

ABSTRACT
Regulation of protein synthesis is an essential step of gene expression. This process is under the control of cis-acting RNA elements and trans-acting factors. Gemin5 is a multifunctional RNA-binding protein organized in distinct domains. The protein bears a non-canonical RNA-binding site, designated RBS1, at the C-terminal end. Among other cellular RNAs, the RBS1 region recognizes a sequence located within the coding region of Gemin5 mRNA, termed H12. Expression of RBS1 stimulates translation of RNA reporters carrying the H12 sequence, counteracting the negative effect of Gemin5 on global protein synthesis. A computational analysis of RBS1 protein and H12 RNA variability across the evolutionary scale predicts coevolving pairs of amino acids and nucleotides. RBS1 footprint and gel-shift assays indicated a positive correlation between the identified coevolving pairs and RNA-protein interaction. The coevolving residues of RBS1 contribute to the recognition of stem-loop SL1, an RNA structural element of H12 that contains the coevolving nucleotides. Indeed, RBS1 proteins carrying substitutions on the coevolving residues P1297 or S1299S1300, drastically reduced SL1-binding. Unlike the wild type RBS1 protein, expression of these mutant proteins in cells failed to enhance translation stimulation of mRNA reporters carrying the H12 sequence. Therefore, the PXSS motif within the RBS1 domain of Gemin5 and the RNA structural motif SL1 of its mRNA appears to play a key role in fine-tuning the expression level of this essential protein.
KEYWORDS: RNA-protein interaction, coevolving pairs, Gemin5, RNA-binding, RNA SHAPE reactivity, translation control
Introduction
RNA-binding proteins (RBPs) play a pivotal role in gene expression control [1]. Studies carried out over the years have established a number of conventional RNA-binding domains (RBDs) according to their structural composition and RNA recognition features [2]. However, recent RNA-capture procedures have increased the list of RBPs harbouring previously unknown RBDs [3,4], often including protein–protein interaction sites as well as intrinsically disordered regions (IDRs) [5,6]. Despite the knowledge gained of novel RBPs, non-conventional RBDs are rather diverse precluding the identification of amino acids directly involved in RNA-binding from the primary sequence of a putative RBD. This heterogeneity hampers the identification of novel RBPs by conventional methodologies.
Gemin5 is an abundant predominantly cytoplasmic protein involved in small nuclear ribonucleoproteins (snRNPs) assembly and translation control [7]. The protein was originally described as the RBP of the survival of motor neuron (SMN) complex [8]; subsequent studies reported a role in translation control [9–11]. Distinct domains of the protein are responsible for the recognition of specific targets, either RNAs or proteins [12]. Beyond the WD40 repeat domain located at the N-terminal region, which is involved in the recognition of snRNAs [13] and in ribosome interaction [14], our group identified a non-conventional RNA-binding site (designated RBS1) on the C-terminal region of the protein (Fig. 1A). The RBS1 domain is responsible for the recognition of viral IRES elements [15], as well as for specific cellular mRNAs [16]. Previous attempts to study the RNA-binding site of RBS1 by NMR indicated that this region of the protein does not adopt a uniform three-dimensional structure [15], consistent with the notion of an intrinsically disordered region.
Figure 1.
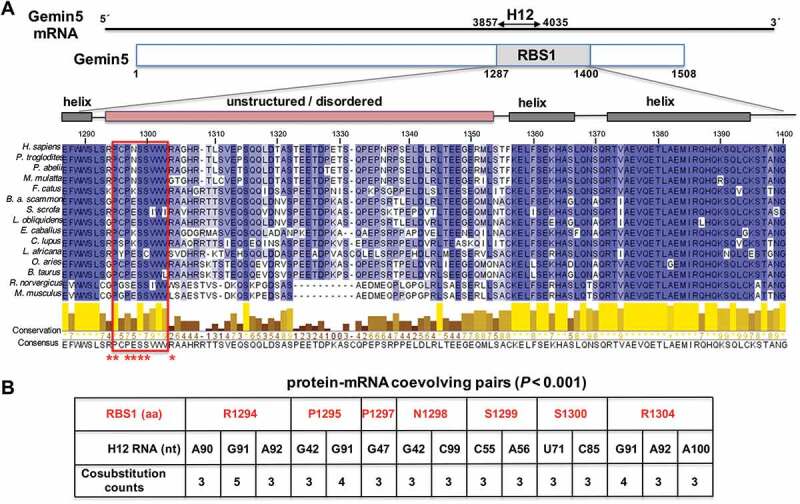
Conservation of RBS1 domain of Gemin5, and coevolving pairs found between RBS1 and the Gemin5 mRNA. (A) Schematic of Gemin5 mRNA and its coding region. Numbers indicate the nucleotides flanking the H12 region on the mRNA and the amino acids flanking the RBS1 domain on the protein. Alignment of 15 Gemin5 sequences from mammals spanning the RBS1 domain (amino acids 1287-1400). Residues are coloured according to their degree of identity. The predicted α-helices and IDR regions are depicted above the sequence. Coevolving residues are marked with red asterisks. The red rectangle across the amino acid sequences depicts the most conserved zone of the IDR. (B) Protein-mRNA coevolving pairs displaying cosubstitution counts ≥3, and p-value < 0.001. RBS1 residues are numbered as in full-length Gemin5. For simplicity, the H12 RNA nucleotides are numbered taking as position 1 the nucleotide 3857 of the mature mRNA.
We have recently shown that the RBS1 domain interacts with cellular RNAs in human cells [16]. Of note, one of the most abundant targets of RBS1 was an internal region of Gemin5 mRNA, which included two hits (designated H12 for hits 1–2) (Fig. 1A). The results obtained from functional analysis provided the basis for a positive feedback loop involving H12 RNA and the RBS1 domain. First, RBS1 stimulates the translation of RNA reporters bearing the H12 sequence. Second, RBS1–mRNA interaction was dependent on RNA structure as a destabilized RNA (H12d) displayed a decreased binding in vitro and in living cells. Third, expression of RBS1 in human cells leads to an increase of Gemin5 mRNA translation [16], counteracting the negative effect of Gemin5 on global protein synthesis. However, the specific RBS1 residues responsible for H12 RNA recognition remain elusive.
Both, specific RNA motifs and secondary RNA structure play a crucial role for protein recognition, as reflected by the impact of synonymous mutations altering the effect of secondary structure on protein expression and protein interaction [17]. Therefore, it follows that together with the amino acid sequence of RBPs, RNA structural motifs constrain the evolutionary selection of mRNA. Indeed, a driving force of coevolution in ribonucleoprotein (RNP) complex depends on biophysical interactions, revealing positional relationships preserved across evolutionary time scales [18]. The development of computational models analysing the association of proteins with extended RNA elements contributed to deciphering the mechanisms discriminating regulated from non-regulated mRNAs, besides revealing novel regulatory motifs [19]. Thus, studying the molecular determinants of the interaction between RBS1 and Gemin5 mRNA could provide hints for understanding the function of Gemin5 in gene expression control.
This possibility prompted us to investigate RNA-protein coevolving pairs between RBS1 domain and H12 RNA. To this end, we took advantage of statistical computational methods that can predict coevolution pairs between a given protein and its RNA partner [20]. Interacting positions between RNA and protein show correlated patterns of sequence evolution due to constraints imposed by the interaction that define epistasis between sites (in this case, amino acids and nucleotides). Here we show that the residues located at the N-terminal region of RBS1 predicted to coevolve with its own mRNA are critical for RNA binding. The coevolving nucleotides are placed in the stem-loop SL1 of H12 RNA, a critical RNA element for the recognition by RBS1. Conversely, mutant RBS1 proteins carrying deletions or substitutions on the predicted coevolving residues drastically reduced RNA-binding capacity. Moreover, expression of the mutant proteins in human cells failed to stimulate translation compared to the RBS1-WT polypeptide when were co-expressed with RNA reporters carrying the Gemin5 sequence H12. Together, our results reveal that selection of variants during RNA-protein coevolution contributed to fine-tune the level of expression of this multifaceted factor.
Results
Protein-mRNA coevolving pairs within the RBS1 domain and the mRNA of Gemin5
As previously reported, the RBS1 domain of Gemin5 contains a robust RNA-binding site able to recognize a coding region of its own mRNA (termed H12) in HEK293 cells [16]. Of note, the specificity of this interaction was observed in the cellular context where RNA competitors are naturally present. To identify the residues involved in RNA recognition, we first analysed its amino acid sequence conservation. Amino acid sequence alignment of the RBS1 domain showed distinct degree of conservation across this region in mammalian species (Fig. 1A). In addition, the predicted structure of Gemin5 suggests that the RBS1 domain consists of a variable intrinsically disordered region flanked by helices, consistent with previous attempts to analyse the structure of this domain by NMR, indicating the lack of a unique three-dimensional structure [15]. Notably, besides the strong conservation of residues predicted to form helical structures, the amino-terminus of the unstructured region contains a relatively conserved tract of amino acids (positions 1295-1303). Another conserved region, rich in acidic residues, is located at the C-terminus of the IDR (residues 1342-1351).
To infer the probable interacting sites between Gemin5 and its own mRNA, we detected coevolutionary signatures between the RBS1 domain of Gemin5 and its mRNA target site (H12) using a statistically robust computational method [21]. Detecting the coevolutionary site pairs involved four steps, (i) generating multiple sequence alignments for both Gemin5 protein and H12 RNA, (ii) predicting a phylogenetic tree using a combined alignment (Gemin5 and its own mRNA), (iii) inferring the evolutionary co-substitutions occurring in the internal branches of the phylogenetic tree and (iv) screening the statistically significant co-substitution pairs (coevolutionary site pairs) from the numerous site pairs obtained from step (iii). For detailed methodologies employed, refer to Materials and Methods. This robust analysis revealed 4506 initially co-substituted pairs, which were further screened through a rigorous computational and statistical pipeline giving finally 15 co-evolving pairs. The predicted 15 coevolving pairs consist of seven amino acids of RBS1 and 11 nucleotides of the H12 region of Gemin5 mRNA (nts 3857-4035 of the mRNA) (p < 0.001) (Fig. 1B). These coevolving pairs may include physically interacting residue or the residues very close to them (± 4 amino acids or ±4 nucleotides of the physically interacting pair). Interestingly, the coevolving amino acids of RBS1 stay close to each other at the N-terminal region of the unstructured moiety (residues RP-PNSS---R, 1294-1304) (Fig. 1A, red asterisks), on the most conserved region of the IDR. Conversely, the coevolving nucleotides of H12 RNA were located between positions G42-A100 (Fig. 1B; for simplicity, position 1 of H12 RNA refers to nt 3857 of Gemin5 mRNA). These coevolving pairs were used as a guide to design various perturbations in the following sections.
RNA reactivity decrease in H12 upon incubation with RBS1 provides evidence for RNA-protein binding
Beyond nucleotide sequence, RNA structure plays a key role for protein recognition. In agreement with this, the hits of RBS1 identified in living cells were predicted to adopt stable secondary structure [16]. Therefore, we sought to analyse experimentally the impact of RNA structure for RBS1 interaction. To this end, we determined the H12 RNA secondary structure in solution by selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) [22]. In this methodology, high reactive positions denote unpaired nucleotides, while non-reactive nucleotides are those forming base pairs in stems or engaged in long-range interactions, as well as those protected by proteins or other RNA ligands [23]. Incorporating the normalized reactivity values into RNAstructure software resulted into the secondary structure model of H12 RNA (Fig. 2A). This model highlights the presence of two stem-loops, designated SL1 (nt 1-123) and SL2 (nt 124-171). SL1 adopts a Y shape, including two hairpins hold by a relative long stem, whereas SL2 folds as an internal bulge flanked by short stems.
Figure 2.
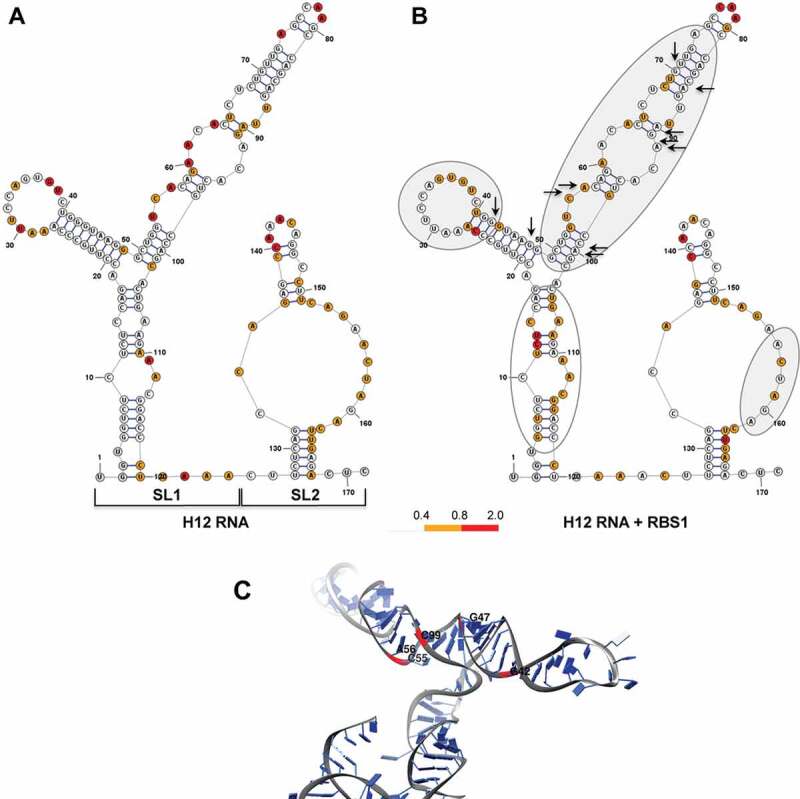
Local flexibility changes on H12 RNA upon incubation with RBS1 protein. (A) Secondary structure of naked H12 RNA according to RNAstructure software [38] incorporating the values of SHAPE reactivity. Nucleotides are coloured according to their reactivity, numbers are indicated every 10 nt. Stem-loops SL1 and SL2 are indicated. (B) Secondary structure of H12 RNA according to SHAPE reactivity upon the incubation with His-RBS1 protein (50 nM). Grey ovals indicate RNA regions with lower reactivity upon RBS1 incubation, while the empty oval represents regions with increased reactivity compared to free RNA. Black arrows mark the position of nucleotides coevolving with the RBS1 region of Gemin5 protein. (C) RNA structure model of SL1 imposing SHAPE reactivity values. Red ribbons and black letters depict nucleotides which are spatially close to each other and are involved in RNA–protein interaction.
Incubation of H12 RNA with purified His-RBS1 protein (50 nM) reduced the SHAPE reactivity of specific nucleotides located in SL1 (grey ovals in Fig. 2B), consistent with the observation that the nucleotides predicted to coevolve with RBS1 (Fig. 1B) are located in SL1. Thereby, there is a strong coincidence between statistically significant co-substitution counts and the SHAPE reactivity decrease on the coevolved nucleotides (±4 positions) upon incubation of the RNA with RBS1 (Fig. 2B, black arrows). Moreover, a three-dimensional structure model of the SL1 region shows proximity of bases involved in RNA-protein coevolution (Fig. 2C).
We also noticed an increase of reactivity in the basal stem of SL1 (compare SHAPE reactivity in Fig. 2A, B), suggesting that upon binding of the RBS1 polypeptide the RNA structure of this region is destabilized. In addition, moderate reactivity changes on SL2 were observed. In summary, these results indicated that RBS1 associates mainly to the SL1 region of H12 RNA coincident with the coevolved nucleotides.
Mapping RBS1 interaction with H12 RNA uncovers two recognition sites
The results derived from the RBS1 footprint prompted us to evaluate the structural motifs found in H12 RNA regarding the capacity to interact with RBS1. To this end, we generated two transcripts, designated SL1 (nt 1-123), and SL2 (nt 124-171) (Fig. 3A). Uniformly labelled probes H12, SL1, and SL2 were incubated with increasing amounts of purified His-RBS1, and the RNA-protein complexes were fractionated in native gels. Quantification of the retarded complexes relative to the free probe indicated that RBS1 showed dose-dependent RNA-binding ability towards H12 RNA (Fig. 3A), in agreement with previous studies [16]. In accordance with the results of the coevolution study and the footprint assay, SL1 probe retained the capacity to interact with RBS1, albeit with lower efficiency than H12 RNA. Similarly, RBS1 exhibited a decreased RNA-binding capacity towards SL2 compared to H12 RNA (Fig. 3A). However, retarded complex formation in the gel-shift assay of RBS1 with probe SL2 was lower than SL1 at high protein concentration, suggesting that SL1 is the preferential motif recognized by RBS1 in H12 RNA.
Figure 3.
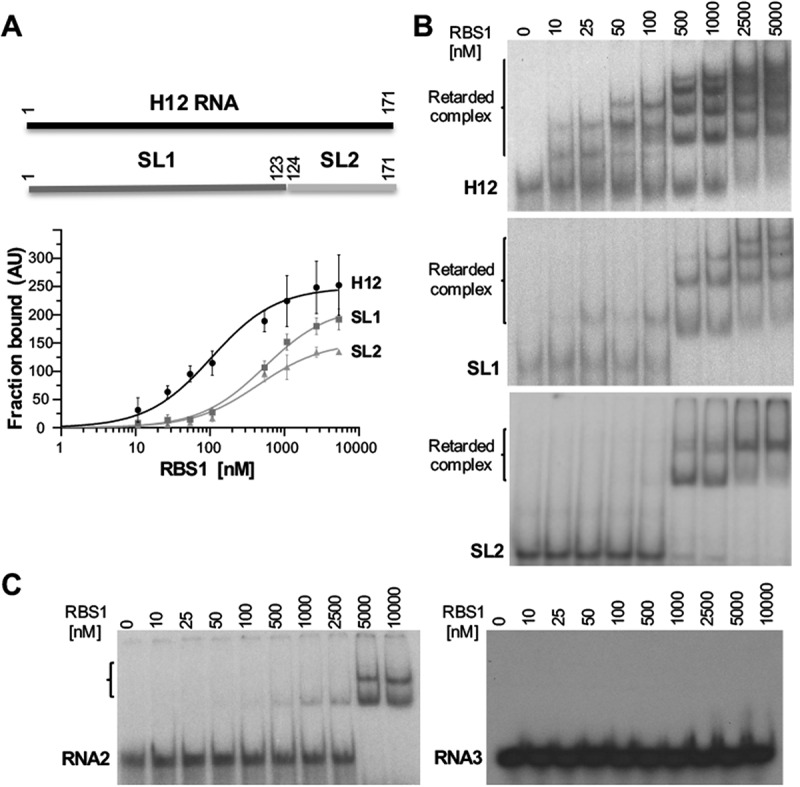
Gel-shift analysis of RNA structural motifs with RBS1. (A) Schematic representing the H12 probes used in the assay (top). (Bottom) Graph representing the adjusted curves obtained from the quantification (mean ± SD) of three independent gel-shift assays using H12, SL1, and SL2 probes incubated with increasing amounts of His-RBS1 protein. (B) Representative examples of the gel-shift assays conducted with labelled H12, SL1 or SL2 RNAs. (C) Gel-shift assays conducted with labelled RNA2 (encompassing nucleotides 4204-4259 of Gemin5 mRNA), and RNA3 (UUUCCUUU synthetic RNA).
Beyond differences in the RNA-binding capacity depending on the RNA counterpart, we noticed that the number of retarded complexes increased in a dose-dependent manner with protein concentration (Fig. 3B). This is especially remarkable for H12 and SL1 RNAs that yield several super-shifts at saturating concentration, while SL2 shows one super-shift. These observations suggest that assembly of RNA-RBS1 complexes could consist of several units of protein/RNA depending on the number of stem-loops present in the RNA and the protein concentration.
Next, to analyse the RNA-binding specificity of RBS1 we used two additional probes, RNA2 and RNA3. Gel shift assays conducted with RNA2 only produced retarded complexes at high protein concentration (5 and 10 µM) (Fig. 3C, left panel), in marked difference with H12, SL1, or SL2 RNAs (Fig. 3B). Moreover, labelled RNA3 failed to produce retarded complexes with RBS1 even at the highest protein concentration (Fig. 3C, right panel). Along this line, previous data have shown that the protein fails to interact with the H34 RNA in vitro [16]. All together, we conclude that the RBS1 protein has the capacity to interact with distinct RNAs with different affinity and specificity.
Deletion of the RBS1 coevolving residues diminished binding with SL1 RNA
Next, we focused our attention to the protein determinants for RNA-binding. The coevolution study identified seven amino acids (RP-PNSS---R, 1294-1304) within the most N-terminal region of RBS1 (Fig. 1B). Thus, we asked if this short region of the protein could be responsible for RNA interaction. Thereby we generated a His-tagged RBS1 N-terminal deletion protein, RBS1ΔPNSSVWVR, which lacks the putative coevolving amino acids (Fig. 4A). Gel-shift assays carried out with H12 probe showed that RBS1ΔPNSSVWVR reduced the binding capacity about twofold at saturating protein concentration (Fig. 4B), indicating that the coevolved residues are important determinants of RNA binding.
Figure 4.
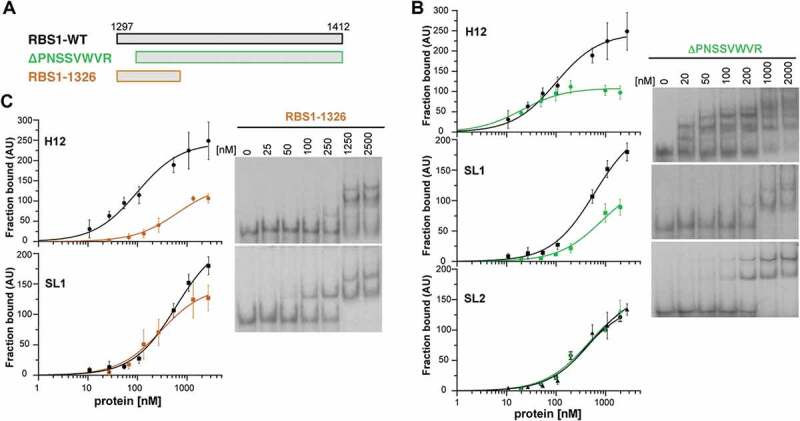
RBS1 constructs carrying deletions of the N-terminal or C-terminal residues exhibit different RNA-binding capacity. (A) Schematic representing different versions of His-RBS1 protein used in gel-shift analysis. (B) Graph representing the adjusted curves obtained from the quantification (mean ± SD) of three independent gel-shift assays using H12, SL1, or SL2 probes incubated with increasing amounts of RBS1-WT (black line) or RBS1ΔPNSSVWVR (green line) proteins. Representative examples of the gel-shift assays conducted with labelled H12, SL1, or SL2 with RBS1ΔPNSSVWVR probes (right panel). (C) Graph representing the adjusted curves obtained from the quantification (mean ± SD) of independent assays using H12 or SL1 probes incubated with increasing amounts of RBS1-WT (black line) or RBS11326 (brown line) proteins. Representative examples of the gel-shift assays conducted with labelled H12 or SL1 probes with RBS11326 (right panel).
To further investigate if this protein region also determines the recognition of the individual H12 RNA stem-loops, we performed gel-shift assays using SL1 and SL2 probes. RBS1ΔPNSSVWVR displayed a decreased retarded complex formation with the SL1 probe compared to the RBS1-WT protein (Fig. 4B), resembling the result observed with H12 RNA. In contrast, no differences in binding to SL2 probe were observed (Fig. 4B), suggesting the existence of other motifs in RBS1 preferentially interacting with SL2.
To explore the possibility that the coevolving residues are the ones responsible for the interaction with SL1, we generated the RBS11326 protein that maintains the putative coevolving residues but lacks the C-terminal region (Fig. 4A). Incubation of RBS11326 with H12 RNA showed a decreased retarded complex formation (Fig. 4C), indicating that other RNA binding motifs in RBS1 could participate in the interaction with H12 RNA, in concordance with the results of the interaction between RBS1ΔPNSSVWVR and SL2. However, the binding of RBS11326 to SL1 RNA is fairly similar to RBS1-WT protein (Fig. 4C), strongly suggesting that the coevolving residues of the protein are responsible for the interaction with SL1.
The PXSS motif of RBS1 determines its RNA-binding capacity
Since the N-terminal domain of RBS1 was sufficient to recognize SL1 RNA, we sought to identify the residues responsible for the interaction. To this end, we analysed the conservation of the region spanning coevolving residues among vertebrata species (Fig. S1). Variant positions such as R1294 and P1295 showed G, T, E, K, or S and N, R, D, H, or S substitutions, respectively, suggesting that the protein can tolerate the presence of different type of amino acids at these positions. Amino acid sequence alignment revealed that N1298 accepted positive and negatively charged residues, as well as L, V, or G; this position is assigned X (for any residue). Likewise, R1304 tolerated substitutions to L, T, A, P, W, G, N, or H. In contrast to these positions, we noticed that P1297, S1299, and S1300 were less tolerant to changes, both in number and type of amino acid (Fig. S1). Taken together, these data pointed to the strongly conserved PXSS motif as the candidate to investigate its relevance for RNA binding.
Hence, we generated two His-tagged RBS1 proteins carrying the sequence ENSS (construct P/E), or PNDD (construct SS/DD) (Fig. 5A). P1297 or S1299S1300 were substituted by E or D, respectively, two residues with chemical properties absent in vertebrata species at these positions. Gel-shift assays carried out with these proteins and H12 RNA revealed clear differences with RBS1-WT. The protein carrying the single P/E substitution showed a decreased affinity for the RNA, resulting in about 50% of retarded probe at the maximum protein concentration (Fig. 5B). Furthermore, the double substitution SS/DD drastically reduced its RNA-binding capacity, requiring higher protein concentration to form retarded complexes, which in all cases were below 50% compared with the wild type protein (Fig. 5B). We conclude that S1299 and S1300 in the PXSS motif play a critical role on the H12-binding capacity of RBS1.
Figure 5.
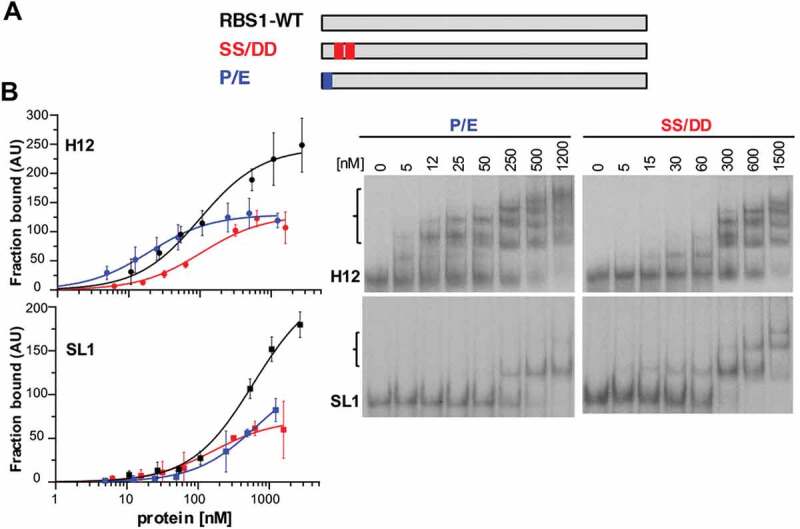
RBS1 substitution mutants display lower RNA-binding capacity. (A) Schematic depicting the substitution His-RBS1 proteins used in gel-shift analysis. (B) Graph representing the adjusted curves obtained from the quantification (mean ± SD) of independent assays using H12 or SL1 probes incubated with increasing amounts of RBS1-WT (black line), P-E (blue line) or SS-DD (red line) proteins. Representative examples of the gel-shift assays conducted with labelled H12 or SL1 with SS-DD or P-E proteins (right panels).
Next, to better understand the influence of the conserved residues P1297, S1299 and S1300 for RNA-binding we performed gel-shift assays using the P/E and SS/DD RBS1 proteins with transcript SL1, which contains all the nucleotides involved in coevolution. The results indicated the same trend with SL1 than with H12, requiring higher protein concentration to achieve 50% of RNA fraction bound (Fig. 5B). These results strongly suggest that PXSS motif forms part of the RNA-binding site of RBS1.
The PXSS motif of RBS1 mediates the counteracting effect of Gemin5 in translation control
We have recently shown that Gemin5 selectively stimulates translation of its own mRNA via recognition of the H12 sequence through the RBS1 domain [16]. This function relies on the RBS1 domain since expression of RBS1 polypeptide in human cells leads to higher levels of Gemin5. Therefore, since the RBS1 mutants SS/DD and P/E showed a marked decrease in RNA-binding affinity, we set up to determine whether the decrease in binding observed in vitro could modify translation efficiency in living cells. To this end, HEK293 cells were co-transfected with different constructs that express RBS1 wild type protein, or the mutant versions SS/DD and P/E fused to the TAP epitope. In addition, we used two reporter mRNAs. The H12-luc contains the RBS1 target sequence H12 at the 3ʹ end of the luciferase, allowing measurement of the effect of the expressed proteins depending on their ability to interact with H12 sequence. The cap-luc construct, which lacks any Gemin5 sequences, serves as a control to normalize the levels of luciferase activity produced from H12-luc (Fig. 6A). The expression of the proteins was verified by immunoblotting, using tubulin as loading control (Fig. 6B). In agreement with previous results [16], the activity of luc-H12 construct was higher than cap-luc (120%) in control cells, only expressing the TAP epitope. Expression of the RBS1 polypeptide enhanced translation of the reporter H12-luc relative to cap-luc, compared to control cells (Fig. 6C). However, expression of the RBS1 mutants SS/DD or P/E failed to stimulate translation of the luc-H12 RNA, relative to the wild type protein (Fig. 6C). Therefore, we conclude that the PXSS motif of RBS1 domain is required to stimulate translation of RNAs that contain the H12 sequence. This effect depends on the recognition of the H12 region of Gemin5 mRNA by the RBS1 domain.
Figure 6.
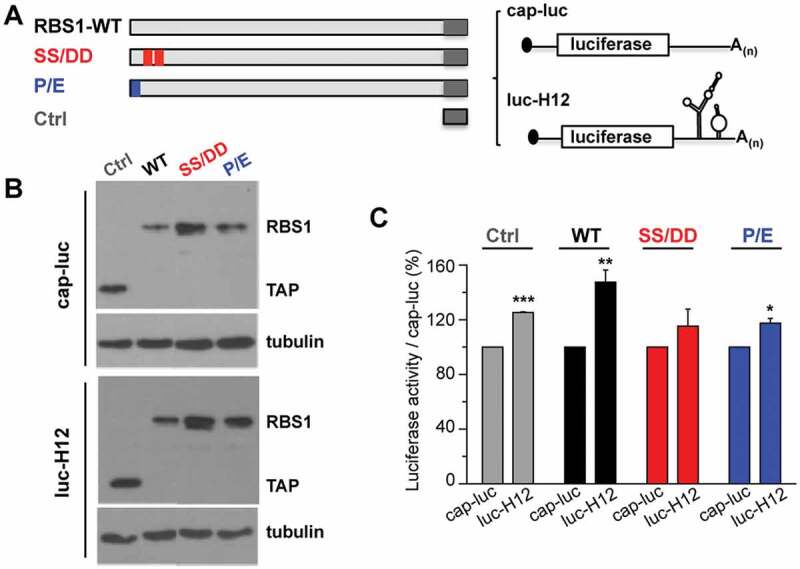
RBS1 substitution mutants lose the stimulatory effect on H12-dependent translation. (A) Schematic representing the proteins and RNA reporters expressed in HEK293 cells. (B) Expression of the TAP fusion peptide (Ctrl), RBS1-WT, SS-DD, and P-E proteins were immunodetected with anti-CBP. Tubulin was used as loading control. (C) Normalized luciferase activity obtained for luc-H12 mRNA relative to cap-luc transcript in cells expressing Ctrl, RBS1-WT, SS-DD, or P-E proteins. Values represent the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t-test).
In summary, our study has identified the coevolving residues of the RBS1 domain of Gemin5 as the residues responsible for the specific recognition of its own mRNA, providing a direct evidence of the coevolution of Gemin5 and its mRNA that, in turn, regulates its own translation.
Discussion
Gemin5 performs different functions depending on the domain of the protein involved and the counterpart targets [7]. Besides its role in the assembly of spliceosomal snRNPs [8], Gemin5 performs an important function in translation control. Thus, Gemin5 associates through its N-terminal domain to the ribosomal 60S subunit, downregulating global protein synthesis [14]. On the other hand, the C-terminal domain harbours a robust RNA-binding site (RBS1) which interacts with its mRNA stimulating its own translation [16]. This interaction has profound implications helping to fine tune the level of Gemin5 protein.
The physical interaction identified between the Gemin5 mRNA and its RBS1 domain led us to hypothesize that these counterparts could have coevolved. Computational analysis identified statistically significant co-substitution counts indicating signatures of coevolving pairs. Furthermore, additional observations support the coevolution of these separate elements. First, the seven amino acids that coevolve with RNA are close to one another, suggesting the existence of an RNA-binding motif located at the N-terminal moiety of RBS1. Consistent with this finding, previous data showed that deletions of 10 or 43 residues in the most RBS1 N-termini produced a drastic reduction in the binding to domain 5 of FMDV IRES [15]. Second, the coevolving RNA nucleotides are situated in the first half of H12 RNA, within SL1 stem-loop. This RNA region contains the bases which exhibited a marked reduction of SHAPE reactivity observed in the RBS1 footprint assay. The proximity of these nucleotides on the three-dimensional structure model of SL1 RNA (Fig. 2C) could imply that there is a selection of nucleotide substitutions within the RBS1 interaction region identified in the footprint assay. Third, LocARNA (http://rna.informatik.uni-freiburg.de) alignment obtained for two RBS1 sequence clades (Primates and Suidae) indicated that predicted base pairs on the RNA secondary structure are highly conserved (data not shown).
Previous work has established that RNA structure plays a critical role in discriminating RNA–protein interactions, including the recognition of its own mRNA as shown for fragile X mental retardation protein (FMRP) [24,25]. In this regard, the RNAs recognized by RBS1 domain do not contain a consensus sequence. On the contrary, the RNA targets were enriched in secondary structured elements [16]. Reinforcing this observation, a destabilized H12d RNA, mapped by SHAPE (Fig. S2), revealed about 30% reduced binding to RBS1 domain in vitro, and also to Gemin5 in living cells [16], relative to H12 RNA. Hence, RNA secondary structure strongly affects RBS1-H12 interaction.
Although the preferential RNA motif inside H12 recognized by RBS1 is SL1, other RNA motifs can contribute to RBS1 interaction. For instance, RBS1 binds to SL2 in gel-shift assays, albeit to lower extent than H12 or SL1 (Fig. 3), and data from RBS1 footprint showed a SHAPE reactivity decrease within SL2 (Fig. 2B). The positions with significant modifications of SHAPE reactivity are largely coincident with the nucleotides of hit 2 observed in the Gemin5 mRNA interacting with RBS1 in living cells [16] (Fig. S3).
RBS1 differs from the IDRs of well-established proteins in the absence of RGG boxes, RS dipeptides, G-rich tracts, as well as high content of aromatic residues (F, W, Y, H) [26]. A prominent feature of the non-conventional RBS1 domain is that all the coevolving amino acids reside on the most conserved moiety of the IDR, suggesting that these residues are important for RNA-binding. Accordingly, deletion of these residues as well as substitution to amino acids with different chemical properties, concur with a strong reduction of RNA-binding capacity. Our study shows that P1297, S1299, and S1300 are crucial for H12 and SL1 binding (Fig. 5), suggesting that the PXSS motif forms part of the RNA-binding site involved in its own mRNA interaction. Expression of the P-E and SS-DD mutant proteins in living cells, did not enhance H12-dependent translation as the RBS1 wild type protein (Fig. 6), indicating that the binding to H12 region in the cell is also reduced, as it occurs in vitro. We envision that understanding of intrinsic features of the non-canonical RBD of RBS1 could provide the basis for the identification of RBPs carrying similar motifs. The coevolving residues identified in this study are evolutionary conserved (Fig. S1), suggesting that the inherent sequence diversity of this region is neutralized by the need for conservation of functional elements.
Our results led us to propose that the PXSS motif of RBS1 and the SL1 stem-loop of H12 form part of key elements involved in regulating Gemin5 cellular levels (Fig. 7A). Modification of one of these elements has an effect on this regulatory mechanism. Indeed, the destabilization of the RNA secondary structure in the stem-loop SL1 of H12 (H12d mutant) hampers the association to RBS1 and fails to stimulate H12-dependent translation (Fig. 7B). Conversely, alterations on the PXSS motif of RBS1 impair the interaction with H12 and SL1, and abrogate the stimulatory effect on translation (Fig. 7C). In summary, the RNA-protein coevolution study of Gemin5 allowed the discovery of the PXSS motif of RBS1 domain as a critical region for RNA-binding. In turn, this motif plays a key role fine-tuning the cellular levels of Gemin5.
Figure 7.
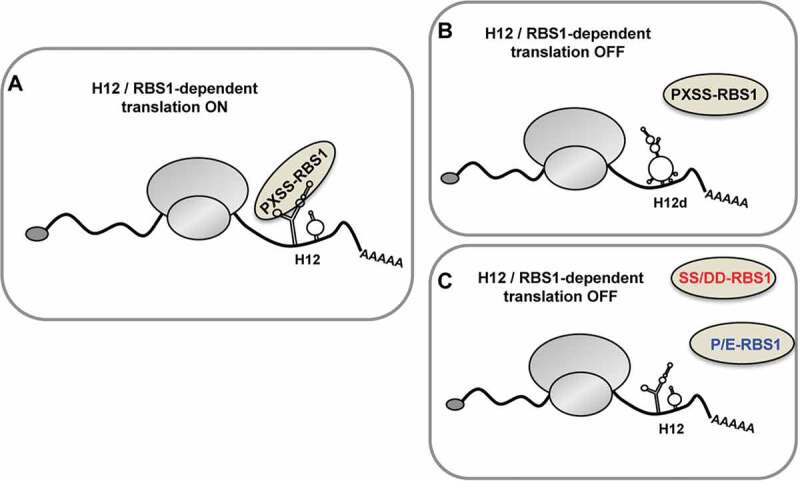
Model depicting the role of RBS1 domain of Gemin5 on H12-dependent translation. The RBS1 domain of Gemin5 recognizes the SL1 stem–loop of H12 sequence in Gemin5 mRNA through the PXSS motif, regulating its translation (A). Alteration on the mutual elements involved in RBS1-H12 interaction, such as the loss of RNA secondary structure in SL1 (H12d) (B) or the substitution of residues PXSS (C), hampers the selective translation stimulation of the Gemin5 mRNA.
Materials and methods
Computational analysis of coevolving pairs
Gemin5 protein and their respective mRNA sequences were downloaded from both Uniprot [27] and NCBI. Partial sequence data or hypothetical entries were discarded. Finally, sequence data from 60 organisms falling under Vertebrata are used for our analysis. The mRNA target region relevant for our study is H12 (nt 3857-4035) and the RBS1 region (aa 1287-1400) of Gemin5. The working pipeline follows four basic steps: (i) generating a multiple alignment using ClustalW [28], (ii) The multiple sequence alignment is used to build a phylogenetic tree using MEGA7 [29], (Maximum Likelihood, BioNJ tree, GTR Model, 1000 bootstrap replicates) (iii) tracing out the evolutionary substitutions in the internal branches, and extraction the statistically significant co-evolving site pairs.
The phylogenetic tree along with the alignment was fetched into BaseML program of PAML package to infer the posterior probabilities of the ancestral nucleotides at the internal nodes of the tree. Nucleotides possessing higher posterior probabilities are used to reckon substitution. The next step was to build a matrix where the columns represent the amino acid sites of Gemin5 protein and the nucleotide sites of Gemin5 mRNA, while the row represents the branches of the phylogenetic tree. If the xth amino acid (nucleotide) of Gemin5 gets substituted at the yth branch of the phylogenetic tree, we have designated else it remains 0. In this way, a matrix is generated where the observed co-substitution counts for for a site pair (x,y) is the count of the number of branches in which i and j is simultaneously substituted. A null distribution is obtained by randomizing the original matrix keeping the sum of the row (imposing similar evolutionary pressure on all the sites for a branch) and column (imposing similar evolutionary pressure on a single site along the divergence time scale) as constants using the Curveball algorithm [30]. Similarly, for a random matrix, we estimate the . After 1000 randomization, we extract out the pairs where is significantly greater than (p < 0.001) using one-tailed T-test. Only those pairs that come out from this stringent filtering criteria [21] are further proceeded with experimental validation. This analysis revealed 4506 initially co-substituted pairs. Further screening through the above mentioned computational and statistical pipeline resulted in 15 co-evolving pairs (cosubstitution counts ≥ 3).
Constructs
The constructs encoding the RBS1 domain of Gemin5 (pETM-11-RBS1, and pcDNA3-RBS1-CTAP), and H12 RNA (pGEM3-H12) were described [16]. The plasmid expressing the N-terminal deletion RBS1ΔPNSSVWVR protein (pET-RBS1ΔPNSSVWVR) was generated inserting the sequence obtained by PCR from pcDNA3-Xpress-G5 [14] in the vector pET-M-11 via NcoI/XhoI, using the oligonucleotides described in Table S1. Constructs expressing RBS11326 (pETM-11-RBS1-1326), and the substitution mutants (pETM-11-RBS1-P/E, pETM-11-RBS1-SS/DD, pcDNA3-RBS1-P/E-TAP, and pcDNA3-RBS1-SS/DD-TAP) were generated by the QuikChange mutagenesis procedure (Agilent Technologies) with the pairs of primers described in Table S1.
Constructs encoding H12 RNA stem-loops (pGEM3-SL1 and pGEM3-SL2) were obtained inserting the corresponding sequence into the pGEM3 vector via EcoRI/BamHI. For pGEM3-SL1, the SL1 sequence was amplified by PCR from pcDNA3-Xpress-G5 using the pair of primers described in Table S1. The constructs pGEM3-SL2 and RNA2 were obtained annealing the primers indicated in the Table S1. Oligonucleotides were purchased from Sigma and Macrogen, and all the plasmids were confirmed by DNA sequencing (Macrogen).
Expression and purification of proteins
Escherichia coli BL21 transformed with plasmids pETM-11-RBS1 (His-RBS1 wild type), pET-RBS1ΔPNSSVWVR, pETM-11-RBS1-1326, pETM-11-RBS1-P/E, and pETM-11-RBS1-SS/DD growing at 37ºC were induced with IsopropylβD-1-thiogalactopyranoside (IPTG) 0.5 mM during 2 h. Bacterial cell lysates were prepared in binding buffer (20 mM NaH2PO4, 500 mM NaCl, 20 mM Imidazole), and cell debris was eliminated by centrifugation at 16,000 g 30 min at 4ºC twice. The clear lysates were loaded in His-GraviTrap columns (HealthCare) and the recombinant proteins were eluted using imidazole 500 mM. Proteins were dialysed against phosphate buffer pH 6.8, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, and stored at −20ºC in 50% glycerol [15]. The purified proteins were analysed by SDS-PAGE.
SHAPE reactivity reactions
Prior to treatment, in vitro synthesized H12 RNA was prefolded by heating at 95°C for 2 min, snap cooling on ice for 2 min, and subsequently incubated in a final volume of 18 μl of folding mix (100 mM HEPES pH 8.0, 100 mM NaCl, 5.25 mM MgCl2) for 20 min at 37°C [31]. Prefolded RNA (2 pmol) was incubated with 6.5 mM NMIA (Thermo Fisher Scientific) for 30 min, at 37ºC [32]. Untreated RNA was incubated with DMSO. Treated and untreated RNAs were precipitated and finally resuspended in 0.5X TE (10 μl). For SHAPE probing of RNA in the presence of RBS1, RNA-protein complexes were assembled during 15 min at 37ºC in folding buffer in the presence of RBS1 (50 nM), according to the RNA–protein interaction data obtained from gel-shift assays.
Treated and untreated RNA (2 pmol) was incubated with the fluorescent primer 5ʹ-NED-TAGCCTTATGCAGTTGCTCTCC (0.1 µM) at 65°C for 5 min, then at 35ºC 5 min, and 4ºC 1 min [33]. Primer extension reactions were conducted in a final volume of 16 μl containing reverse transcriptase (RT) buffer (50 mM Tris–HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 7.5 mM DTT), 10 U RNase OUT (Thermo-Fisher Scientific), 1 mM each dNTP and 60 U of Superscript III RT (Thermo-Fisher Scientific). Reverse transcriptions were performed during 30 min at 52°C, followed by 15 min at 70ºC. Primer extension products were resolved by capillary electrophoresis. The 5ʹ-FAM-TAGCCTTATGCAGTTGCTCTCC primer was used for the sequencing ladder, using 2.5 pmol of untreated RNA in the presence of 0.1 mM ddCTP, 30 min at 52°C RT reaction [34].
SHAPE reactivity data analysis
SHAPE electropherograms were analysed using QuSHAPE software [35]. The reactivity values obtained for each untreated RNA (DMSO) were subtracted from the NMIA treated RNA to obtain the net reactivity for each nucleotide. Quantitative SHAPE reactivity for individual datasets were normalized to a scale spanning 0 to 2 in which 0 indicates an unreactive nucleotide and the average intensity at highly reactive nucleotides is set to 1. Data from two independent assays were used to calculate the mean SHAPE reactivity. For footprint analysis of RBS1 on H12 RNA, the normalized mean SHAPE reactivity obtained for the free RNA was subtracted to the mean reactivity obtained for each extract. RNA secondary structures were modelled with RNAstructure and edited with StructureEditor (http://rna.urmc.rochester.edu/). The RNA composer (http://rnacomposer.cs.put.poznan.pl/) edited with UCSF CHIMERA (https://www.cgl.ucsf.edu/chimera/) was used to generate the three-dimensional RNA model.
RNA electrophoretic mobility shift assay
RNA probes were prepared as described [36]. Briefly, transcripts were uniformly labelled using α32P-CTP (500 Ci/mmol), T7 RNA polymerase (10 U), and linearized plasmid (1 µg). RNA was extracted with phenol-chloroform, ethanol precipitated and resuspended in TE to a concentration of 0.04 pmol/µl. RNA integrity and mobility as a single band was examined in 6% acrylamide 7 M urea denaturing gel electrophoresis [37]. RNA3 (5´-UUUCCUUU-3´) was labelled at the 5´ using T4-polynucleotide kinase and γ-ATP.
RNA-binding reactions were carried out in 10 µl of RNA-binding buffer (40 mM Tris-HCl pH 7.5, 250 mM NaCl, 0.1% (w/v) βME) for 15 min at room temperature. Increasing amounts of protein were incubated with a constant concentration of 32P-labelled RNA (∼2 nM), prepared in a master mix sufficient for all points of the curve. Electrophoresis was performed in native 6.0% (29:1) polyacrylamide gels. The gels were run in TBE buffer (90 mM Tris-HCl pH 8.4, 64.6 mM boric acid, 2.5 mM EDTA) at 100 V in the cold room. The 32P-labelled RNA and retarded complexes were detected by autoradiography of dried gels. The percentage of the retarded complex was calculated relative to the free probe, run in parallel. Graphs representing the adjusted curves obtained from the quantification of at least three independent gel-shift assays were represented using GraphPad PRISM 6.01.
Gemin5 polypeptides expression and luciferase activity assays
HEK293 cell monolayers (2 × 105) were cotransfected with a plasmid expressing luciferase, with or without H12 sequence (pCAP-luc-H12, pCAP-luc) [16], and a plasmid expressing the TAP peptide, RBS1-WT, RBS1-SS/DD, or RBS1-P/E (pcDNA3-CTAP, pcDNA3-RBS1-TAP, pcDNA3-RBS1-P/E-TAP, and pcDNA3-RBS1-SS/DD-TAP) side by side using lipofectamine LTX (Thermo Fisher Scientific).
Cell lysates were prepared 24 h post-transfection in 100 µl lysis buffer (50 mM Tris-HCl pH 7.8, 100 mM NaCl, 0.5% NP40). The protein concentration in the lysate was determined by Bradford assay. Equal amounts of protein were loaded in SDS-PAGE and processed for western blotting to determine the expression of the polypeptides. RBS1 proteins were immunodetected using anti-CBP (Thermo Fisher Scientific) antibody. Immunodetection of tubulin (Sigma) was used as loading control. Secondary antibodies (Thermo Fisher Scientific) were used according to the manufacturer’s instructions. The signal detected was done in the linear range of the antibodies.
Luciferase activity (RLU)/µg of total protein was internally normalized to the value obtained with the empty vector performed side by side. Each experiment was repeated independently three times. Values represent the mean ± SEM. We computed P values for a difference in distribution between two samples with the unpaired two-tailed Student’s t-test. Differences were considered significant when P < 0.05.
Supplementary Material
Acknowledgments
We thank Gloria Lozano for advice with SHAPE analysis, Francisco Gomez for early contribution to this work, Jorge Ramajo for technical assistance, and Ugo Bastolla for valuable comments on the manuscript. This work was supported by Ministerio de Economia y Competitividad (MINECO) (grant BFU2017-84492-R), Comunidad de Madrid (B2017/BMD-3770) and an Institutional grant from Fundación Ramón Areces.
Funding Statement
This work was supported by the Comunidad de Madrid [B2017/BMD-3770]; and Ministerio de Economia y Competividad (MINECO) [BFU2017-84492-R].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Gehring NH, Wahle E, Fischer U.. Deciphering the mRNP code: RNA-bound determinants of post-transcriptional gene regulation. Trends Biochem Sci. 2017;42:369–382. [DOI] [PubMed] [Google Scholar]
- [2].Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Castello A, Fischer B, Frese CK, et al. Comprehensive identification of RNA-binding domains in human cells. Mol Cell. 2016;63:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Trendel J, Schwarzl T, Horos R, et al. The human RNA-binding proteome and its dynamics during translational arrest. Cell. 2019;176:391–403 e319. [DOI] [PubMed] [Google Scholar]
- [5].Jarvelin AI, Noerenberg M, Davis I, et al. The new (dis)order in RNA regulation. Cell Commun Signal. 2016;14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Francisco-Velilla R, Embarc-Buh A, Martinez-Salas E. Impact of RNA-protein interaction modes on translation control: the versatile multidomain protein Gemin5. Bioessays. 2019;41:e1800241. [DOI] [PubMed] [Google Scholar]
- [8].Battle DJ, Lau CK, Wan L, et al. The Gemin5 protein of the SMN complex identifies snRNAs. Mol Cell. 2006;23:273–279. [DOI] [PubMed] [Google Scholar]
- [9].Pacheco A, Lopez de Quinto S, Ramajo J, et al. A novel role for Gemin5 in mRNA translation. Nucleic Acids Res. 2009;37:582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Workman E, Kalda C, Patel A, et al. Gemin5 binds to the survival motor neuron mRNA to regulate SMN expression. J Biol Chem. 2015;5:528–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pineiro D, Fernandez N, Ramajo J, et al. Gemin5 promotes IRES interaction and translation control through its C-terminal region. Nucleic Acids Res. 2013;41:1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moreno-Morcillo M, Francisco-Velilla R, Embarc-Buh A, et al. Structural basis for the dimerization of Gemin5 and its role in protein recruitment and translation control. Nucleic Acids Res. 2020;48:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yong J, Kasim M, Bachorik JL, et al. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell. 2010;38:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Francisco-Velilla R, Fernandez-Chamorro J, Ramajo J, et al. The RNA-binding protein Gemin5 binds directly to the ribosome and regulates global translation. Nucleic Acids Res. 2016;44:8335–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fernandez-Chamorro J, Pineiro D, Gordon JM, et al. Identification of novel non-canonical RNA-binding sites in Gemin5 involved in internal initiation of translation. Nucleic Acids Res. 2014;42:5742–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Francisco-Velilla R, Fernandez-Chamorro J, Dotu I, et al. The landscape of the non-canonical RNA-binding site of Gemin5 unveils a feedback loop counteracting the negative effect on translation. Nucleic Acids Res. 2018;46:7339–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sharma Y, Miladi M, Dukare S, et al. A pan-cancer analysis of synonymous mutations. Nat Commun. 2019;10:2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Karakostis K, Fahraeus R. Shaping the regulation of the p53 mRNA tumour suppressor: the co-evolution of genetic signatures. BMC Cancer. 2019;19:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou Q, Kunder N, De la Paz JA, et al. Global pairwise RNA interaction landscapes reveal core features of protein recognition. Nat Commun. 2018;9:2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mallik S, Kundu S. Coevolutionary constraints in the sequence-space of macromolecular complexes reflect their self-assembly pathways. Proteins. 2017;85:1183–1189. [DOI] [PubMed] [Google Scholar]
- [21].Mallik S, Basu S, Hait S, et al. Translational regulation of ribosomal protein S15 drives characteristic patterns of protein-mRNA epistasis. Proteins. 2018;86:827–832. [DOI] [PubMed] [Google Scholar]
- [22].Lozano G, Fernandez N, Martinez-Salas E. Magnesium-dependent folding of a picornavirus IRES element modulates RNA conformation and eIF4G interaction. FEBS J. 2014;281:3685–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Weeks KM, Mauger DM. Exploring RNA structural codes with SHAPE chemistry. Acc Chem Res. 2011;44:1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Didiot MC, Tian Z, Schaeffer C, et al. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 2008;36:4902–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Taliaferro JM, Lambert NJ, Sudmant PH, et al. RNA sequence context effects measured in vitro predict in vivo protein binding and regulation. Mol Cell. 2016;64:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Choi JM, Holehouse AS, et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018;174:688–699 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Strona G, Nappo D, Boccacci F, et al. A fast and unbiased procedure to randomize ecological binary matrices with fixed row and column totals. Nat Commun. 2014;5:4114. [DOI] [PubMed] [Google Scholar]
- [31].Lozano G, Trapote A, Ramajo J, et al. Local RNA flexibility perturbation of the IRES element induced by a novel ligand inhibits viral RNA translation. RNA Biol. 2015;12:555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wilkinson KA, Merino EJ, Weeks KM. Selective 2ʹ-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. [DOI] [PubMed] [Google Scholar]
- [33].Dotu I, Lozano G, Clote P, et al. Using RNA inverse folding to identify IRES-like structural subdomains. RNA Biol. 2013;10:1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lozano G, Francisco-Velilla R, Martinez-Salas E. Ribosome-dependent conformational flexibility changes and RNA dynamics of IRES domains revealed by differential SHAPE. Sci Rep. 2018;8:5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Karabiber F, McGinnis JL, Favorov OV, et al. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA. 2013;19:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pineiro D, Ramajo J, Bradrick SS, et al. Gemin5 proteolysis reveals a novel motif to identify L protease targets. Nucleic Acids Res. 2012;40:4942–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fernandez-Chamorro J, Francisco-Velilla R, Ramajo J, et al. Rab1b and ARF5 are novel RNA-binding proteins involved in FMDV IRES-driven RNA localization. Life Sci Alliance. 2019;2(1):pii: e201800131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.