

ABSTRACT
The ability to rapidly respond to applied force underpins cell/tissue homeostasis. This response is mediated by mechanotransduction pathways that regulate remodeling and tension of the actomyosin cytoskeleton to counterbalance external forces. Enhanced extracellular matrix tension hyper-activates mechanotransduction and characterizes diseased states such as cancer, but is also required for normal epidermal regeneration. While the impact of extracellular matrix tension on signaling and cell biology are well appreciated, that of acute compressive force is under-studied. We show here that acute compressive force applied to cells and tissues in a native 3-dimensional context elevates RHOA-GTP levels and increases regulatory myosin phosphorylation, actomyosin contractility and tension via ROCK. In consequence, cell proliferation was increased, as was the expression of regulators of epithelial-mesenchymal transition. Pharmacological inhibition of ROCK abrogated myosin phosphorylation, but not RHOA activation. Our results strongly suggest that acute compressive stress impairs cellular homeostasis in a RHO/ROCK-dependent manner, with implications for disease states such as cancer.
KEYWORDS: RHOA, ROCK, actomyosin tension, cytoskeleton, compressive stress, extracellular matrix (ECM), mechano-reciprocity, mechanical signaling
Introduction
Most tissues encounter external mechanical forces throughout their lifetime. However, aberrantly increased external force may be experienced in pathological conditions such as edema or tumor growth. Tissue integrity and function rely on rapidly counterbalancing these forces. Mechanical forces exerted by the extracellular matrix (ECM) upon cells, be they shear, compressive or tensile forces, are rapidly counteracted by cytoskeletal forces generated within the cell in a phenomenon termed mechano-reciprocity [1,2]. These forces are generated through the contractility or elongation of the filaments of the actomyosin cytoskeleton, which is regulated by defined biochemical signaling pathways initiated by mechano-sensitive receptors such as integrins on the cell surface [3].
Understanding the phenomenon of mechano-reciprocity and its regulation during homeostasis is necessary for investigating how these processes become corrupted in disease. Mechanical force can significantly influence tumor growth and metastasis [4], and application of tension to cells can stimulate diverse phenotypic responses [5]. We have previously shown that normal epithelial cells and those of tumor origin can enhance ECM tension by causing the increased production of ECM components [6]. This increased ECM tension is in turn sensed by cells, leading to a vicious cycle of persistent and pro-tumorigenic mechano-reciprocity. As enhanced mechano-reciprocity is a significant feature of cancer progression within solid tumors [2], identifying the specific players and pathways that respond to mechanical stress is of clinical significance.
The main regulator of cytoskeletal dynamics in non-muscle cells is myosin II. Its regulatory subunit, myosin regulatory light chain-2 (MLC2), can be directly activated by calcium-dependent MLC kinase (MLCK), Rho-associated coiled-coil-containing protein kinase (ROCK) [7], and others [8–10]. Myosin II activity is also controlled indirectly through inhibition of its negative regulators. ROCK, protein kinase C, p21-activated kinase and integrin-linked kinase can all independently deactivate myosin phosphatase (MLCP) through phosphorylation and inhibition of its targeting subunit (MYPT1) [11,12]. Actomyosin tension within the cytoskeleton is therefore tightly regulated.
Serine/threonine kinases ROCK1 and ROCK2 contain a RHO-binding domain and regulate both actin polymerization and myosin contractility [13]. They are mainly activated by the small GTPase RHOA when in its active GTP-bound form [14]; however they may also be directly activated by other second messengers including arachidonic acid [15] and sphingosylphosphorylcholine [16].
It was recently reported that pMlc2 and pMypt1 levels were markedly and acutely reduced despite increased activation of RhoA in murine skeletal myoblast cells cultured in two dimensions and compressed laterally along the culture plane [17]. Under these conditions, RhoA was reported to be phosphorylated by protein kinase A, thereby precluding its association with, and activation of Rock. We therefore sought to determine whether acute compression of cells within collagen matrices in a more physiological, three-dimensional context, or ex vivo compression of freshly dissected tissues exhibited similar non-canonical signaling downstream of RhoA.
Here we show that applying acute compressive force to cells cultured in three-dimensional matrices, or to freshly isolated, intact epithelial tissues, engages the canonical RHO/ROCK signaling pathway to regulate actomyosin tension, with consequent functional outcomes known to determine cell fate.
Results
RHOA is activated by compressive stress
Active RHOA-GTP binds to and activates the actomyosin regulator ROCK [18], downstream of varied extracellular stimuli including ECM tension [19]. To determine if compression activated RHOA in a three-dimensional (3D) context, HEK-293T cells cultured within collagen matrices were placed on BioPress™ compression culture plates, and a Flexcell® FX-5000™ Compression System was used to apply static mechanical stress at 20 kPa (Fig. 1A), which is within the physiological range for tissues [20]. Compressive stress significantly and persistently increased the number of cells with GTP-bound, active RHOA by ∼9-fold after only 2 minutes of compression (Fig. 1B-C) as detected by quantitative immunofluorescence analysis using a conformation-specific antibody raised against the active form of RHOA [21–23] (Supp. Fig. 1). These observations are consistent with previous reports of experiments undertaken in two-dimensional cell culture [17].
Figure 1.
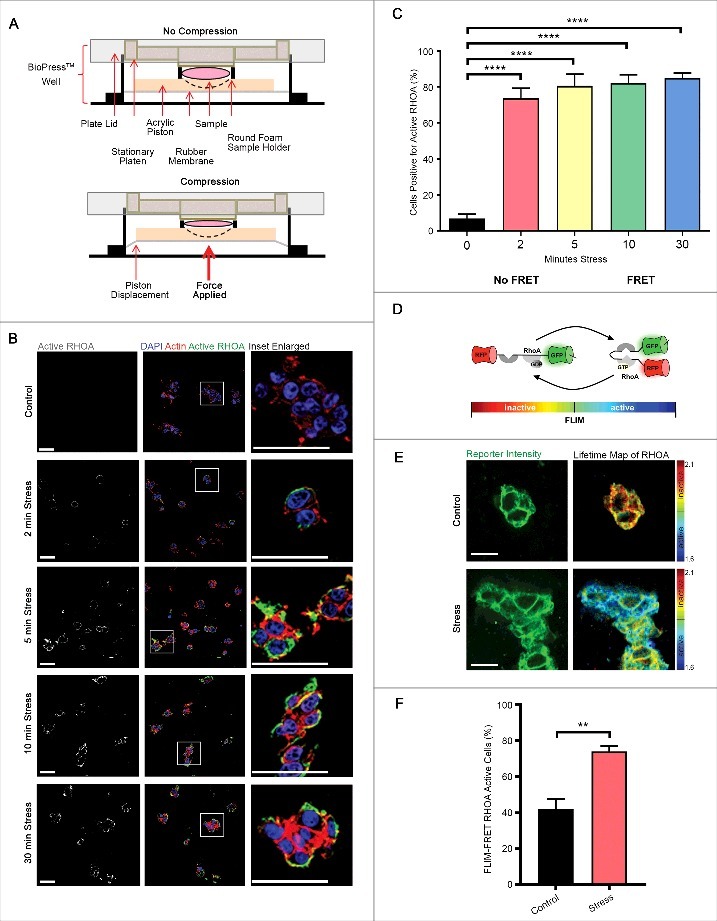
RHOA is activated by compressive stress. (A) Diagram of how compressive stress was applied to cells embedded in collagen matrices, and to whole mouse tissues. (B-C) Immunofluorescence analysis of GTP-bound RHOA (white in monochrome and green in merge), as detected by a conformation-specific anti-Active RHOA antibody, in HEK-293T cells embedded in collagen followed by application of compressive stress for times as specified. F-actin is labelled with phalloidin and cell nuclei with DAPI (red and blue in merge). Scale Bars: 50 µm. Column graph shows percentage of cells positive. n = 5 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. ****p < 0.0001. (D) Schematic representation of the principle of Förster resonance energy transfer (FRET) using the Raichu RHOA-FRET biosensor. (E) Representative images of collagen-embedded HEK-293T-RHOA-FRET cells (green) following compressive stress for 10 minutes, with corresponding lifetime map of RHOA FRET. Scale Bars: 20 µm. (F) HEK-293T-RHOA-FRET cells were subjected to compressive stress for 10 minutes and percent of FLIM-FRET RHOA active cells quantified. n = 3 collagen matrices per analysis and data (mean+SEM) were analyzed by unpaired t-test. **p < 0.01.
In order to independently confirm that compressive stress activated RHOA, we took advantage of a Raichu RHOA-FRET biosensor (Fig. 1D) that we have adapted and characterized previously [24–26]. Cells in which this biosensor had been expressed exhibited decreased fluorescence lifetimes as a result of FRET following compression, indicating increased RHOA activity (Fig. 1E-F), consistent with our observation (Fig. 1B-C) from immunofluorescence analysis using the antibody raised against active RHOA-GTP, that RHOA is present in its active conformation in these cells.
Compressive stress increases actomyosin tension
We next sought to determine whether externally applied compressive forces that activate RHOA can similarly increase the intracellular actomyosin adaptive force by activating the regulatory myosin light chain MLC2. HEK-293T cells were embedded within collagen matrices and subjected to compression at 20 kPa. Cells exhibited activating phosphorylation of MLC2 at Ser19 (Fig. 2A-B) [27] in an incremental manner as well as inhibitory phosphorylation of the myosin phosphatase target subunit MYPT1 at Thr696 (Fig. 2D-E), thereby preventing the inactivation of MLC2 by myosin phosphatase [28]. Furthermore, phosphorylation levels of both proteins, measured by integrated density, were also elevated over time (Fig. 2C, F). Following 30 minutes of compression, phosphorylation of these proteins was observed in approximately 80% of cells. Interestingly, our analysis revealed a step-wise temporal progression of signaling, with compression first triggering activation of RHO followed by phosphorylation of MLC2 and then of MYPT1 (Fig. 2G, data taken from Fig. 1C, Fig. 2B, E). The activation of MLC2 and the inhibition of MYPT1 are strongly correlated with actomyosin contractility and enhanced actomyosin force generation [13]. These data therefore suggest that acute compression rapidly increases force generation by the actomyosin cytoskeleton.
Figure 2.
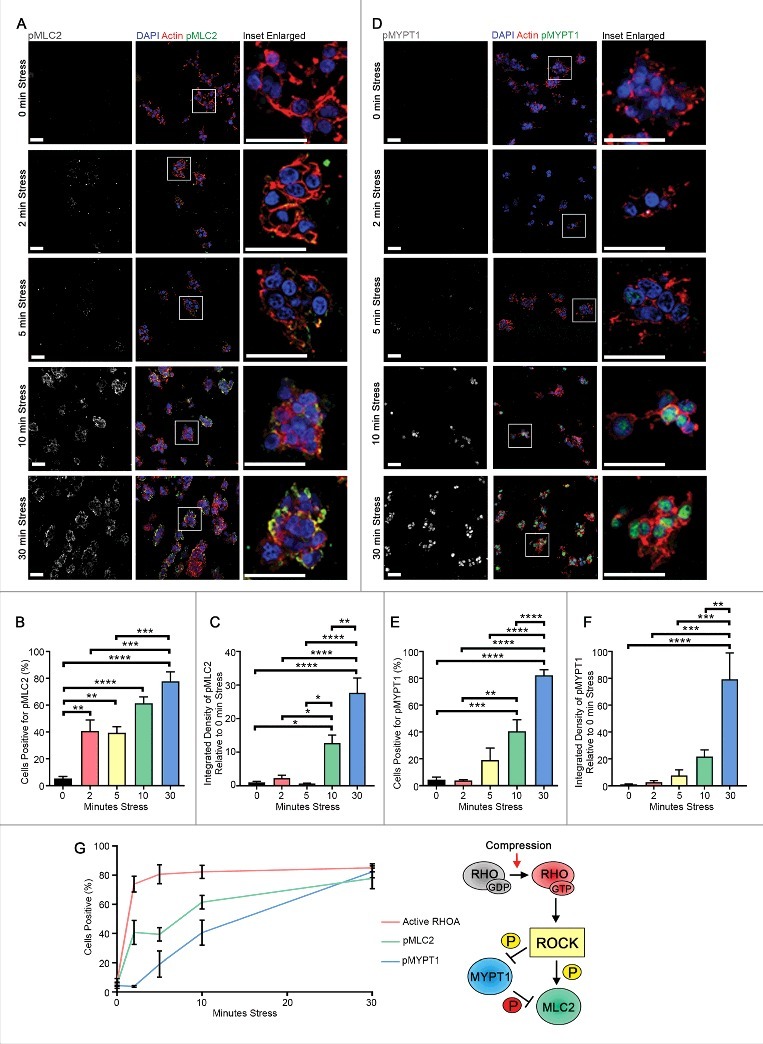
Compressive stress progressively elevates actomyosin tension. Immunofluorescence analysis of p(Ser19)MLC2 (A-C) and p(Thr696)MYPT1 (D-F) (white in monochrome and green in merge) of HEK-293T cells embedded in collagen followed by application of compressive stress for time points as indicated. F-actin is labelled with phalloidin and cell nuclei with DAPI (red and blue in merge). Scale Bars: 50 µm. Column graphs show percentage of cells positive and level of integrated density relative to no stress. n = 5-7 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (G) Time-course of RHO/ROCK signaling activation (mean±SEM) upon compression and inferred sequence of signaling events.
We next investigated whether compression-mediated enhancement of actomyosin tension is a feature of tissues. The gastrointestinal tract is exposed to a number of physical stresses, including peristalsis and muscle contraction, the movement of food boluses and cyclical strain due to motile villi [29]. We therefore subjected freshly dissected murine proximal small and large intestinal tissues to compressive stress to determine whether a frequently stressed tissue is nevertheless capable of responding to acute compressive stress in a similar manner to cells cultured in 3D collagen matrices. Both small intestine (Fig. 3A-D) and large intestine (Fig. 3E-H) tissues subjected to compressive stress exhibited significantly higher Mlc2 and Mypt1 phosphorylation compared to uncompressed tissues, assessed by either Western analysis of epithelial lysates (Fig. 3A, E) or quantitative immunofluorescence of tissues (Fig. 3B-C, 3F-G).
Figure 3.
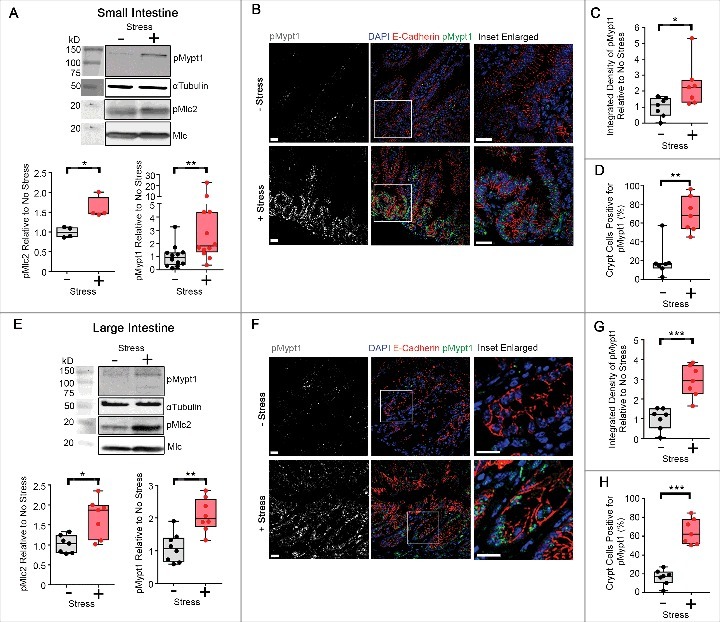
Compressive stress elevates actomyosin tension in freshly dissected mouse intestinal tissues. (A) Western analysis of p(Thr18/Ser19)Mlc2 (n = 4 mice) and p(Thr696)Mypt1 (n = 12 mice) in epithelial cell lysates derived from proximal murine small intestinal tissue subjected to compressive stress. Box and whisker plots show relative band intensities. (B-D) Immunofluorescence analysis (B) of p(Thr696)Mypt1 (white in monochrome and green in merge) in small intestinal tissues (n = 7 mice) subjected to compressive stress. Cell junctions are labelled with E-Cadherin and cell nuclei are labelled with DAPI (red and blue in merge). Scale Bars: 20 µm. Box and whisker plots show level of integrated density relative to no stress (C) and percentage of crypt cells positive (D), and were graphed by averaging multiple fields of view. (E) Western analysis of p(Thr18/Ser19)Mlc2 (n = 7 mice) and p(Thr696)Mypt1 (n = 8 mice) in epithelial cell lysates derived from murine large intestinal tissue subjected to compressive stress. Box and whisker plots show relative band intensities. (F-H) Immunofluorescence analysis (F) of p(Thr696)Mypt1 (white in monochrome and green in merge) in large intestinal tissues (n = 7 mice) subjected to compressive stress. Cell junctions are labelled with E-Cadherin and cell nuclei are labelled with DAPI (red and blue in merge). Scale Bars: 20 µm. Box and whisker plots show level of integrated density relative to no stress (G) and percentage of crypt cells positive (H), and were graphed by averaging multiple fields of view. (A-H) Data (median±IQR) were analyzed by the Mann-Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001.
Interestingly, we observed that compression-induced Mypt1 phosphorylation was highest within the crypts of Lieberkühn (Fig. 3D, H), and quantification of the proportion of cells positive for pMypt1 within the crypts revealed a highly significant difference between compressed and uncompressed tissues. This suggests that intestinal crypts are more sensitive to changes in externally applied stress than the lateral epithelial surfaces of the intestine.
Next, we assessed the consequences of acute compressive stress on skin, an organ frequently subjected to such forces by interaction with the external environment. Following the application of acute compressive stress upon murine dorsal skin, we observed an overall increase in Mlc2 and Mypt1 phosphorylation, indicating increased actomyosin-exerted force (Fig. 4A).
Figure 4.
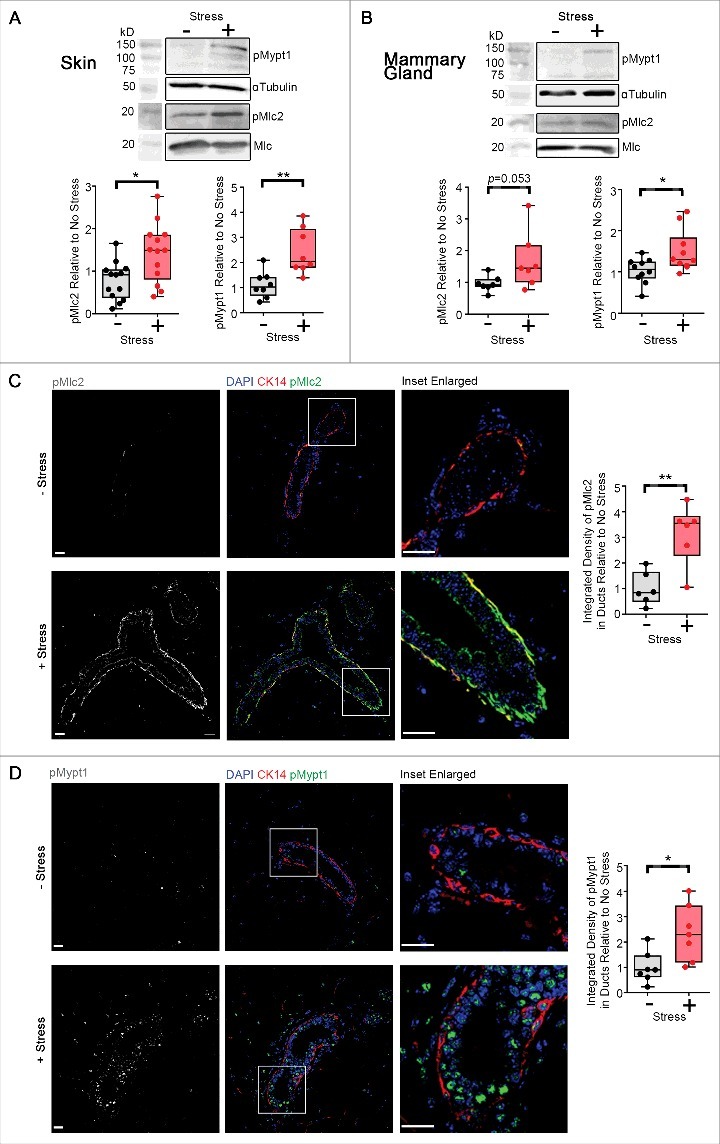
Compressive stress elevates actomyosin tension in freshly dissected mouse skin and mammary tissues. (A) Western analysis of p(Thr18/Ser19)Mlc2 (n = 13 mice) and p(Thr696)Mypt1 (n = 8 mice) in skin tissue subjected to compressive stress. Box and whisker plots show relative band intensities. (B) Western analysis of p(Thr18/Ser19)Mlc2 (n = 7 mice) and p(Thr696)Mypt1 (n = 10 mice) in mammary gland tissue subjected to compressive stress. Box and whisker plots show relative band intensities. (C-D) Immunofluorescence analysis of p(Ser19)Mlc2 (n = 6 mice) (C) and p(Thr696)Mypt1 (n = 7 mice) (D) (white in monochrome and green in merge) in mammary gland tissue subjected to compressive stress. Basal cells are labelled with cytokeratin-14 and cell nuclei are labelled with DAPI (red and blue in merge). Scale Bars: 20 µm. Box and whisker plots show level of integrated density relative to no stress, and were graphed by averaging multiple fields of view. (A-D) Data (median±IQR) were analyzed by the Mann-Whitney test. *p < 0.05, **p < 0.01.
The mammary gland encounters a multitude of external stresses, during ductal morphogenesis, lactation and involution [30]. Consistent with the need to counteract forces arising from these processes, mammary ducts exhibited greater Mypt1 phosphorylation when subjected to acute compressive stress as measured by Western analysis on total mammary gland lysates (Fig. 4B), or within mammary ductal structures as measured by immunofluorescence analysis (Fig. 4D), and greater Mlc2 phosphorylation within mammary ductal structures as measured by immunofluorescence analysis (Fig. 4C), relative to uncompressed samples in a manner that is similar to that demonstrated above in intestinal and skin tissue. Western analysis of whole mammary fat pad lysates showed a trend towards increased phosphorylation of Mlc2 in samples subjected to compressive stress (Fig. 4B), compared to uncompressed tissues, although this difference was not statistically significant.
Compression-induced enhancement of actomyosin tension requires ROCK kinase activity
While MLC2 can be activated by multiple signaling pathways [10,27], we have previously shown that Rho-associated kinase (ROCK) integrates biochemical and biomechanical signaling by forcing the equilibrium between the phosphorylated active and non-phosphorylated inactive forms of MLC2 towards the active form in vivo [6,31,32]. To determine whether MLC2 and MYPT1 phosphorylation downstream of compressive stress was mediated by ROCK, we pre-treated collagen-embedded HEK-293T cells with Y27632 or Fasudil, which are selective pharmacologic inhibitors of ROCK kinase activity that are structurally unrelated. Inhibiting ROCK kinase activity with either inhibitor strongly attenuated compressive stress-induced MLC2 (Fig. 5A-C) and MYPT1 (Fig. 5D-F) phosphorylation, suggesting that the induction of actomyosin tension downstream of compressive stress is via the ROCK pathway, and that this response can be inhibited pharmacologically. Pre-treatment of cells with Y27632 or Fasudil before compression did not affect RHOA activation, as measured by Active RHOA fluorescence analysis (Fig. 6A-B) or RHOA-FLIM-FRET (Fig 6C-D), but pre-treatment with a RHO inhibitor (RHOi – a cell-permeable version of the exoenzyme C3 Transferase from Clostridium botulinum) depleted both RHOA-GTP (Fig. 6A-B) and downstream phosphorylation of MLC2 (Fig. 6E-F). Taken together, these data strongly suggest that actomyosin contractility is regulated by ROCK activity downstream of RHOA activation, following the application of compressive stress.
Figure 5.
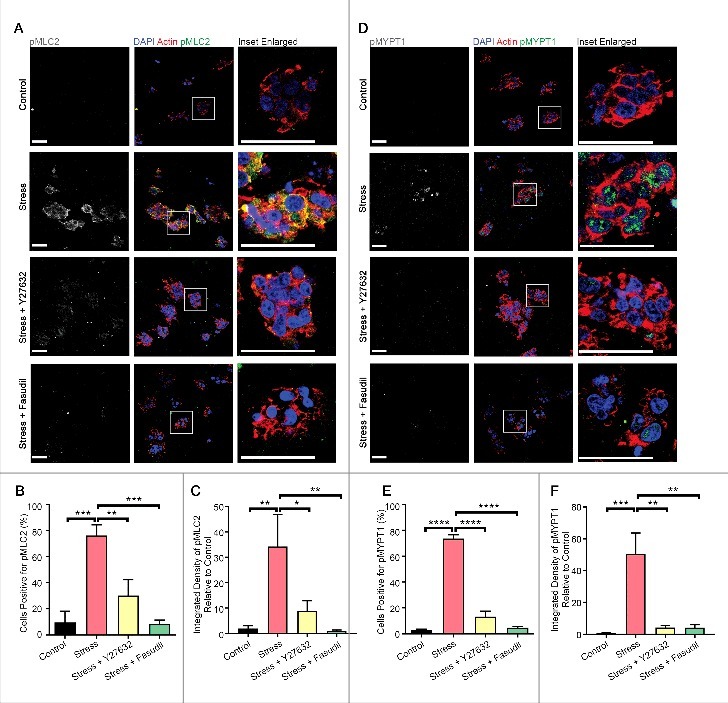
Compression-induced enhancement of actomyosin tension requires ROCK kinase activity. Immunofluorescence analysis of p(Ser19)MLC2 (A-C) and p(Thr696)MYPT1 (D-F) (white in monochrome and green in merge) in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with the ROCK-specific kinase inhibitors Y27632 and Fasudil. F-actin is labelled with phalloidin and cell nuclei with DAPI (red and blue in merge). Scale Bars: 50 µm. Column graphs show percentage of cells positive and level of integrated density relative to no stress. n = 5-7 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 6.
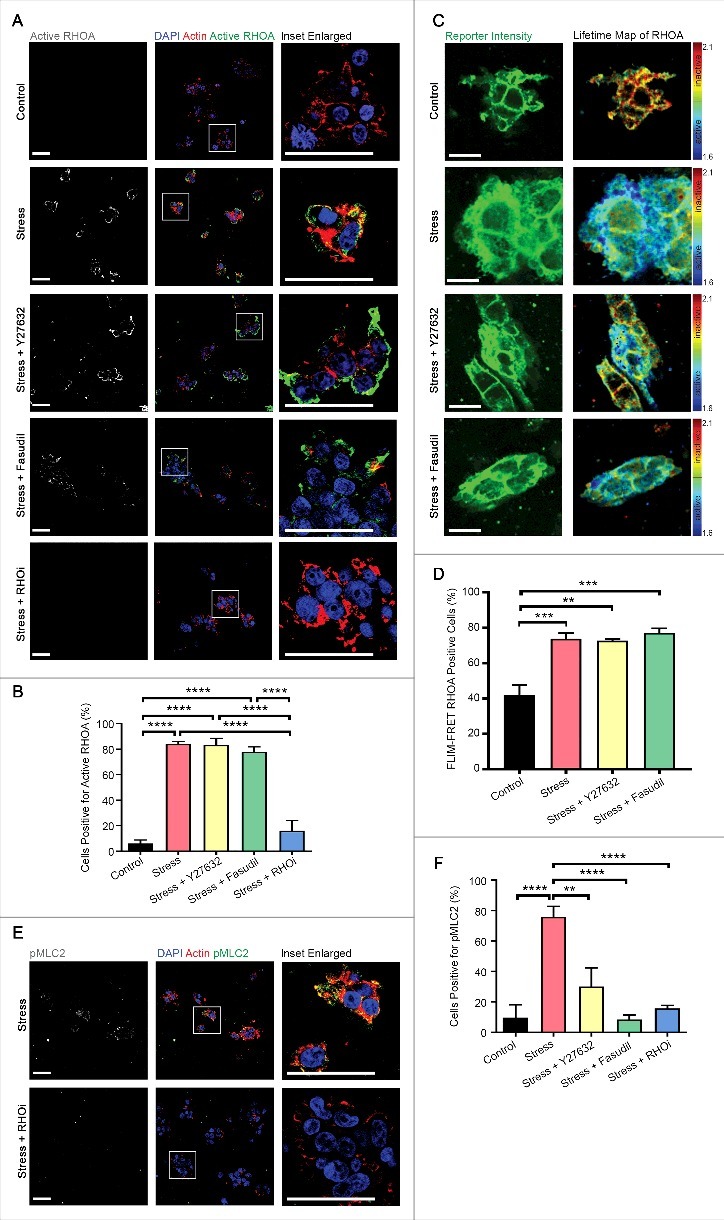
Compression-induced enhancement of RHO signaling can be blocked pharmacologically. (A-B) Immunofluorescence analysis of GTP-bound active RHOA (white in monochrome and green in merge), as detected by a conformation-specific anti-Active RHOA antibody, in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil, and a RHO-specific inhibitor (RHOi). F-actin is labelled with phalloidin and cell nuclei with DAPI (red and blue in merge). Scale Bars: 50 µm. Column graph shows percentage of cells positive. n = 5-7 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. ****p < 0.0001. (C) Representative images of collagen-embedded HEK-293T-RHOA-FRET cells (green) followed by application of compressive stress for 10 minutes with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil, with corresponding lifetime map of RHOA FRET. Scale Bars: 20 µm. (D) HEK-293T-RHOA-FRET cells were subjected to compressive stress for 10 minutes with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil and percent of FLIM-FRET RHOA active cells quantified. Data is combined with that in Fig. 1F. n = 3 collagen matrices per analysis and data (mean+SEM) were analyzed by one-way ANOVA. **p < 0.01. ***p < 0.001. (E-F) p(Ser19)MLC2 (white in monochrome and green in merge) in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with RHOi. Data in F is combined with that in Fig. 5B. F-actin is labelled with phalloidin and cell nuclei with DAPI (red and blue in merge). Scale Bars: 50 µm. Column graph shows percentage of cells positive. n = 5-7 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. **p < 0.01, ****p < 0.0001.
Actomyosin tension caused by compressive stress induces persistent cell physiological effects
Signaling downstream of RHOA is known to regulate diverse cellular processes including proliferation [33,34], cell migration [35] and epithelial-mesenchymal transition (EMT) [36] in a variety of cell types. We therefore next investigated whether RHO/ROCK pathway activation caused by acute compressive stress to cells cultured in collagen matrices caused persistent changes in cellular physiology and function. HEK-293T cells were embedded in collagen matrices, were then either subjected to compression or not compressed, placed back in culture under serum starvation conditions for a further 16 hours and pulsed with Bromodeoxyuridine (BrdU) for an hour prior to fixation. Cells subjected to compressive stress exhibited over 5-fold more cells that had incorporated BrdU relative to cells that had not been subjected to acute compression (Fig. 7). Furthermore, pretreatment before compression with the ROCK inhibitors Y27632 or Fasudil, or the RHO inhibitor (RHOi), largely abolished BrdU incorporation (Fig. 7). These data demonstrate that acute compressive stress strongly induces cell proliferation and that this is mediated by signaling through RHO and ROCK.
Figure 7.
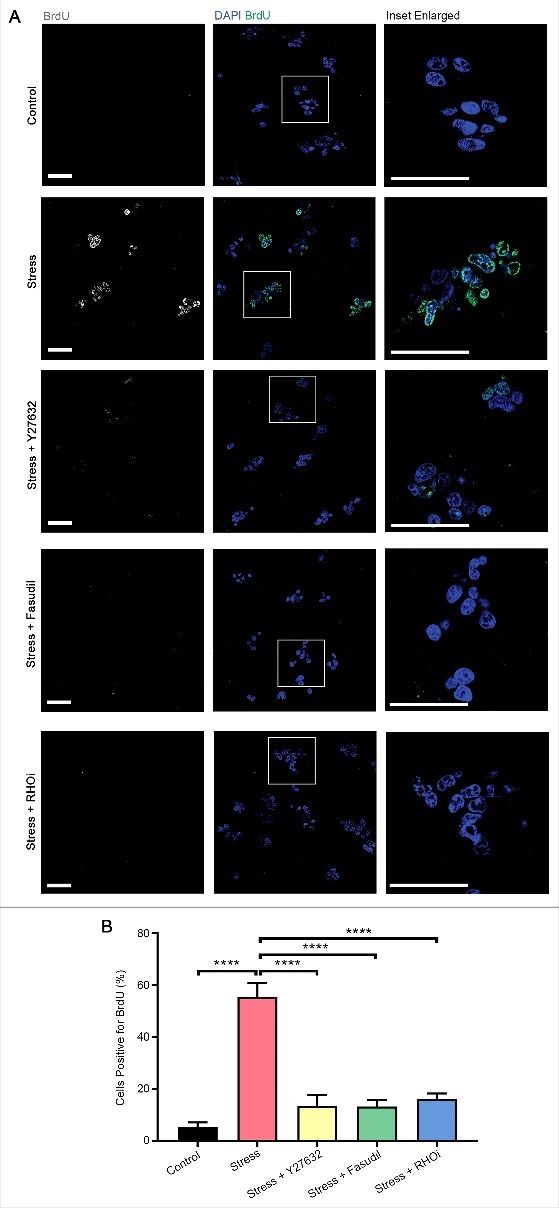
Compression induces long-term effects on cellular proliferation. (A) Immunofluorescence analysis of bromodeoxyuridine (BrdU, white in monochrome and green in merge) incorporation in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil and a RHO-specific inhibitor (RHOi). Cells were cultured in starvation conditions for a further 16 hours before pulsing with BrdU for analysis. Cell nuclei are labelled with DAPI (blue in merge). Scale Bars: 50 µm. (B) Column graph shows percentage of cells positive for BrdU incorporation. n = 8 collagen matrices per analysis and data (mean+SEM) were graphed by averaging multiple fields of view and analyzed by one-way ANOVA. ****p < 0.0001.
We next assessed whether acute compressive stress was capable of inducing genes that regulate EMT, a feature of cancer progression that is associated with increased invasiveness. Collagen-embedded HEK-293T cells were subjected to acute compressive stress, and then analyzed 2 hours later for changes in expression of genes, or 16 hours later for changes in protein expression, associated with an EMT. Whilst mRNA expression of the epithelial marker CDH1 (E-Cadherin) was unchanged (Fig. 8A), possibly reflecting transcript stability in this time-frame, mesenchymal marker VIM (Vimentin) was significantly upregulated at both the gene and protein levels (Fig. 8B-D, Supp. Fig. 2), and gene expression of key regulators of the mesenchymal phenotype SNAI2 (Slug) and ZEB1 were significantly upregulated upon application of acute compressive stress (Fig. 8E-F). Upregulation of VIM/Vimentin, SNAI2 and ZEB1 were abolished by pretreatment with Y27632, Fasudil or RHO inhibitor prior to the application of compressive force (Fig. 8B-F, Supp. Fig. 2). Taken together, our results suggest that compressive stress induces RHO/ROCK pathway-mediated cell proliferation and changes in gene and protein expression that are associated with an EMT, with implications for the role of enhanced compressive force in tissue homeostasis and its dysregulation in disease states such as cancer.
Figure 8.
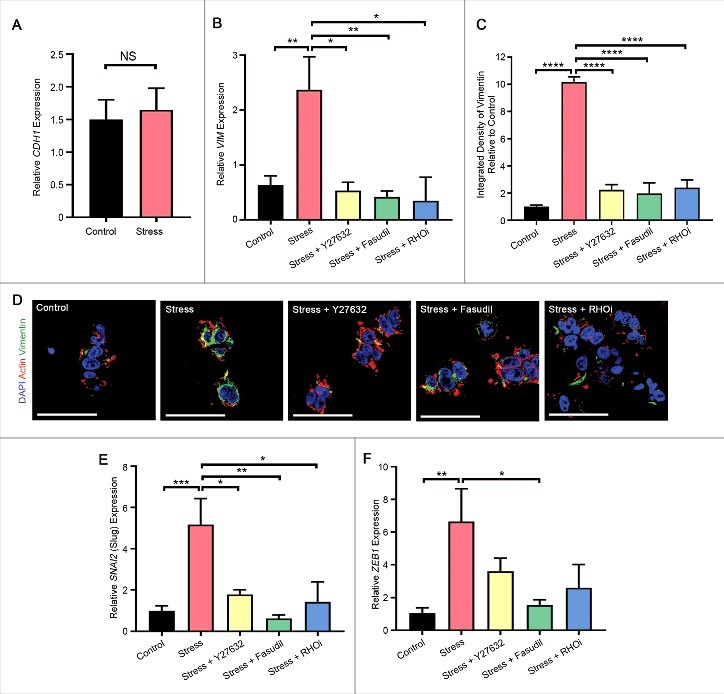
Compression induces long-term effects on mesenchymal gene and protein expression. (A) Gene expression (assessed by quantitative PCR) of epithelial marker CDH1 (E-Cadherin) in HEK-293T cells embedded in collagen followed by application of compressive stress. Data were analyzed by unpaired t-test. (B-D) Gene (assessed by quantitative PCR) (B) and protein (assessed by immunofluorescence analysis) (C-D) expression of mesenchymal marker VIM/Vimentin in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil, and a RHO inhibitor (RHOi). For immunofluorescence analysis, cells were cultured a further 16 hours to detect changes in protein (shown in green, with F-actin labelled with phalloidin and cell nuclei with DAPI (red and blue)). Scale Bars: 50 µm, n = 3 collagen matrices per analysis. Data were analyzed by one-way ANOVA. *p < 0.05, **p < 0.01, ****p < 0.0001. (E-F) Gene expression of mesenchymal markers SNAI2 (Slug) (E) and ZEB1 (F) in HEK-293T cells embedded in collagen followed by application of compressive stress with and without pre-treatment with the ROCK-specific inhibitors Y27632 and Fasudil, and a RHO inhibitor (RHOi). Data were analyzed by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. (A-B, E-F) Cells were cultured for a further 2 hours to allow detection of changes in gene expression and are normalized to ribosomal 18S (n = 10 collagen matrices per analysis), All data (mean+SEM) are relative to no stress.
Discussion
In this study we sought to determine the mechanisms adopted by cells to withstand compressive stress and to identify some of the physiological effects of compressive stress on cells and tissues. Understanding these phenomena, which are relatively understudied, is necessary to determine how mechanical signaling in this context can become corrupted and lead to diseased states. We have previously demonstrated that the Rho/ROCK signaling pathway is dysregulated in cancer, resulting in enhanced tissue ECM stiffness that perpetuates cell proliferation and tumor progression [6]. Several molecular mediators have been shown to transduce mechanical signals arising from the extracellular environment into bio-molecular outcomes within the cell, including YAP/TAZ – effectors of the hippo signaling pathway [37], β-catenin – effector of the Wnt signaling pathway [6], the serum response factor (SRF) [38] and NFκB [39]. We have since discovered that the Rho/ROCK mechanotransduction pathway is closely regulated to maintain normal tissue homeostasis [32]. In this study, we first show that the RHO/ROCK signaling pathway has a key role in regulating the cellular response to compressive stress and that it responds robustly in short time-frames following the application of compressive stress. We then go on to demonstrate that the transient activation of RHO/ROCK signaling increases cell proliferation and induces the expression of the regulators of epithelial-mesenchymal transition (EMT).
The activation of MLC2 and the inhibition of MYPT1 are well known to regulate actomyosin contractility and enhanced actomyosin force generation [13]. Our discovery that compressive stress significantly and rapidly increased the levels of phosphorylated MLC2 and MYPT1, suggests that compression induces biochemical signaling that results in compensatory actomyosin force generation. This activation of signaling could be blocked by the pre-treatment of cells with selective inhibitors of ROCK (Y27632 and Fasudil) or a RHO inhibitor (Fig. 6F). Importantly, in primary mouse tissues, compression significantly increased levels of both pMlc2 and pMypt1, demonstrating that this mechanism is active in the context of whole tissues. Another key feature of this system is the clear evidence of progressive activation of signaling molecules along the RHOA-ROCK-MLC2 signaling axis (Fig. 2G), which provides confidence that compensatory actomyosin force generation following the application of compressive stress is regulated by signaling from RHOA to ROCK and then MLC2.
Interestingly, we found that in intestinal tissues the activation of RhoA and downstream signaling was particularly high within the crypts of Lieberkühn. The base of the crypt encompasses the intestinal stem cell niche and part of the zone occupied by transit amplifying cells [40]. Crypt stem cells and transit amplifying cells have both been demonstrated as cells-of-origin of intestinal cancers [41–43]. Recently, Farge and colleagues reported that force generated by magnets applied against the body wall in mice, to attract ultra-magnetic liposomes injected intravenously, recapitulated the stress encountered by colonic tissues in the early stages of tumorigenesis. This stress significantly enhanced tumor growth in vivo via the Wnt/β-catenin mechanotransduction pathway. β-catenin signaling was enhanced in peri-tumoral crypts, suggesting that mechanical stress can engender a positive-feedback loop to promote tumor growth [44].
Our data suggest that intestinal crypts are particularly sensitive to compression-mediated enhancement of actomyosin tension and may activate RhoA signaling to compensate against compression. Furthermore, we have previously shown that Rac, another small GTPase, is higher in cells at the base of crypts and is required for cellular transformation leading to intestinal cancer via regulating expansion of Lgr5+ intestinal stem cells within epithelial crypts [45,46]. Taken together with those of Farge and colleagues, our observations support the hypothesis that mechanical stress induces within intestinal crypt cells, key pathways involved in mechanotransduction and cellular tension, that have also been implicated in tumor progression [47].
Our data obtained using primary skin samples suggest that compression-induced enhancement of actomyosin tension also occurs within this tissue. Given the significance of the canonical RhoA signaling pathway in epidermal homeostasis and wound healing in particular [32], this observation may have useful clinical implications for diseases of homeostasis characterized by enhanced mechano-reciprocity and Rho/ROCK pathway activation such as cutaneous squamous cell carcinoma progression and chronic non-healing wounds.
Enhanced mechano-reciprocity between mammary tumor cells and the ECM can promote invasiveness and malignancy of cancer cells [48], and compressive stress applied to normal epithelial or cancer cell lines specifically enhances the motility of highly invasive breast cancer cells through actomyosin contractility [49]. Our data suggest that susceptibility to compression-induced actomyosin tension is also a feature of normal mammary tissue in vivo. Taken together with previous studies, it is possible that compression forces may therefore contribute to increased actomyosin tension and consequently enhance mechano-reciprocity in breast cancer progression.
We found that compression significantly and rapidly activated RHOA – as revealed by analysis using a conformation-specific antibody against GTP-bound, active RHOA and decreased fluorescence lifetime of a RHOA-FRET biosensor – which was not affected by pre-treatment with ROCK-specific inhibitors but could be abrogated by the RHO inhibitor. Downstream of this, compression increased phosphorylation of ROCK targets MLC2 and MYPT1. Our data contrast with those of Takemoto et al., who demonstrated rapidly reduced actomyosin contractility downstream of RhoA activation in cells, arising from an inhibitory phosphorylation event on RhoA precluding its association with ROCK [17]. These experiments were carried out on cells grown in two dimensions with stress applied along the cell culture plane. We propose that our data are relevant to the physiological, three-dimensional context of tissues subjected to compressive force across a plane. Our results support the contention that within epithelial cells and tissues, compression-induced stress converts RHOA to the active GTP-bound form, which then activates its effector kinase ROCK leading to regulation of actomyosin tension via phosphorylation of MLC2 and MYPT1.
Notably, compressive stress also significantly affected the longer-term physiology of cells cultured in three dimensions. Cells became more proliferative (a 5-fold increase in proliferating cells was observed) and exhibited increased expression of key regulators of epithelial-mesenchymal transition (VIM, SNAI2 and ZEB1), signifying an acquisition of a replicative and mesenchymal phenotype. Combined with what is already known about feed-forward mechano-reciprocal signaling during cancer progression, our results therefore have clear implications for the involvement of mechanical signaling during tumor progression. Its direct implication in cell proliferation and EMT raises the possibility that compressive stress resulting from tumor growth within a constricted space could play a role in tumor progression. These observations complement our previous work in which we showed that signaling through ROCK is frequently and progressively upregulated in tumors [6]. While the Rho/ROCK pathway is likely co-opted by growth factor signaling pathways, this work provides mechanistic insight into a novel non-biochemical mechanism by which Rho/ROCK signaling can be upregulated and demonstrates the functional implications for tissue homeostasis. Given recent advances in our understanding of how inhibiting ROCK activity influences tissue biology and chemotherapy efficacy [50], we now have a further rationale for targeting the Rho/ROCK signaling axis in disease states including cancer.
Materials and methods
Cell culture
HEK-293T cells were obtained from the American Type Culture Collection in 2011 (ATCC® CRL-3216™) and were maintained in Dulbecco's modified Eagle's medium (DMEM, ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS). Cells are regularly authenticated by morphology observation at high and low density, mycoplasma detection and growth curve analysis.
Stable HEK-293T-RHOA FRET cells were generated as described previously [50]. Briefly, the RHOA-FRET biosensor [24] was cloned into the pPB-CAG.EBNXN vector [51] by NdeI/SalI excision from its pcDNA3.1 backbone. pPB-CAG.EBNXN-RHOA-FRET was then co-transfected with pCMV-hyPBase [52]. Stably expressing RHO-FRET biosensor cells were subsequently purified by fluorescence-activated cell sorting and expanded in culture.
Compression
BioPress™ compression culture plates and the Flexcell® FX-5000™ Compression System were from Flexcell Int. Corp. (Hillsborough). Static mechanical compression was applied at 20 kPa for 30 minutes unless otherwise specified.
HEK-293T collagen matrix preparation
A single cell preparation of HEK-293T cells was embedded in a cocktail of rat tail collagen (0.8 mg/ml in 17.5 mM acetic acid), 1x minimal essential medium (ThermoFisher Scientific) and 20% FBS at 2.5 × 105 cells per ml. The pH was adjusted using 0.22 M NaOH to approximately 7 as indicated by phenol red color change. Collagen matrices were allowed to set at 37°C before overlaying with DMEM/10% FBS. Before compression, cells were starved of serum for 6 hours ± ROCK1/2 inhibitors Y27632 (10 µM; Tocris Bioscience) or Fasudil (50 µM; SelleckChem), or a RHO inhibitor (CT04, 1 µg/ml; Cytoskeleton Incorporated).
siRNA knockdown of RHOA
RHOA expression was silenced using a RHOA-specific siRNA (Life Technologies). Silencer Select negative control siRNAs were used as a control (Life Technologies). HEK-293T cells were transfected with either RHOA or control siRNA at 5 nM using Lipofectamine RNAiMAX (Life Technologies) according to manufacturer's guidelines. Cells were analyzed at 72 hours post-transfection.
Primary mouse tissue preparation
All mice used were 7.5–9.5 week old wild-type FVB/n females and procedures were performed under appropriate licenses and with the oversight of the institutional animal ethics committee constituted according to the Animal Welfare Act 1985 of SA.
Large and proximal small intestines were dissected and flushed twice with PBS, then separated into two, and one portion compressed. Dorsal skin was depilated and a 2 cm × 1 cm section dissected out, separated into two 1 cm × 1 cm portions, and one portion compressed with the epidermis against the stationary platen. The fourth inguinal mammary glands were dissected with removal of the lymph node, and one gland was compressed.
Bromodeoxyuridine incorporation
Following compression, cells in collagen matrices were cultured a further 16 hours in DMEM and pulsed with BrdU (Cell Proliferation Labeling Reagent, GE Healthcare) for 1 hour.
Immunofluorescence analysis
For active RHOA-GTP, pMLC2, pMYPT1 and Vimentin immunofluorescence of HEK-293T cells within collagen matrices, matrices were fixed for 72 hours in 0.25% formaldehyde in PBS at 4°C before permeabilization at room temperature (RT) for 20 minutes in 0.2% Triton-X and blocking of non-specific binding for 24 hours in PBS/0.2% bovine serum albumin (BSA) at 4°C. Primary antibodies were added overnight in PBS/0.2% BSA at 4°C. Antibodies used were, mouse anti-Active RHOA-GTP (NewEast Biosciences #26904, 1:100), rabbit anti-p(Thr696)MYPT1 (Millipore #ABS45, 1:100), mouse anti-p(Ser19)MLC2 (Cell Signaling Technologies #3675, 1:50), and mouse anti-Vimentin (Sigma-Aldrich #V2258, 1:100). Matrices were washed for 20 minutes at RT in 0.1% Triton-X, and secondary antibody (Alexa Fluor®-488 goat anti-rabbit/mouse, 1:400) and Alexa Fluor®-594 phalloidin (1:250) (all from Thermo Fisher Scientific) were added for 1 hour at RT in the dark.
For detection of BrdU incorporation in HEK-293T cells within collagen matrices, matrices were fixed in formalin (freshly-prepared 10% formaldehyde in PBS) for 30 minutes at RT, incubated for 20 minutes at RT in denaturing solution (3N HCl/0.5% Tween-20), washed for 5 minutes in PBS/0.5% BSA, then 0.1 M sodium borate (pH 8.5) was added for 2 minutes at RT to neutralize the acid. Matrices were washed as before and mouse anti-BrdU primary antibody (Beckton Dickson #347580, 1:200) was added overnight in PBS/0.5% BSA at 4°C. Matrices were washed again before addition of secondary antibody (Alexa Fluor®-488 goat anti-mouse, Thermo Fisher Scientific, 1:400) for 1 hour at RT in the dark.
DAPI (2 µg/ml in PBS) was added to all collagen matrices in PBS. Matrices were spread onto coverslips using forceps immediately prior to image acquisition.
Primary mouse tissues for immunofluorescence analysis were fixed in formalin at 4°C overnight and embedded in paraffin before sectioning at 4 µm. Sections were de-waxed in xylene and rehydrated through 1 minute changes of graduated ethanol (100% x2, 70% x1, 50% x1) followed by a 2-minute wash in distilled water. Antigen retrieval was performed by boiling slides in a pressure cooker in either 10 mM citrate buffer (pH 6) or 10 mM Tris 1 mM EDTA buffer (pH 9) (see below for specifications). Sections were blocked in normal goat serum in CAS-Block (Thermo Fisher Scientific) for at least 20 minutes in a humidified chamber at RT then primary antibodies were added overnight in blocking buffer at 4°C. Primary antibodies used were, rabbit anti-p(Thr696)Mypt1 (Millipore #ABS45, 1:100, citrate buffer), rabbit anti-p(Ser19)Mlc2 (Cell Signaling Tech #3671, 1:50, Tris Buffer), mouse anti-ECadherin (Becton Dickson #610182, 1:100) and mouse anti-Cytokeratin 14 (Leica Biosystems #NCL-L-LL002, 1:50). Sections were washed in distilled water and secondary antibodies (Alexa Fluor®-488 or Alexa Fluor®-594 goat anti-rabbit/mouse, Thermo Fisher Scientific, 1:400) were added for 1 hour at RT in the humidified chamber in the dark. Slides were washed in distilled water and mounted in Vectashield Mounting Medium containing DAPI (Vector Laboratories Inc.).
Images were acquired using 40× (Apo 40×/1.2W DICIII, collagen and sections) and 63x (Apo 63x/1.2W DICII, sections) objectives at RT on a Zeiss LSM 700 confocal system and recorded using an Axio Observer.Z1 camera and ZEN 2011 (Black Edition) software (Carl Zeiss Microscopy). All quantification (cells positive and integrated density [a measure of signal intensity]) was performed using ImageJ (National Institutes of Health (NIH)), thresholding on the corresponding isotype control, and a minimum of three fields of view were averaged for every biological replicate.
Flim-fret microscopy
FLIM-FRET data was acquired as described previously [26,50] using an inverted Leica DMI 6000 SP8 confocal microscope (Leica Camera AG) with a 25 × 0.95 NA water immersion objective. A Titanium:Sapphire femtosecond laser (Chameleon Ultra II, Coherent Inc.), operating at 80 MHz, was used as an excitation source at 890 nm. The EGFP signal was recorded with RLD HyD detectors (Leica Camera AG) using a 525/50 nm emission filter (Chroma Technology Corp.). FLIM data was acquired using a Picoharp 300 TCSPC FLIM system (PicoQuant). For each sample, 5 representative regions of interest were imaged at a pixel depth of 512 × 512.
FLIM data was analyzed as described previously [53,54], using FLIMfit (5.0.3), recording average single cell lifetimes of the EGFP donor. Cells were further grouped into RHOA “active” and “inactive” by the presence or absence of regions of high FRET within the cell membrane.
Western analysis
Primary mouse tissues were prepared for Western analysis as follows: Intestinal tissue was opened up longitudinally and incubated without agitation for 1 hour at 4°C in chelation buffer (3 mM EDTA, 0.5 mM DTT in PBS). Tissue was transferred to PBS and intestinal epithelium separated from the underlying lamina propria by shaking vigorously in 15 second increments. Epithelial cells were collected by centrifuging at 700 rpm and suspended in RIPA lysis buffer (10 mM Tris-Cl (pH 7.5), 5 mM EDTA, 1% v/v NP-40, 0.5% w/v sodium deoxycholate, 40 mM sodium tetrapyrophosphate, 1 mM sodium vanadate, 50 mM sodium fluoride, 1 mM PMSF, 0.025% w/v SDS, 150 mM sodium chloride) containing protease and phosphatase inhibitors. Skin tissue was snap-frozen, diced and homogenized in RIPA buffer using a FastPrep-24™ homogenizer (MP Bio). Mammary gland tissue was snap-frozen and homogenized in RIPA buffer. HEK-293T cells from adherent culture were lysed 72 hours post-transfection in RIPA lysis buffer.
Proteins were separated by 10% or 12% SDS-PAGE and immobilized onto nitrocellulose membrane. Western analysis was performed by blocking the membrane in either 5% skim milk in Tris buffered saline (TBS)/Tween-20 (TBS-T) or TBS-T/5% BSA (see below for specifications) for 1 hour at RT followed by addition of primary antibodies overnight at 4°C. Primary antibodies used were, rabbit anti-p(Thr696)Mypt1 (Millipore #ABS45, 1:1,000, 5% skim milk), rabbit anti-p(Thr18/Ser19)Mlc2 (Cell Signaling Tech #3674, 1:1,000, 5% BSA), mouse anti-Mlc (Sigma-Aldrich #M4401, 1:1,000, 5% skim milk), mouse anti-RHOA (NewEast Biosciences #26007, 1:1,000, 5% skim milk), and mouse anti-αTubulin (Abcam #AB7291, 1:1,000, 5% skim milk). Membranes were washed 3x in TBS-T then incubated for 1 hour at RT with secondary antibodies (horseradish peroxidase-conjugated goat anti-rabbit/mouse, Rockland Inc.). Membranes were washed 2x in TBS-T and 1x in TBS. Bands were visualized using a ChemiDoc™ Imaging System (Bio-Rad Laboratories Pty. Ltd.) using either Pierce® ECL or Supersignal Femto Western blotting substrates (ThermoFisher Scientific). Lane densities were quantified using ImageJ (NIH).
Quantitative real-time PCR analysis
HEK-293T cells in collagen matrices were homogenized using a FastPrep-24™ homogenizer (MP Bio) in TRIzol Reagent (ThermoFisher Scientific). Adherent HEK-293T cells were lysed on the plate in TRIzol Reagent. Total RNA was extracted and reverse-transcribed using the Quantitect Reverse Transcription Kit (Qiagen), followed by quantitative PCR analysis using the SYBR Green method. Samples were normalized to endogenous ribosomal protein 18S. qPCR primer sequences are as follows:
|
Gene |
Forward Primer 5′-3′ |
Reverse Primer 5′-3′ |
| CDH1 | CCCACCACGTACAAGGGTC | CTGGGGTATTGGGGGCATC |
| SNAI2 | AAGCATTTCAACGCCTCCAAA | AGGATCTCTGGTTGTGGTATGAC |
| VIM | GAACGCCAGATGCGTGAAATG | CCAGAGGGAGTGAATCCAGATTA |
| ZEB1 | TTCAAACCCATAGTGGTTGCT | TGGGAGATACCAAACCAACTG |
| 18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| RHOA | Quantitect® Primer Assay Hs_RHOA_1_SG (Qiagen) #QT000447723 | |
Statistical analyses
Statistical significance was assessed using GraphPad Prism (GraphPad Software). Data is presented either as mean ± standard error of the mean (SEM) or median ± interquartile range (IQR). Tests used are indicated in the figure legends. *p < 0.05 **p < 0.01 ***p < 0.001 ****p < 0.0001.
Supplementary Material
Acknowledgments
The authors thank Pauline Mélénec for generating the cell line expressing the RHOA-FRET biosensor and Prof. Angel Lopez for his critique and advice on the manuscript. The authors thank donors to the Health Services Charitable Gifts Board of SA and the Australian Cancer Research Foundation for funding the imaging equipment used.
Abbreviations
- CK14
Cytokeratin-14
- FLIM
Fluorescence-lifetime imaging microscopy
- FRET
Förster resonance energy transfer
- kPa
Kilopascal
- MLC2/Mlc2
Myosin regulatory light chain-2
- MYPT1/Mypt1
Myosin phosphatase targeting subunit-1
- ROCK
Rho-associated coiled-coil-containing protein kinase.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding details
The authors are supported by National Health and Medical Research Council (APP1021323, APP1103712 and APP1103713 to MSS), Cancer Council South Australia (APP1077800 to MSS), Australian Research Council (Future Fellowship FT120100132 to MSS. This project was undertaken by STB whilst holding a Royal Adelaide Hospital Early Career Fellowship.
Author contributions
Sarah T. Boyle designed and carried out the majority of experiments, analyzed results and wrote the manuscript. Jasreen Kular performed RHOA experiments. Max Nobis performed FLIM-FRET microscopy and analysis. Andrew Ruszkiewicz provided intellectual input to assist with interpretation of results. Paul Timpson provided significant intellectual input and supervised the FLIM-FRET aspect of the study. Michael S. Samuel conceived of the study, supervised the study, designed experiments and wrote the manuscript.
References
- [1].Boyle ST, Samuel MS.. Mechano-reciprocity is maintained between physiological boundaries by tuning signal flux through the Rho-associated protein kinase. Small GTPases. 2016;7(3):139–46. doi: 10.1080/21541248.2016.117377110.1080/21541248.2016.1173771. PMID:27168253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–22. doi: 10.1038/nrc254410.1038/nrc2544. PMID:19165226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carvalho RS, Scott JE, Yen EH. The effects of mechanical stimulation on the distribution of beta 1 integrin and expression of beta 1-integrin mRNA in TE-85 human osteosarcoma cells. Arch Oral Biol. 1995;40:257–64. doi: 10.1016/0003-9969(95)98814-F10.1016/0003-9969(95)98814-F. PMID:7541624 [DOI] [PubMed] [Google Scholar]
- [4].Cohen HJ, Laszlo J. Influence of trauma on the unusual distribution of metastases from carcinoma of the larynx. Cancer. 1972;29:466–71. doi:. PMID:5013548 [DOI] [PubMed] [Google Scholar]
- [5].De Witt MT, Handley CJ, Oakes BW, et al. In vitro response of chondrocytes to mechanical loading. The effect of short term mechanical tension. Connective Tissue Res. 1984;12:97–109. doi: 10.3109/0300820840899277510.3109/03008208408992775. [DOI] [PubMed] [Google Scholar]
- [6].Samuel MS, Lopez JI, McGhee EJ, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–91. doi: 10.1016/j.ccr.2011.05.00810.1016/j.ccr.2011.05.008. PMID:21665151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Totsukawa G, Yamakita Y, Yamashiro S, et al. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.79710.1083/jcb.150.4.797. PMID:10953004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamashiro S, Totsukawa G, Yamakita Y, et al. Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol Biol Cell. 2003;14:1745–56. doi: 10.1091/mbc.E02-07-042710.1091/mbc.E02-07-0427. PMID:12802051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jiang Y, Wang Y, Wang T, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014;5:5566. doi: 10.1038/ncomms656610.1038/ncomms6566. PMID:25412762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chew TL, Masaracchia RA, Goeckeler ZM, et al. Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (gamma-PAK). J Muscle Res Cell Motility. 1998;19:839–54. doi: 10.1023/A:100541792658510.1023/A:1005417926585. [DOI] [PubMed] [Google Scholar]
- [11].Strassheim D, May LG, Varker KA, et al. M3 muscarinic acetylcholine receptors regulate cytoplasmic myosin by a process involving RhoA and requiring conventional protein kinase C isoforms. J Biol Chem. 1999;274:18675–85. doi: 10.1074/jbc.274.26.1867510.1074/jbc.274.26.18675. PMID:10373480 [DOI] [PubMed] [Google Scholar]
- [12].Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005;15:371–7. doi: 10.1016/j.tcb.2005.05.00410.1016/j.tcb.2005.05.004. PMID:15935670 [DOI] [PubMed] [Google Scholar]
- [13].Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm112810.1038/nrm1128. PMID:12778124 [DOI] [PubMed] [Google Scholar]
- [14].Bhadriraju K, Yang M, Alom Ruiz S, et al. Activation of ROCK by RhoA is regulated by cell adhesion, shape, and cytoskeletal tension. Exp Cell Res . 2007;313:3616–23. doi: 10.1016/j.yexcr.2007.07.00210.1016/j.yexcr.2007.07.002. PMID:17673200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Araki S, Ito M, Kureishi Y, et al. Arachidonic acid-induced Ca2+ sensitization of smooth muscle contraction through activation of Rho-kinase. Pflugers Archiv: Eur J Physiol. 2001;441:596–603. doi: 10.1007/s00424000046210.1007/s004240000462. [DOI] [PubMed] [Google Scholar]
- [16].Shirao S, Kashiwagi S, Sato M, et al. Sphingosylphosphorylcholine is a novel messenger for Rho-kinase-mediated Ca2+ sensitization in the bovine cerebral artery: unimportant role for protein kinase C. Circulation Res. 2002;91:112–9. doi: 10.1161/01.RES.0000026057.13161.4210.1161/01.RES.0000026057.13161.42. PMID:12142343 [DOI] [PubMed] [Google Scholar]
- [17].Takemoto K, Ishihara S, Mizutani T, et al. Compressive stress induces dephosphorylation of the myosin regulatory light chain via RhoA phosphorylation by the adenylyl cyclase/protein kinase A signaling pathway. PloS One. 2015;10:e0117937. doi: 10.1371/journal.pone.011793710.1371/journal.pone.0117937. PMID:25734240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Matsui T, Amano M, Yamamoto T, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–16. PMID:8641286 [PMC free article] [PubMed] [Google Scholar]
- [19].Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases. 2014;5:e27958. doi: 10.4161/sgtp.2795810.4161/sgtp.27958. PMID:24607953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Y, Marshall KL, Baba Y, et al. Hyperelastic material properties of mouse skin under compression. PloS One. 2013;8:e67439. doi: 10.1371/journal.pone.006743910.1371/journal.pone.0067439. PMID:23825661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu G, Sengupta PK, Jamal B, et al. N-glycosylation induces the CTHRC1 protein and drives oral cancer cell migration. J Biol Chem. 2013;288:20217–27. doi: 10.1074/jbc.M113.47378510.1074/jbc.M113.473785. PMID:23703614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuipers D, Mehonic A, Kajita M, et al. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J Cell Sci. 2014;127:1229–41. doi: 10.1242/jcs.13828910.1242/jcs.138289. PMID:24463819 [DOI] [PubMed] [Google Scholar]
- [23].Lim HC, Couchman JR. Syndecan-2 regulation of morphology in breast carcinoma cells is dependent on RhoGTPases. Biochim Et Biophysica Acta. 2014;1840:2482–90. doi: 10.1016/j.bbagen.2014.01.01810.1016/j.bbagen.2014.01.018. [DOI] [PubMed] [Google Scholar]
- [24].Timpson P, McGhee EJ, Morton JP, et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71:747–57. doi: 10.1158/0008-5472.CAN-10-226710.1158/0008-5472.CAN-10-2267. PMID:21266354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yoshizaki H, Ohba Y, Kurokawa K, et al. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223–32. doi: 10.1083/jcb.20021204910.1083/jcb.200212049. PMID:12860967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nobis M, Herrmann D, Warren SC, et al. A RhoA-FRET biosensor mouse for intravital imaging in normal tissue homeostasis and disease contexts. Cell Reports. 2017;21(1):274–288. doi: 10.1016/j.celrep.2017.09.02210.1016/j.celrep.2017.09.022. PMID:28978480 [DOI] [PubMed] [Google Scholar]
- [27].Amano M, Ito M, Kimura K, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.2024610.1074/jbc.271.34.20246. PMID:8702756 [DOI] [PubMed] [Google Scholar]
- [28].Feng J, Ito M, Ichikawa K, et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–90. doi: 10.1074/jbc.274.52.3738510.1074/jbc.274.52.37385. PMID:10601309 [DOI] [PubMed] [Google Scholar]
- [29].Gayer CP, Basson MD. The effects of mechanical forces on intestinal physiology and pathology. Cell Signal. 2009;21:1237–44. doi: 10.1016/j.cellsig.2009.02.01110.1016/j.cellsig.2009.02.011. PMID:19249356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harbor Perspectives Biol. 2011;3:a003228. doi: 10.1101/cshperspect.a00322810.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ibbetson SJ, Pyne NT, Pollard AN, et al. Mechanotransduction pathways promoting tumor progression are activated in invasive human squamous cell carcinoma. Am J Pathol. 2013;183:930–7. doi: 10.1016/j.ajpath.2013.05.01410.1016/j.ajpath.2013.05.014. PMID:23830873 [DOI] [PubMed] [Google Scholar]
- [32].Kular J, Scheer KG, Pyne NT, et al. A negative regulatory mechanism involving 14-3-3zeta limits signaling downstream of ROCK to regulate tissue stiffness in epidermal homeostasis. Dev Cell. 2015;35:759–74. doi: 10.1016/j.devcel.2015.11.02610.1016/j.devcel.2015.11.026. PMID:26702834 [DOI] [PubMed] [Google Scholar]
- [33].Kim JG, Kim MJ, Choi WJ, et al. Wnt3A induces GSK-3beta phosphorylation and beta-Catenin accumulation through RhoA/ROCK. J Cell Physiol. 2017;232:1104–13. doi: 10.1002/jcp.2557210.1002/jcp.25572. PMID:27575935 [DOI] [PubMed] [Google Scholar]
- [34].Pirone DM, Liu WF, Ruiz SA, et al. An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA-ROCK signaling. J Cell Biol. 2006;174:277–88. doi: 10.1083/jcb.20051006210.1083/jcb.200510062. PMID:16847103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Petrie RJ, Gavara N, Chadwick RS, et al. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. 2012;197:439–55. doi: 10.1083/jcb.20120112410.1083/jcb.201201124. PMID:22547408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Salvi A, Thanabalu T. WIP promotes in-vitro invasion ability, anchorage independent growth and EMT progression of A549 lung adenocarcinoma cells by regulating RhoA levels. Biochem Biophys Res Commun. 2017;482:1353–9. doi: 10.1016/j.bbrc.2016.12.04010.1016/j.bbrc.2016.12.040. PMID:27939884 [DOI] [PubMed] [Google Scholar]
- [37].Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. doi: 10.1038/nature1013710.1038/nature10137. PMID:21654799 [DOI] [PubMed] [Google Scholar]
- [38].Connelly JT, Gautrot JE, Trappmann B, et al. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat Cell Biol. 2010;12:711–8. doi: 10.1038/ncb207410.1038/ncb2074. PMID:20581838 [DOI] [PubMed] [Google Scholar]
- [39].Sero JE, Sailem HZ, Ardy RC, et al. Cell shape and the microenvironment regulate nuclear translocation of NF-kappaB in breast epithelial and tumor cells. Mol Syst Biol. 2015;11:790. doi: 10.15252/msb.2014564410.15252/msb.20145644. PMID:26148352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–84. doi: 10.1016/j.cell.2013.07.00410.1016/j.cell.2013.07.004. PMID:23870119 [DOI] [PubMed] [Google Scholar]
- [41].Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11. doi: 10.1038/nature0760210.1038/nature07602. PMID:19092804 [DOI] [PubMed] [Google Scholar]
- [42].Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–7. doi: 10.1038/nature0758910.1038/nature07589. PMID:19092805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–20. doi: 10.1038/ng.16510.1038/ng.165. PMID:18536716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fernandez-Sanchez ME, Barbier S, Whitehead J, et al. Mechanical induction of the tumorigenic beta-catenin pathway by tumour growth pressure. Nature. 2015;523:92–5. doi: 10.1038/nature1432910.1038/nature14329. PMID:25970250 [DOI] [PubMed] [Google Scholar]
- [45].Myant KB, Cammareri P, McGhee EJ, et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–73. doi: 10.1016/j.stem.2013.04.00610.1016/j.stem.2013.04.006. PMID:23665120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Johnsson AK, Dai Y, Nobis M, et al. The Rac-FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues. Cell Reports. 2014;6:1153–64. doi: 10.1016/j.celrep.2014.02.02410.1016/j.celrep.2014.02.024. PMID:24630994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Samuel MS, Olson MF. Actomyosin contractililty: force power drives tumor growth. Cell Cycle. 2011;10:3409–10. doi: 10.4161/cc.10.20.1772210.4161/cc.10.20.17722. PMID:22067650 [DOI] [PubMed] [Google Scholar]
- [48].Chaudhuri O, Koshy ST, Branco da Cunha C, et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Materials. 2014;13:970–8. doi: 10.1038/nmat400910.1038/nmat4009. PMID:24930031 [DOI] [PubMed] [Google Scholar]
- [49].Tse JM, Cheng G, Tyrrell JA, et al. Mechanical compression drives cancer cells toward invasive phenotype. Proc Natl Acad Sci U S A. 2012;109:911–6. doi: 10.1073/pnas.111891010910.1073/pnas.1118910109. PMID:22203958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vennin C, Chin VT, Warren SC, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Science Translational Med. 2017;9:eaai8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yusa K, Rad R, Takeda J, et al. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods. 2009;6:363–9. doi: 10.1038/nmeth.132310.1038/nmeth.1323. PMID:19337237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yusa K, Zhou L, Li MA, et al. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A. 2011;108:1531–6. doi: 10.1073/pnas.100832210810.1073/pnas.1008322108. PMID:21205896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Conway JRW, Warren SC, Timpson P. Context-dependent intravital imaging of therapeutic response using intramolecular FRET biosensors. Methods. 2017;128:78–94. doi: 10.1016/j.ymeth.2017.04.01410.1016/j.ymeth.2017.04.014. PMID:28435000 [DOI] [PubMed] [Google Scholar]
- [54].Warren SC, Margineanu A, Alibhai D, et al. Rapid global fitting of large fluorescence lifetime imaging microscopy datasets. PloS One. 2013;8:e70687. doi: 10.1371/journal.pone.007068710.1371/journal.pone.0070687. PMID:23940626 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.