4-HA strain as the parent line for Pfmtr4-Ty1-Ribo transfection [33]. For those transfection lines of Pfdis3-Ty1-Ribo, Pfrrp6-Ty1-Ribo and Pfmtr4-Ty1-Ribo, we modified the plasmid pL6-gfp by replacing the gfp box with a ~ 1 kb homologue sequence flanking the C-terminus of the targeted gene containing the glucosamine (GlcN)-inducible glms ribozyme sequence, and inserting a guide RNA sequence specific to the Pfrrp6 (PF3D7_1449700), Pfdis3 (PF3D7_1359300), and Pfmtr4 (PF3D7_0602100), respectively (Additional file 2: Supplemental Table S9). The resulting plasmids pL6-Pfrrp6- Ty1-Ribo, pL6-Pfdis3-Ty1-Ribo and pL6-Pfmtr4-Ty1-Ribo were transfected into 3D7-G7 clone or Pfrrp4-HA line (dual-labelling transfection) respectively, together with the plasmid pUF1-Cas9-infusion carrying Cas9 expression cassette.
Western-blot analysis
Total parasite extract, nuclear or cytoplasmic extracts were resuspended in 1x SDS-loading buffer, then separated on 8-12% SDS-PAGE gel (Bio-rad), and subjected to Western blot analysis. The commercial antibodies used for immunodetection were mouse anti-HA (1:2000, Roche), mouse anti-Ty1 (1:500, Sigma, SAB4800032), rabbit anti-Histone 3 (1:1000, Abcam, ab1791), and rabbit anti-PfAldolase (1:1000, Abcam, 169,544). ECLTM Prime Western Blotting Detected Reagent (GE healthcare) was used to develop blots.
Nascent RNA labelling
Parasites synchronized at four time points (TP10, TP20, TP30 and TP40) were centrifuged at 800 g for 5 min in 15 ml tubes. The parasite pellets were washed in 10 ml pre-warmed RPMI-1640 (Gibco, 31800105) containing 5 U/ml RNase Inhibitor (Invitrogen, 10777019) and centrifuged as above. All the pellets were gently resuspended in 10 ml pre-warmed permeabilization solution (5 U/ml RNase inhibitor and 0.02% saponin (Sigma-Aldrich, 84510) in RPMI-1640), mixed by inversion and incubated in 37°C water bath for up to 5 min or until the suspension became translucent. Then, the supernatants were centrifuged (3200 g for 5 min) and discarded. Parasite pellets were resuspended in pre-warmed RPMI-1640 medium containing 5 U/ml RNase Inhibitor and transferred into a new 1.5 ml tube followed by centrifuging and two washing steps. Then, the pellets were gently resuspended in 500 μl pre-warmed transcription buffer (50 mM HEPES, 100 mM KCl, 5 mM MgCl2, 0.5 mM EGTA in water (The Ambion® Buffer Kit, AM9010)) with 5 mM DTT (Thermo Scientific™, R0861), 0.5 mM PMSF (Roche, 10837091001), 100 U/ml RNase inhibitors, 2 mM ATP, 1 mM CTP, 1 mM GTP (NEB, N0450 L) and 0.5 mM EU (Click-iT Nascent RNA Capture Kit, Thermo Fisher) and incubated at 37°C for 30 min with 300 g shaking (block heating shaker). After incubation, total RNAs were extracted from the pellet using Trizol reagents (Invitrogen, 15596026) followed by phenol-chloroform treatment. Then, the isolated RNAs were subjected to biotinylation reaction in 50 μl Click-iT reaction cocktail according to the manufacturer’s instructions. After that, 1 μl UltraPureTM Glycogen (Invitrogen, 10814010), 50 μl 7.5 M NaCH4, and 700 μl pre-chilled 100% ethanol were added to each cocktail reactions. The tube contents were mixed by vortex, and incubated overnight at −80°C. Next, the tube was centrifuged at 13,000 g for 20 min at 4°C. Then, the supernatant was removed without disturbing the RNA pellet. After washing twice with 700 μl 75% ethanol followed by centrifugation at 13,000 g for 5 min, the pellet was dried for 5 ~ 10 min at room temperature, then resolved in 50 μl pre-warmed RNase-free water (Invitrogen, 10977015). The biotin labelled nascent RNA was purified by Dynabeads MyOne Streptavidin T1 magnetic beads (Invitrogen, 65602). Beads with purified nascent RNA were resuspended in Click-iT reaction wash buffer for downstream RNA-seq library preparation.
Co-immunoprecipitation (Co-IP) and immunofluorescence assay (IFA)
Co-IP was performed as described previously [11]. In brief, parasite cultures were collected and treated with 0.15% saponin and washed three times with 1 x PBS. The released parasites were resuspended in three volumes of lysis buffer (25 mM Tris-KCl pH 7.5, 100 mM KCl, 2 mM EDTA, 0.05% NP-40, 0.5 mM phenylmethylsulphonyl fluoride, 1x protease inhibitor cocktail (Thermo Scientific™, 78439)) and lysed for 30 min on ice, then subjected to sonication for 4 min at 30 s intervals on the highest power setting with a sonicator (BioruptorTM UCD-200). The supernatants of the lysates were isolated and immediately incubated with PierceTM Anti-HA Magnetic Beads (Thermo Scientific™, 88837) at 4°C overnight. After the beads washed twice with IPP500 (500 mM NaCl, 10 mM Tris-HCl pH 8.0, 0.05% NP-40), bounded proteins were eluted with 100 μl Elution buffer (0.1 M glycine, pH 2.0) and neutralized with 15 μl Neutralization buffer (1 M Tris-HCl, pH 8.5), and then subjected to western blot analysis. For IFA, the synchronous parasites were fixed with 4% paraformaldehyde (Thermo Scientific™, 28906) in 1 x PBS, and then verified by Immunofluorescence assay using the method of Droll et al. [33]. The antibody dilution for mouse anti-Ty1 was 1:300, and second antibodies of Alexa-Fluor-488-conjugated anti-rabbit were 1:2000.
RNA-seq library preparation and sequencing
To construct nascent RNA-seq libraries, the captured nascent RNA pellets at 4 time points (TP10, TP20, TP30 and TP40) prepared from the above steps were used. In addition, for the steady-state RNAs, we have divided all the parasite cultures into two equal parts (one for capturing nascent RNA and the other for constructing the steady-state RNA-seq libraries) before nascent RNA labelling step. For RNA-seq libraries of transfection lines (Pfdis3-Ty1-Ribo, Pfrrp6-Ty1-Ribo and Pfmtr4-Ty1-Ribo) as well as their wild type strain 3D7-G7 clone, we have collected the parasites of early ring stage (TP10) in order to be able to compare with nascent RNA-seq and steady-state RNA-seq. All the RNA-seq libraries have two independent biological replicates.
Each RNA-seq libraries were prepared using KAPA Stranded RNA-seq Kit with RiboErase (HMR) (KAPA Biosystems, KR1151) according to the manufacturer’s instructions except the PCR application step in order to adapt to the high AT-rich of P. falciparum genome (reducing the extension temperature from 72°C to 62°C). In other words, all libraries were amplified for 14 PCR cycles using the following conditions: 45 s at 98°C for initial denaturation, 14 cycles of (15 s at 98°C for denaturation, 30 s at 60°C for annealing and 30 s at 62°C for extension) and 5 min at 62°C for final extension. Libraries purified and adaptor dimers depleted by 1 x volume of Agencourt AMPure XP beads (Beckman, A63881) and finally sequenced on Illumina HiSeq X Ten (Illumina, San Diego, CA, USA) to acquire 150 bp paired-end sequence reads.
Strand-specific RNA-seq data analysis
The RNA-seq data analysis procedures include raw data processing, reads alignment and filtering, transcripts assembly, and gene expression normalization (no significant discrepancy between two replicates).
Raw data processing: The raw reads were first evaluated using FastQC software [42]. Adaptor sequences and unpaired reads were removed using trim_glore tool (0.4.4_dev). The last five bases, reads with mean quality score across each base position below 25 or Ns, and reads shorter than 18 bases were filtered out until matching the quality score threshold using trim_glore.
Reads mapping: All clean paired reads were mapped to P. falciparum 3D7 genome from PlasmoDB release 36 (https://plasmodb.org) using HISAT2 software (version 2.1.0) with the options ‘–dta -p 30 – rna-strandness RF’ for dUTP stranded RNA-seq libraries and compatible with the stringtie software [43]. The multiple mapping reads, unmapped reads, and reads of mapping quality score below 25 were filtered out by samtools (version 1.7) [44]. And reads were separated according to the strand they mapped to using the samtools FLAG mark (FLAG 16 for sense strand and FLAG 0 for antisense strand). Bigwig files were generated by deeptools (version 3.1.3) and visualized in the IGV genome browser (version 2.4.13). The mapped SAM files were converted into BAM files using samtools view subcommand. Spearman correlations between replicates were calculated using multiBigwigSummary of deeptools. Downstream analyses were performed using the data from replicate 1 of each RNA-seq library.
Transcripts assembly: To generate RNA abundance Each filtered mapped reads were feed into the stringtie (version 1.3.4d) with the options ‘-p 20 – rf’ for dUTP strand-specific RNA-seq libraries [45]. The GFF annotation file (with the same release of genome) used by stringtie was obtained from the PlasmoDB release 36. All assembled transcripts were merged together using merge function of stringtie based on the default options without any adjustment.
Normalization: To generate the normalized expression profile for RNA libraries per transcripts, we adopted the normalization method according to the pervious published literature [15]. In short, for each transcript, we calculated the transcripts abundance (Transcripts Per Kilobase of exon model per Million mapped reads, TPM) by stringtie first. Then, we used a stage-specific scaling factor to accurately show transcriptional activity at each stage (Supplemental Table S2(A)). The principle of scaling factor is to consider the amount of RNA yield per parasite (for details, see normalizing method of Lu et al.) [15]. To calculate the final transcript abundance per transcript, the TPM value per transcript was divided by the scaling factor of their stage separately.
Gene classification: 5712 annotated genes (PlasmoDB release 36) were divided into three groups (constantly active, constantly silence and post-transcriptional-regulated) according to their gene abundance. We first sorted the gene abundance of steady-state RNA samples separately and rearranged the gene order in nascent RNA samples according to the order of steady-state. Genes with the final abundance value <15% of the median at each stage of steady-state RNA samples were considered as lowly or not expressed genes (constantly silence, CS); >2 folds of median were considered as the highly expressed genes (constantly active, CA); and the remains were considered as the post-transcriptional regulated genes [15]. In order to better reflect gene expression abundance, we adopted the original normalized gene abundance values without any transformation to generate the heatmaps (Fig. 1B) using the seaborn package (version 0.9.0).
Identification and classification of ns-ncRNAs
Since ns-ncRNAs were referred to the cryptic unstable RNA population, here we defined the ns-ncRNAs as those nascent transcripts with higher transcriptional abundance than the steady-state RNA by >2-fold. Therefore, two kinds of ns-ncRNAs would be identified, i.e., the fully cryptic ncRNAs which were not observed in steady-state RNAs, and the relatively cryptic ncRNAs with higher transcriptional abundance in nascent RNA samples.
ncRNA candidates were predicted from each stranded RNA-seq libraries. In brief, each assembled transcripts file (GTF format) generated by stringtie software above was feed to FEELnc tools [46]. The prediction and classification of ncRNAs using FEELnc tool could be divided into three steps. The FEELnc_filter.pl module removed the protein-coding transcripts first; the coding potential score (CPS) of filtered transcripts was calculated using the FEELnc_codpot.pl script of FEELnc tool; Finally, FEELnc_classifier.pl script was used to classification of identified lncRNAs.
Because of the very closed distance between most of the genes across from the whole genome (mean distance between two adjacent genes is 1547.5 bp), we redefined the ncRNA classifications in our annotation step based on the FEELnc_classifier.pl results. ncRNAs marked as ‘divergent’ and the distance (to their nearest annotated mRNA transcripts) <2.5 kb were defined as ‘Divergent ncRNAs’; ncRNAs marked as ‘antisense’ and the distance <0.5 kb were defined as ‘Antisense ncRNAs’; ncRNAs: 1) marked as ‘divergent’ and the distance >2.5 kb or 2) non-divergent marked but the distance >0.5 kb were defined as ‘Intergenic ncRNAs’; ncRNAs marked as ‘sense’ and the distance <0.5 kb were defined as ‘Sense ncRNAs’ (Fig. 2A, top panel).
ATAC-seq library preparation
ATAC-seq library preparation used in this manuscript was based on the published ATAC-seq method with some fine-tuning modifications for transposing reaction time (modified to 37°C for 37 min) and PCR amplification (using KAPA HiFi Hotstart Ready Mix (2X) and reducing the extension temperature to 62°C) [47]. In brief, 1 × 105 highly synchronized parasites of different stages (TP10, TP20, TP30, TP40) were lysed with 0.1% saponin in nuclease-free 1 x PBS. Then, the lysed parasites were isolated and washed once with nuclease-free 1xPBS, and permeabilized with 50 μl cold ATAC-Resuspension Buffer (ATAC-RSB) (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP40, 0.1% Tween-20 and 0.01% digitonin) for 3 min on ice. Followed by adding 1 ml wash buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20), the mixture was spun at 1800 g for 5 min to remove the supernatant. The pellet proceeded to transpose immediately by adding 50 μl transposition mixture containing 25 μl 2 x TD buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 20% dimethyl formamide), 2.5 μl transpose (100 nM final), 16.5 μl nuclease-free 1xPBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20 and 5 μl nuclease-free H2O. Reactions were incubated at 37°C for 37 min with continuous mixing and cleaned up with E.Z.N.A Cycle Pure Kit (OMGA bio-tek, D6492-02) after incubation. Libraries were amplified using KAPA HiFi Hotstart Ready Mix (2X) (KAPA Biosystems, KM2612) with following PCR condition: 72°C for 5 min; 98°C for 30 s; 13 cycles of 98°C for 20 s, 62°C for 3 min; following by 62°C for 5 min. Finally, libraries were purified using 1.8 x AMPure beads. Each library was sequenced by illumina HiSeq X Ten platform to obtain 150 bp paired-end reads.
ATAC-seq data analysis
The raw reads were evaluated using FastQC software before reads trimming. Adaptor sequences, low-quality reads and Ns were processed as the same protocol described previously in RNA-seq data analysis. Then, clean reads were mapped to P. falciparum 3D7 genome from PlasmoDB release 36 using default parameters of Bowtie2 (version 2.3.4.3) with two options modified (-X 2000, -p 30) [48]. Reads that mapped to the apicoplast and mitochondrial DNA, unmapped reads and mapping quality <25 were removed by samtools. PCR duplicated reads were filtered out using MarkDuplicates subcommand of Picard Tools (2.18.14-SNAPSHOT, http://broadinstitute. github.io/picard/). All the filtered mapped files were converted into BAM files by samtools. The reads insert sizes between 50 bp and 150 bp were selected for downstream analysis (between 39.1 and 46.1 million for all samples). All the reads were shifted 4 bp for ‘+’ strand and 5 bp for ‘-’ strand before computing the pileup signal using Linux bash shell code (‘awk’ command). The visualization files of IGV browser were generated by deeptools with the normalization of Reads Per Kilobase per Million mapped reads (RPKM). In addition, to obtain the genome background corrected tracks, the RPKM normalized coverage tracks in each library were divided by the coverage of their gDNA control (+0.1 offset).
For ATAC-seq peaks calling step, MACS2 (version 2.1.2) was used as the peak caller [49]. The local enriched peaks were called with macs2 callpeak subcommand using the gDNA bam files as background control with the following options: ‘-t -c -f BAM – nomodel – shift −75 – extsize 150’. Peaks in each stage were filtered by p value < 0.05 and fold change > 1.5 (1213, 2899, 6723 and 6656 peaks for each stage, see Supplemental Table S5).
To compare methodological consistency, we have compared the peak similarity with published datasets (GSE104075 and GSE109599) by Toenhake et al. and Ruiz JL et al. [31,32]. We reanalysed the downloaded datasets (here named ‘Toenhake data’ and ‘Ruiz data’) using our ATAC-seq datasets (here named ‘Yin data’) analysis pipeline without any parameter adjustment. Peaks for the same stage were combined together by bedtools (version v2.27.1, https://bedtools.readthedocs.io/en/latest/) intersect function. Finally, we performed Pearson correlation analysis and generated scatter diagrams of each merged peaks by their related fold changed scores (log2 transformed) for each stage.
Chromatin immunoprecipitation (CHIP-seq) and data analysis
For H3K4me3 and H3K9ac modifications, the datasets were obtained from NCBI GEO database by the accession number GSE23787 and were reanalysed [30]. For all ChIP-seq data sets, raw reads were cleaned as RNA-seq and ATAC-seq libraries in this manuscript. Then, clean reads were mapped to P. falciparum 3D7 genome from PlasmoDB release 36 using the tool of Bowtie2. Unmapped and low mapping quality reads (mapping quality score < 25) was removed using samtools. PCR duplicates were filtered out using the MarkDuplicates subcommand of Picard Tools.
To make the reads mean coverage plots of specific gene list (e.g., genes with antisense ns-ncRNAs and genes without antisense ns-ncRNAs in this manuscript), the mapped reads were first normalized by coverage depth (reads of gene model per million mapped reads, RPM) and were expressed as the ratio of between ChIP and Input. Then, we have averaged the normalized reads coverage of each gene in gene list and generated the line plot in 2 kb upstream of ATG (start codon), gene body and 2 kb downstream of TAA (stop codon) using metaseq (version 0.5.5.4) package [50].
Data visualization and GO enrichment analysis
GO enrichment analysis and functional visualization of gene or gene clusters were performed using R package ClusterProfile [51]. The FDR (false discovery rate) <0.05 was considered for a significant GO function. For data visualization, all the reads mean coverage line plots (ATAC-seq, ChIP-seq and stranded RNA-seq) were generated using metaseq package as described in previous section. All the heatmaps with k-means clustering were first calculated the gene clusters using z-scored expression values. In order to show results better, expression values at the four different stages were z-scored, followed by k-means clustering with a maximum of 1000 iterations. The number of clusters (k value for k-means) is guided by elbow method, which makes an elbow curve (k for x-axis and y-axis for SSE (sum of the squared errors)) and wherever the change in slope is highest, that is the optimum number of clusters. Circos plot was made by Circos (version 0.69) module in Perl platform (v5.22.2) [52]. The other figures such as violin plots were all generated by seaborn package.
Supplementary Material
Funding Statement
This work was supported by the National Key R&D Program of China Grant [2018YFA0507300] and the National Natural Science Foundation of China (NSFC) [81630063, 31671353 to Q.Z., 31771455 to L.J].
Availability of Data
The ChIP-seq, ATAC-seq and RNA-seq data generated in this study have been submitted to the NCBI Bioproject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA524965. The Genome Browser compatible files of the nascent RNA-seq and steady-state RNA-seq throughout the blood-stage cycles have been shown in Additional file 3.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Authors Contribution
S.Y., L. J., and Q. Z. conceived experiments. S.Y., Y. F, X.H., G. W., Y.Z., M.S., J. W., H.C., and J.H. performed experiments. S.Y., and Y.W. performed bioinformatics analysis. S.Y., L.J., and Q.Z. wrote the manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Baum J, Papenfuss AT, Mair GR, et al. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 2009;37:3788–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Duraisingh MT, Voss TS, Marty AJ, et al. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell. 2005;121:13–24. [DOI] [PubMed] [Google Scholar]
- [3].Duraisingh MT, Horn D.. Epigenetic regulation of virulence gene expression in parasitic protozoa. Cell Host Microbe. 2016;19:629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, et al. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell. 2005;121:25–36. [DOI] [PubMed] [Google Scholar]
- [5].Voss TS, Healer J, Marty AJ, et al. A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature. 2006;439:1004–1008. [DOI] [PubMed] [Google Scholar]
- [6].Josling GA, Williamson KC, Llinás M. Regulation of sexual commitment and gametocytogenesis in malaria parasites. Annu Rev Microbiol. 2018;72:501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Santos JM, Josling G, Ross P, et al. Red blood cell invasion by the malaria parasite is coordinated by the PfAP2-I transcription factor. Cell Host Microbe. 2017;21:731–741.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kafsack BFC, Rovira-Graells N, Clark TG, et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014;507:248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gómez-Díaz E, Yerbanga RS, Lefèvre T, et al. Epigenetic regulation of Plasmodium falciparum clonally variant gene expression during development in Anopheles gambiae. Sci Rep. 2017;7:40655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rai R, Zhu L, Chen H, et al. Genome-wide analysis in Plasmodium falciparum reveals early and late phases of RNA polymerase II occupancy during the infectious cycle. BMC Genomics. 2014;15:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang Q, Siegel TN, Martins RM, et al. Exonuclease-mediated degradation of nascent RNA silences genes linked to severe malaria. Nature. 2014;513:431–435. [DOI] [PubMed] [Google Scholar]
- [12].Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mahat DB, Kwak H, Booth GT, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc. 2016;11:1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Churchman LS, Weissman JS. Native elongating transcript sequencing (NET-seq). Curr Protoc Mol Biol. 2012;14: 1–17. Chapter 4, Unit 4. [DOI] [PubMed] [Google Scholar]
- [15].Lu XM, Batugedara G, Lee M, et al. Nascent RNA sequencing reveals mechanisms of gene regulation in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2017;45:7825–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Painter HJ, Chung NC, Sebastian A, et al. Genome-wide real-time in vivo transcriptional dynamics during Plasmodium falciparum blood-stage development. Nat Commun. 2018;9:2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wyers F, Rougemaille M, Badis G, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. [DOI] [PubMed] [Google Scholar]
- [18].LaCava J, Houseley J, Saveanu C, et al. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell. 2005;121:713–724. [DOI] [PubMed] [Google Scholar]
- [19].Neil H, Malabat C, d’Aubenton-Carafa Y, et al. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. [DOI] [PubMed] [Google Scholar]
- [20].Xu Z, Wei W, Gagneur J, et al. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Preker P, Nielsen J, Kammler S, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. [DOI] [PubMed] [Google Scholar]
- [22].Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Amit-Avraham I, Pozner G, Eshar S, et al. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. Proc Natl Acad Sci USA. 2015;112:E982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mourier T, Carret C, Kyes S, et al. Genome-wide discovery and verification of novel structured RNAs in Plasmodium falciparum. Genome Res. 2008;18:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Siegel TN, Hon -C-C, Zhang Q, et al. Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC Genomics. 2014;15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Broadbent KM, Broadbent JC, Ribacke U, et al. Strand-specific RNA sequencing in Plasmodium falciparum malaria identifies developmentally regulated long non-coding RNA and circular RNA. BMC Genomics. 2015;16:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. [DOI] [PubMed] [Google Scholar]
- [28].Liao Q, Shen J, Liu J, et al. Genome-wide identification and functional annotation of Plasmodium falciparum long noncoding RNAs from RNA-seq data. Parasitol Res. 2014;113:1269–1281. [DOI] [PubMed] [Google Scholar]
- [29].Seila AC, Calabrese JM, Levine SS, et al. Divergent transcription from active promoters. Science. 2008;322:1849–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bártfai R, Hoeijmakers WAM, Salcedo-Amaya AM, et al. H2A.Z demarcates intergenic regions of the plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010;6:e1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ruiz JL, Tena JJ, Bancells C, et al. Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum. Nucleic Acids Res. 2018;46:9414–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Toenhake CG, Fraschka SA-K, Vijayabaskar MS, et al. Chromatin accessibility-based characterization of the gene regulatory network underlying plasmodium falciparum blood-stage development. Cell Host Microbe. 2018;23:557–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Droll D, Wei G, Guo G, et al. Disruption of the RNA exosome reveals the hidden face of the malaria parasite transcriptome. RNA Biol. 2018;15:1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Arigo JT, Carroll KL, Ames JM, et al. Regulation of yeast NRD1 expression by premature transcription termination. Mol Cell. 2006;21:641–651. [DOI] [PubMed] [Google Scholar]
- [35].Castelnuovo M, Rahman S, Guffanti E, et al. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat Struct Mol Biol. 2013;20:851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vera JM, Dowell RD. Survey of cryptic unstable transcripts in yeast. BMC Genomics. 2016;17:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Su XZ, Heatwole VM, Wertheimer SP, et al. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. [DOI] [PubMed] [Google Scholar]
- [38].Calderwood MS, Gannoun-Zaki L, Wellems TE, et al. Plasmodium falciparum var genes are regulated by two regions with separate promoters, one upstream of the coding region and a second within the intron. J Biol Chem. 2003;278:34125–34132. [DOI] [PubMed] [Google Scholar]
- [39].Januszyk K, Lima CD. The eukaryotic RNA exosome. Curr Opin Struct Biol. 2014;24:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kilchert C, Wittmann S, Vasiljeva L. The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol. 2016;17:227–239. [DOI] [PubMed] [Google Scholar]
- [41].Davis CA, Ares M. Accumulation of unstable promoter-associated transcripts upon loss of the nuclear exosome subunit Rrp6p in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2006;103:3262–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Conesa A, Madrigal P, Tarazona S , et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li H, Handsaker B, Wysoker A, et al. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pertea M, Kim D, Pertea GM, et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, Stringtie And Ballgown. Nat Protoc. 2016;11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wucher V, Legeai F, Hédan B, et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017;45:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dale RK, Matzat LH, Lei EP. metaseq: a Python package for integrative genome-wide analysis reveals relationships between chromatin insulators and associated nuclear mRNA. Nucleic Acids Res. 2014;42:9158–9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. 2012; 16:284–287. https://homeliebertpubcom/omi [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Krzywinski M, Schein J, Birol İ, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-seq, ATAC-seq and RNA-seq data generated in this study have been submitted to the NCBI Bioproject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA524965. The Genome Browser compatible files of the nascent RNA-seq and steady-state RNA-seq throughout the blood-stage cycles have been shown in Additional file 3.