

ABSTRACT
Epac1 and Rap1 mediate cAMP-induced tightening of endothelial junctions. We have previously found that one of the mechanisms is the inhibition of Rho-mediated tension in radial stress fibers by recruiting the RhoGAP ArhGAP29 in a complex containing the Rap1 effectors Rasip1 and Radil. However, other mechanisms have been proposed as well, most notably the induction of tension in circumferential actin cables by Cdc42 and its GEF FGD5. Here, we have investigated how Rap1 controls FGD5/Cdc42 and how this interconnects with Radil/Rasip1/ArhGAP29. Using endothelial barrier measurements, we show that Rho inhibition is not sufficient to explain the barrier stimulating effect of Rap1. Indeed, Cdc42-mediated tension is induced at cell-cell contacts upon Rap1 activation and this is required for endothelial barrier function. Depletion of potential Rap1 effectors identifies AF6 to mediate Rap1 enhanced tension and concomitant Rho-independent barrier function. When overexpressed in HEK293T cells, AF6 is found in a complex with FGD5 and Radil. From these results we conclude that Rap1 utilizes multiple pathways to control tightening of endothelial junctions, possibly through a multiprotein effector complex, in which AF6 functions to induce tension in circumferential actin cables.
KEYWORDS: AF6; Circumferential Actin Cables; cytoskeletal tension; endothelial barrier; FGD5; junctional actin; Radil, Rap1
Introduction
The small G-protein Rap1 is a key regulator of endothelial barrier function [1,2]. It does so by cycling between an inactive GDP-bound state and an active GTP-bound state, the latter strengthening endothelial cell-cell adhesion. Various Guanine nucleotide Exchange Factors (GEFs) induce Rap1 activity, thereby ensuring that endothelial cells maintain the appropriate amount of endothelial barrier [2]. Most notably, high levels of endothelial barrier are produced by cAMP inducing agents, which stimulate Rap1 activation via the cAMP responsive GEF Epac1 [3–5]. Activated Rap1 ensures a tight endothelial barrier, but the molecular pathways which mediate this effect remain a matter of debate, with multiple effector pathways being suggested. First, we and others have shown that Rap1 relieves radial tension that is exerted on cell-cell junctions by radial stress fibers. To this end, Rap1 controls the localization of its effectors Radil and Rasip1, which together control the RhoGAP ArhGAP29, ensuring low Rho activity and thereby less ROCK-mediated phosphorylation of Myosin Light Chain 2 (MLC2) on T18 and S19 [6–8]. The Rap1 effector KRIT1 also decreases ROCK-mediated radial tension [9–11], but how it does so and how it cooperates with Radil/Rasip1 has yet to be determined. Second, Rap1 increases tension in Circumferential Actin Bundles (CABs). This is mediated by junctional recruitment of the RhoGEF FGD5, an activator of Cdc42. The Cdc42 effector MRCK then induces phosphorylation of MLC2 on S19 to increase tension [12]. Importantly, FGD5 lacks an RA or RBD domain that would allow it to be directly regulated by active Rap1. Therefore, a yet unidentified Rap1 effector presumably controls FGD5. Third, the Rap1 effector AF6 mediates Rap1-induced barrier tightening by enhancing the physical interaction between the Adherens Junction (AJ) protein p120-catenin and the Tight Junction (TJ) protein ZO-1 via an unknown mechanism [13]. Either this or another function of AF6 affects cytoskeletal tension, as AF6 knockdown cells show prolonged phosphorylation of MLC2 after thrombin stimulation [14]. Altogether, various labs have reported Rap1 effector pathways that control endothelial barrier function, but it is unclear whether and how these pathways function together. Here we show that at least two pathways cooperate in Rap1-mediated tightening of endothelial barrier: ArhGAP29-mediated inhibition of Rho to decrease radial tension, and activation of Cdc42 to induce tension in CABs, most likely through the FGD family of Cdc42GEFs and further involving the Rap1 effector AF6, and possibly Radil.
Results
We have previously shown that ArhGAP29-mediated inhibition of Rho is essential for Rap1-induced barrier function. A pivotal part of the data included the observation that depletion of ArhGAP29 completely blocked the increased endothelial barrier induced by an active Rap1 mutant, but did not affect the increased endothelial barrier caused by simultaneous depletion of the Rho isoforms RhoA, RhoB and RhoC [7]. To test whether alternative, Rho independent pathways mediate Rap1-induced barrier function, we measured the endothelial barrier function of HUVEC monolayers depleted of Rap1 and/or Rho (Fig. 1). As shown previously, combined depletion of the Rap1 isoforms Rap1A and Rap1B (siRap1) decreases endothelial barrier [15], whereas combined depletion of the Rho isoforms RhoA, RhoB and RhoC (siRho) increases endothelial barrier [7]. Depletion of both Rap1 and Rho results in barrier levels that are higher than upon depletion of Rap1 only, showing that part of the siRap1 effect is Rho mediated, but also substantially lower than upon depletion of Rho only. This implies that one or more pathways additional to Rho inhibition are used by Rap1 to control endothelial barrier function.
Figure 1.

Rap1-mediated endothelial barrier regulation is partially Rho independent. Endothelial barrier (Rb) of control HUVEC monolayers (siScr) and HUVEC monolayers depleted of Rap1A and Rap1B (siRap1) and/or RhoA, RhoB and RhoC (siRho). Different colors represent independent experiments (n = 4). Averages are indicated by the black lines. Knockdown efficiencies are shown in supplemental figure 1A.
One pathway reported to mediate Rap1-induced endothelial barrier that could potentially function parallel to Rho inhibition is Rap1-induced junctional translocation of the Cdc42GEF FGD5, which activates Cdc42 and its effector MRCK [12]. Indeed, junctional staining of FGD5 is increased when Rap1 is activated with the Epac1 specific cAMP analogue 007-AM (Fig. 2A) [16,17]. However, in our system knockdown of FGD5 does result in only a slight, non-significant reduction of endothelial barrier after 007-AM stimulation (Fig. 2B), despite obtaining good depletion (Fig. 2C). FGD5 is a member of the FGD family of proteins, which consists of six members that all contain a central catalytic DH-PH domain tandem and C-terminal lipid anchoring FYVE and PH domains [18]. Apart from FGD5, endothelial cells express FGD1 and FGD6 [19,20]. QPCR analysis confirms that FGD1 and FGD6 are expressed in our HUVECs (Fig. 2C). These isoforms are functionally redundant with FGD5, since depletion of all three proteins results in a very mild, but significant decrease in 007-AM-induced endothelial barrier (Fig. 2B). Concomitant with these Cdc42GEFs being required for Rap1-induced endothelial barrier, we observe that Cdc42 is required as well, both before and after stimulation with 007-AM (Fig. 2D). This effect is much stronger than upon depletion of the FGD proteins, indicating that either FGD depletion is insufficient or that additional Cdc42 regulation is involved. Either way, these data confirm the requirement for FGD regulated Cdc42 for Rap1-induced endothelial barrier.
Figure 2.
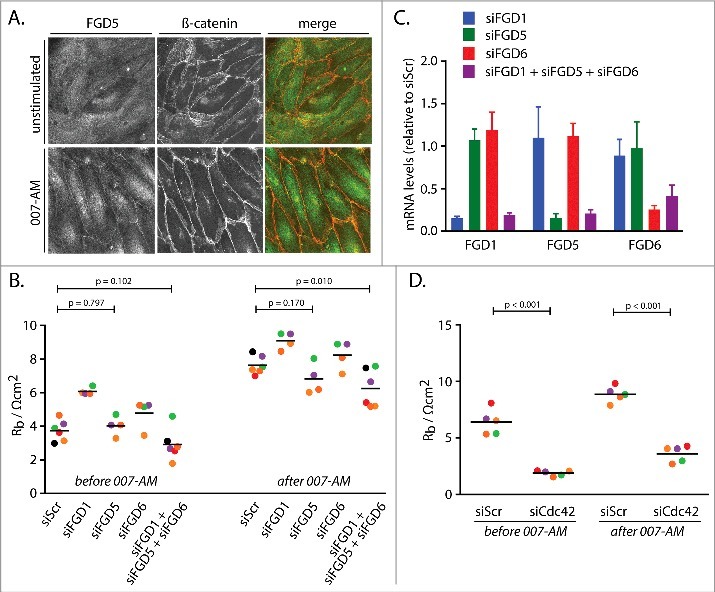
Cdc42GEFs of the FGD family and Cdc42 mediate Rap1-induced barrier function. (A) Immunofluorescence of HUVEC monolayers either not stimulated or stimulated with 007-AM 15 minutes prior to fixation. The cells were stained for FGD5 and β-catenin. The merged image depicts FGD5 in green and β-catenin in red. (B) Endothelial barrier (Rb) of control HUVEC monolayers (siScr) and HUVEC monolayers depleted of FGD1 (siFGD1), FGD5 (siFGD5), FGD6 (siFGD6) or the three FGDs combined (siFGD1 + siFGD5 + siFGD6), either before or 45 minutes after stimulation with 1 µM 007-AM. Different colors represent independent experiments (n > 3). Averages are indicated by the black lines. (C) mRNA levels of FGD1, FGD5 and FGD6 in HUVEC monolayers depleted of FGD1 (siFGD1, blue bars), FGD5 (siFGD5, green bars), FGD6 (siFGD6, red bars) or the three FGDs combined (siFGD1 + siFGD5 + siFGD6, purple bars), represented as expression relative to monolayers transfected with a control siRNA. The bars show averages of independent experiments (n = 4). Error bars indicate standard deviation. (D) Endothelial barrier (Rb) of control HUVEC monolayers (siScr) and HUVEC monolayers depleted of Cdc42 (siCdc42), either before or 45 minutes after stimulation with 1 µM 007-AM. Different colors represent independent experiments (n = 5). Averages are indicated by the black lines. Knockdown efficiencies are shown in supplemental figure 1B.
Rap1-induced junctional recruitment of FGD5 and concomitant activation of Cdc42 are reported to cause increased tension in CABs [12]. Indeed, 007-AM induces a clear junctional staining of MLC2 phosphorylated on S19 (pS19-MLC) (Fig. 3A). In contrast, in unstimulated cells pS19-MLC staining is mainly present on actin stress fibers and much less so on cell-cell junctions. The induction of junctional pS19-MLC by 007-AM is not abolished by depletion of the FGD proteins (Fig. 3A), compatible with the notion that its effect on 007-AM-induced barrier function is also very mild. In contrast, siRNAs targeting Rap1 or Cdc42, which have a stronger effect on 007-AM-induced barrier function, also diminish 007-AM-induced junctional pS19-MLC (Fig. 3B). This suggest that Rap1 indeed induces Cdc42-mediated junctional tension. Just as the decreased Rho-mediated tension [6,7], this increased Cdc42-mediated tension is required for endothelial barrier function, as blocking Myosin function with Blebbistatin decreases 007-AM induced barrier function (Fig. 3C). We therefore conclude, in agreement with the report of Mochizuki and coworkers, that Cdc42 mediates Rap1-induced barrier function by inducing the phosphorylation of MLC2 on S19, and that members of FGD family serve, at least in part, as GEFs for Cdc42 in this process.
Figure 3.
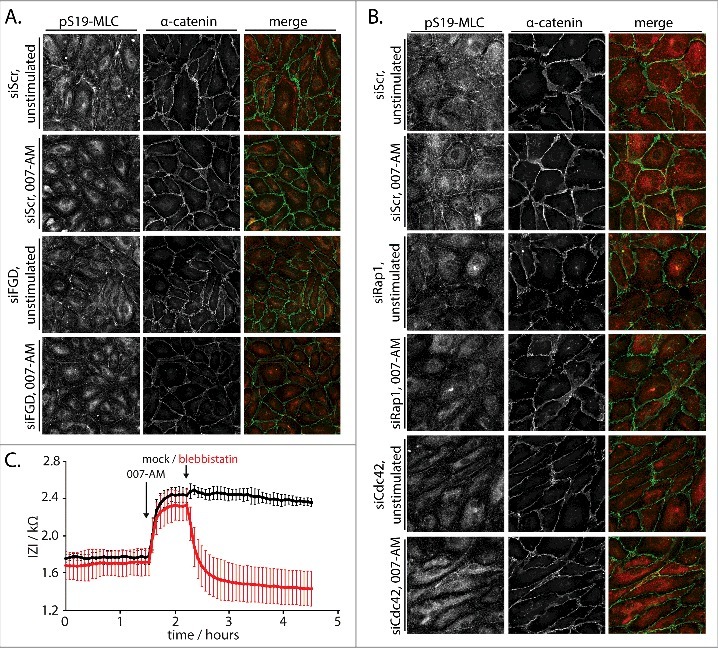
Rap1 induces Cdc42-dependent junctional tension to enhance barrier function. (A) Immunofluorescence of HUVEC monolayers transfected with control siRNA (siScr) or siRNAs targeting FGD1, FGD5 and FGD6 (siFGD), either not stimulated or stimulated with 1 µM 007-AM 15 minutes prior to fixation. The cells were stained for pS19-MLC and α-catenin. The merged image depicts pS19-MLC in red and α-catenin in green. Knockdown efficiencies are shown in supplemental figure 1C. (B) Immunofluorescence of HUVEC monolayers transfected with control siRNA (siScr), siRNAs targeting Rap1A and Rap1B (siRap1) or siRNA targeting Cdc42 (siCdc42), either not stimulated or stimulated with 1 µM 007-AM 15 minutes prior to fixation. The cells were stained for pS19-MLC and α-catenin. The merged image depicts pS19-MLC in red and α-catenin in green. Knockdown efficiencies are shown in supplemental figure 1D. (C) Real-time measurement of the endothelial barrier of HUVEC monolayers. Absolute values of the impedance (Z) at 4000 Hz are plotted over time. 1 µM 007-AM was added to all wells when indicated, followed by mock (black line) or 100 µM Blebbistatin (red line) treatment as indicated. The lines show average values of four technical replicates within one representative experiment. Error bars indicate standard deviation.
We next sought to identify which Rap1 effector protein mediates the induction of junctional tension. Both Radil and Rasip1 clearly mediate Rap1-induced barrier function in our system [6,7], but depleting these proteins does not affect 007-AM-induced junctional pS19-MLC (Fig. 4A). AF6 is another Rap1 effector protein that regulates cell-cell junctions [21]. When AF6 is depleted from HUVEC, 007-AM-induced pS19-MLC is greatly reduced, suggesting AF6 mediates Rap1-induced junctional tension (Fig. 4A). Concomitantly, AF6 is required for endothelial barrier function, as both basal and 007-AM-induced barrier function are decreased by its depletion (Fig. 4B). This inhibitory effect of AF6 depletion acts downstream of Rap1 rather than a Rap1-independent function of Epac1 [22,23], as barrier induction by an active mutant of Rap1 (G12VRap1A) is abolished by AF6 depletion (Fig. 4C). Mechanistically, AF6 does not control endothelial barrier by inhibiting Rho-mediated radial tension, as Rho depletion does not desensitize endothelial barrier to depletion of AF6 (Fig. 4D). Together, these data suggest that AF6 is required for Rap1-induced junctional tension and that this AF6 effect, rather than inhibition of Rho-mediated radial tension, conveys Rap1-induced endothelial barrier function.
Figure 4.
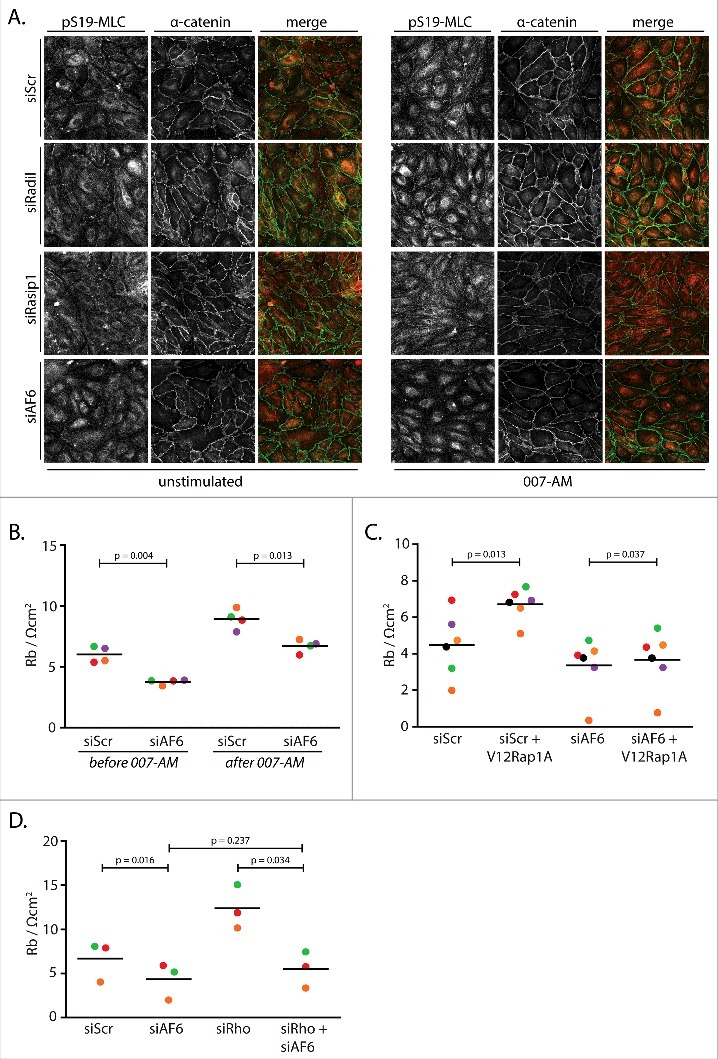
AF6 mediates Rap1-induced junctional tension and concomitant barrier function. (A) Immunofluorescence of HUVEC monolayers transfected with control siRNA (siScr), siRNA targeting Radil (siRadil), Rasip1 (siRasip1) or AF6 (siAF6), either not stimulated or stimulated with 1 µM 007-AM 15 minutes prior to fixation. The cells were stained for pS19-MLC and α-catenin. The merged image depicts pS19-MLC in red and α-catenin in green. Knockdown efficiencies are shown in supplemental figure 1E. (B) Endothelial barrier (Rb) of control HUVEC monolayers (siScr) and HUVEC monolayers depleted of AF6 (siAF6), either before or 45 minutes after stimulation with 1 µM 007-AM. Different colors represent independent experiments (n = 4). Averages are indicated by the black lines. Knockdown efficiencies are shown in supplemental figure 1F. (C) Endothelial barrier (Rb) of control HUVEC monolayers (siC) and HUVEC monolayers depleted AF6 (siAF6), transduced with control lentivirus or G12VRap1A containing lentivirus. Different colors represent independent experiments (n = 6). Averages are indicated by the black lines. Knockdown efficiencies are shown in supplemental figure 1G. (D) Endothelial barrier (Rb) of control HUVEC monolayers (siScr) and HUVEC monolayers depleted of AF6 (siAF6) and/or RhoA, RhoB and RhoC (siRho). Different colors represent independent experiments (n = 3). Averages are indicated by the black lines. Knockdown efficiencies are shown in supplemental figure 1H.
To investigate whether AF6 controls junctional tension by direct regulation of FGD proteins, we tested whether these proteins coimmunoprecipitate when expressed in HEK293T cells. No direct interaction between AF6 and FGD5 could be detected (Fig. 5B). However, during the course of these experiments we observed that Radil does interact with FGD5 (Fig. 5A). This interaction is increased when an active Rap1 mutant is coexpressed. Intriguingly, in contrast to FGD5 alone, the complex of Radil and FGD5 does interact with AF6 (Fig. 5B). Hence, similar to our previous observation that Radil and Rasip1 form a complex to regulate radial tension, a complex of Radil and AF6 may mediate Rap1-induced junctional tension in CABs.
Figure 5.
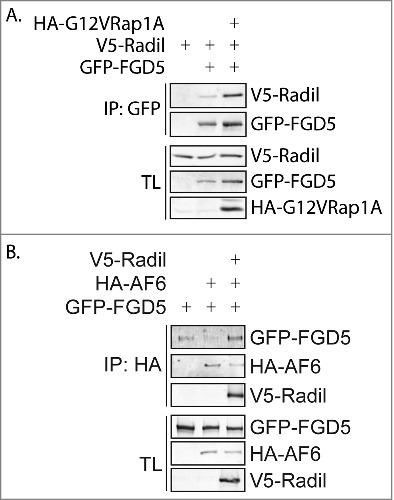
Radil interacts with FGD5 and AF6. (A) Coimmunoprecipitation of V5-Radil with GFP-FGD5 in the absence (lane 2) or presence (lane 3) of HA-G12VRap1A in HEK293T cells. (B) Coimmunoprecipitation of GFP-FGD5 with HA-AF6 in the absence (lane 2) or presence (lane 3) of V5-Radil in HEK293T cells.
Discussion
We have previously shown that Rap1 controls the endothelial barrier by decreasing Rho-mediated radial tension on cell-cell junctions via its effectors Radil and Rasip1 and their binding partner ArhGAP29 [6,7]. However, induction of tension in CABs has also been shown to be essential [12]. Indeed, we now show that Rho inhibition is not sufficient for the full effect of Rap1 to tighten the barrier and confirm that induction of tension is required as well. This junctional tension in CABs requires AF6, Cdc42 and possibly the FGD family of Cdc42GEFs. These data independently confirm the Rap1-induced junctional tension mechanism proposed by Mochizuki and coworkers and show the importance of this mechanism in a system that is also dependent on Radil, Rasip1 and ArhGAP29. As such, these two mechanisms are very likely to function simultaneously.
There are intrinsic flaws in our experiments which caution us to make firm conclusions. Firstly, the effect of depleting FGD5 and its close relatives on both endothelial barrier function and phosphorylation of MLC2 is limited. Perhaps siRNA may not be sufficient to inhibit pathways completely. This could be solved by CRISPR technology, but this technology cannot be used in short lived HUVEC cells, particularly when disruption of several genes is required. Alternatively, FGD proteins do not play an important role in this process. This would be in contrast to the strong effects of FGD5 observed by Mochizuki and coworkers. Our observations that 007-AM induces the translocation of FGD5 to cell-cell junctions and that active Rap1 stimulates the interaction between FGD5 and one of the effectors of Rap1, Radil, provides confidence that the FGD5 pathway is a genuine pathway used by Rap1 to regulate Cdc42 and subsequent junctional tension in CABs. Furthermore, the novel FGD5-Radil interaction than fills the gap between Rap1 and FGD5. Secondly, our interaction studies were performed in HEK293T cells and due to the lack of proper antibodies could not be performed with endogenous proteins in HUVEC. However, the observation that the interaction of FGD5 with Radil is induced by active Rap1 suggests to us that this interaction is of particular relevance.
AF6 is well known to function in cell-cell junctions of epithelial cells, fibroblasts and neuronal cells [21]. In the endothelium, AF6 mediates signaling by VEGF and S1P during angiogenesis [24]. Furthermore, knockout and knockdown experiments show that AF6 relieves Rho-mediated cytoskeletal tension [14,25]. We now add that AF6 is required for the Rap1-induced phosphorylation of S19-MLC, a hallmark of Cdc42-mediated induction of junctional tension. A major question remains how phosphorylation of junctional pS19-MLC is regulated by AF6. A possible link is the complex formation with Radil and FGD5, although their, arguably imperfect, depletions does not affect 007-AM-induced junctional pS19-MLC. Furthermore, we did not observe an effect on 007-AM-induced junctional translocation of FGD5 upon depleting Radil or AF6 (data not shown), suggesting that AF6 would control FGD5 by other means than localization and another Rap1 effector would be responsible for localizing FGD5. Such a mechanism is not unprecedented, as ArhGAP29 is regulated by Rap1 via two effectors, only one of which controls ArhGAP29 localization [6]. Alternatively, AF6 controls junctional pS19-MLC in a manner independent of its interactors Radil and FGD5. Either way, additional components mediating this pathway remain to be identified.
Materials and methods
Cell culture and transfections
HEK293T cells were cultured at 37°C and 6% CO2 in Dulbecco's Modified Eagle Medium containing 4.5 g/L glucose, supplemented with 10% heat inactivated Fetal Bovine Serum, 2 mM L-Glutamine and antibiotics. HUVECs (Lonza) were grown at 37°C and 6% CO2 on tissue culture dishes coated with 0.5% Gelatin in EBM-2 medium (Lonza) supplemented with EGM-2 SingleQuots (EGF, Hydrocortisone, Fetal Bovine Serum, VEGF, FGF-B, R3-IGF-1, Ascorbic acid, GA-100 and Heparin) (Lonza). HUVECs were cultured at most 14 days before experiments. siRNA transfections were performed 72 hours before experiments with 50 nM ON-TARGETplus SMARTpool (Dharmacon Inc.) targeting indicated proteins using Dharmafect-1 (Dharmacon Inc.). Overexpression of G12VRap1 in HUVEC was established by lentiviral transduction, for which lentivirus was produced by transfecting HEK293T cells, growing in EBM-2 medium, with pLV-CMV-V5-G12VRap1A-bc-GFP and third generation packaging constructs using X-tremeGene 9 (Sigma-Aldrich). HUVECs were infected with the undiluted growth medium of virus producing HEK293Ts, supplemented with 8 µg/mL Polybrene 48 hours before experiments.
Plasmids and reagents
pLV-CMV-V5-G12VRap1A-bc-GFP [15], pMT2-HA-G12VRap1A [26], pCDNA3-V5-Radil [6] and pCDNA3-HA-AF6 [27] have been described previously. pEGFP-C1-FGD5 [12] was very kindly provided by Naoki Mochizuki (NCVC, Osaka, Japan). 007-AM (8-pCPT-2’-O-Me-cAMP-AM) was from Biolog Life Sciences and used at a concentration of 1 uM [17]. Blebbistatin was obtained from EMD Millipore and used at a concentration of 100 µM. Antibodies were obtained from Sigma-Aldrich (FGD5), BD Bioscience (α-catenin and β-catenin), Covance (HA), Roche (GFP), Cell Signaling Technology (pS19-MLC2) and Invitrogen (V5, HRP- and Alexa-conjugated secondary antibodies).
Immunofluorescence
48 hours after siRNA transfection, HUVECs were plated onto Fibronectin-coated glass coverslips in 24-well plates (2 × 105 cells/well) and grown to confluency for 24 hours. Cells were stimulated with 1 µM 007-AM or left untreated for 15 minutes and fixed (4% formaldehyde, 20 minutes), permeabilized (0.1% TX-100, 3 minutes) and blocked (1% BSA, at least 2 hours). After incubation with primary antibodies (overnight) and with secondary antibodies (30 minutes), coverslips were mounted onto glass slides, which were examined after drying on a Zeiss LSM880 microscope.
Endothelial barrier measurements
48 hours after siRNA transfection and/or 24 hours after lentiviral transduction, HUVECs were plated at a density of 1 × 105 cells/well onto 8W10E electrodes (Applied Biophysics) that had been reduced with L-cysteine and coated with 5 µg/mL Fibronectin (Sigma-Aldrich). Cells were allowed to settle on the electrodes for 24 hours. Impedance was measured at 37°C and 6% CO2 using a 1600R Electrical Cell Impedance Sensing (ECIS) system (Applied Biophysics). For time course experiments, impedance was measured at 4000 Hz. For endothelial barrier resistance (Rb) measurements, frequency scans were performed within the range of 62.5 Hz and 16000 Hz and Rb was calculated using ECIS software (v1.2.73.0 PC) from Applied Biophysics. When indicated, monolayers were stimulated with 1 µM 007-AM for 45 minutes and frequency scans were repeated to obtain Rb levels after 007-AM. Graphs were created using Graphpad Prism 7, showing multiple independent experiments represented by different colors. Black lines indicate mean values. P values were calculated using Student's t-test (two-tailed, paired).
Co-immunoprecipitations
HEK293T cells were transfected using X-tremeGENE 9 and lysed 48 hours after transfection using a buffer containing 0.5% NP-40, 20 mM Tris (pH 7.5), 150 mM NaCl, 20 mM MgCl2, 10% glycerol and protease and phosphatase inhibitors. Cell lysates were cleared by centrifugation and lysates were incubated with protein A-agarose beads (Pharmacia) coupled to GFP or HA antibody, as indicated. After extensive washing with lysis buffer, bound proteins were eluted using Laemmli sample buffer and analyzed by SDS-PAGE.
QPCR
48 hours after siRNA transfection, HUVEC were plated onto 6 cm dishes coated with 5 µg/mL Fibronectin (Sigma-Aldrich) at the density of 8 × 105 cells/dish and allowed to settle for 24 hours. Total RNA was isolated using the RNeasy Mini Kit (Qiagen) and reverse transcribed into cDNA using the iScript cDNA synthesis kit from Bio-Rad Laboratories, Inc. cDNA levels were measured by SYBR green real-time PCR on a C1000 Thermal Cycler (Bio-Rad Laboratories Inc.) using comercially available KiCqStart® SYBR® Green primers (Sigma-Aldrich) for indicated cDNAs. Nonspecific signals were excluded based on non-template control samples. Amplification of HPRT cDNA was used a control for sample loading. The graph shows averages of 4 independent experiments in which expression levels were normalized to the scrambled siRNA control. Error bars indicate standard deviation.
Supplementary Material
Acknowledgements
We thank all members of our lab for continuous support and discussion. This research was supported by the gravitation program CancerGenomiCs.nl of the Netherlands Organisation of Scientific Research (NWO).
Funding Statement
This work was supported by the gravitation program CancerGenomiCs.nl of the Netherlands Organisation of Scientific Research (NWO).
Abbreviations
- AJ
Adherens Junction
- GEF
Guanine nucleotide Exchange Factor
- HUVEC
Human Umbilical Vein Endothelial Cell
- MLC2
Myosin Light Chain 2
- TJ
Tight Junction
- QPCR
Quantitative Polymerase Chain Reaction
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Chrzanowska-Wodnicka M. Rap1 in endothelial biology. Curr Opin Hematol. 2017;24:248–55. doi: 10.1097/MOH.0000000000000332. PMID:28178039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pannekoek WJ, Post A, Bos JL. Rap1 signaling in endothelial barrier control. Cell Adh Migr. 2014;8:100–7. doi: 10.4161/cam.27352. PMID:24714377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fukuhara S, Sakurai A, Sano H, et al. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–46. doi: 10.1128/MCB.25.1.136-146.2005. PMID:15601837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kooistra MR, Corada M, Dejana E, et al. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–72. doi: 10.1016/j.febslet.2005.07.080. PMID:16115630 [DOI] [PubMed] [Google Scholar]
- [5].Wittchen ES, Worthylake RA, Kelly P, et al. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem. 2005;280:11675–82. doi: 10.1074/jbc.M412595200. PMID:15661741 [DOI] [PubMed] [Google Scholar]
- [6].Post A, Pannekoek WJ, Ponsioen B, et al. Rap1 Spatially Controls ArhGAP29 To Inhibit Rho Signaling during Endothelial Barrier Regulation. Mol Cell Biol. 2015;35:2495–502. doi: 10.1128/MCB.01453-14. PMID:25963656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Post A, Pannekoek WJ, Ross SH, et al. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc Natl Acad Sci U S A. 2013;110:11427–32. doi: 10.1073/pnas.1306595110. PMID:23798437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wilson CW, Parker LH, Hall CJ, et al. Rasip1 regulates vertebrate vascular endothelial junction stability through Epac1-Rap1 signaling. Blood. 2013;122:3678–90. doi: 10.1182/blood-2013-02-483156. PMID:23886837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Glading A, Han J, Stockton RA, et al. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–54. doi: 10.1083/jcb.200705175. PMID:17954608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stockton RA, Shenkar R, Awad IA, et al. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J Exp Med. 2010;207:881–96. doi: 10.1084/jem.20091258. PMID:20308363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Whitehead KJ, Chan AC, Navankasattusas S, et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177–84. doi: 10.1038/nm.1911. PMID:19151728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ando K, Fukuhara S, Moriya T, et al. Rap1 potentiates endothelial cell junctions by spatially controlling myosin II activity and actin organization. J Cell Biol. 2013;202:901–16. doi: 10.1083/jcb.201301115. PMID:24019534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Birukova AA, Fu P, Wu T, et al. Afadin controls p120-catenin-ZO-1 interactions leading to endothelial barrier enhancement by oxidized phospholipids. J Cell Physiol. 2012;227:1883–90. doi: 10.1002/jcp.22916. PMID:21732359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Birukova AA, Tian X, Tian Y, et al. Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol Biol Cell. 2013;24:2678–88. doi: 10.1091/mbc.E13-02-0098. PMID:23864716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pannekoek WJ, Linnemann JR, Brouwer PM, et al. Rap1 and Rap2 antagonistically control endothelial barrier resistance. PLoS One. 2013;8:e57903. doi: 10.1371/journal.pone.0057903. PMID:23469100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Enserink JM, Christensen AE, de Rooij J, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–6. doi: 10.1038/ncb874. PMID:12402047 [DOI] [PubMed] [Google Scholar]
- [17].Vliem MJ, Ponsioen B, Schwede F, et al. 8- pCPT-2'-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem. 2008;9:2052–4. doi: 10.1002/cbic.200800216. PMID:18633951 [DOI] [PubMed] [Google Scholar]
- [18].Nakanishi H, Takai Y. Frabin and other related Cdc42-specific guanine nucleotide exchange factors couple the actin cytoskeleton with the plasma membrane. J Cell Mol Med. 2008;12:1169–76. doi: 10.1111/j.1582-4934.2008.00345.x. PMID:18410521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Daubon T, Buccione R, Genot E. The Aarskog-Scott syndrome protein Fgd1 regulates podosome formation and extracellular matrix remodeling in transforming growth factor beta-stimulated aortic endothelial cells. Mol Cell Biol. 2011;31:4430–41. doi: 10.1128/MCB.05474-11. PMID:21911474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].van Buul JD, Geerts D, Huveneers S. Rho GAPs and GEFs: controling switches in endothelial cell adhesion. Cell Adh Migr. 2014;8:108–24. doi: 10.4161/cam.27599. PMID:24622613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mandai K, Rikitake Y, Shimono Y, et al. Afadin/AF-6 and canoe: roles in cell adhesion and beyond. Prog Mol Biol Transl Sci. 2013;116:433–54. doi: 10.1016/B978-0-12-394311-8.00019-4. PMID:23481206 [DOI] [PubMed] [Google Scholar]
- [22].Sehrawat S, Cullere X, Patel S, et al. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell. 2008;19:1261–70. doi: 10.1091/mbc.E06-10-0972. PMID:18172027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sehrawat S, Ernandez T, Cullere X, et al. AKAP9 regulation of microtubule dynamics promotes Epac1-induced endothelial barrier properties. Blood. 2011;117:708–18. doi: 10.1182/blood-2010-02-268870. PMID:20952690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zankov DP, Ogita H. Actin-tethered junctional complexes in angiogenesis and lymphangiogenesis in association with vascular endothelial growth factor. Biomed Res Int. 2015;2015:314178. doi: 10.1155/2015/314178. PMID:25883953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Majima T, Takeuchi K, Sano K, et al. An Adaptor Molecule Afadin Regulates Lymphangiogenesis by Modulating RhoA Activity in the Developing Mouse Embryo. PLoS One. 2013;8:e68134. doi: 10.1371/journal.pone.0068134. PMID:23840823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zwartkruis FJ, Wolthuis RM, Nabben NM, et al. Extracellular signal- regulated activation of Rap1 fails to interfere in Ras effector signalling. EMBO J. 1998;17:5905–12. doi: 10.1093/emboj/17.20.5905. PMID:9774335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Z, Rehmann H, Price LS, et al. AF6 negatively regulates Rap1- induced cell adhesion. J Biol Chem. 2005;280:33200–5. doi: 10.1074/jbc.M505057200. PMID:16051602 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.