

ABSTRACT
The Rho GTPases were discovered more than 30 years ago, and they were for a long time considered to follow simple cycling between GDP-bound and GTP-bound conformations, as for the Ras subfamily of small GTPases. The Rho GTPases consist of 20 members, but at least 10 of these do not follow this classical GTPase cycle. Thus, based on their kinetic properties, these Rho GTPases can instead be classified as atypical. Some of these atypical Rho GTPases do not hydrolyze GTP, and some have significantly increased intrinsic GDP/GTP exchange activity. This review focuses on this latter category of atypical Rho GTPases, the so-called ‘fast-cycling’ Rho GTPases. The different members of these fast-cycling atypical Rho GTPases are described in more detail here, along with their potential regulatory mechanisms. Finally, some insights are provided into the involvement of the atypical Rho GTPases in human pathologies.
KEYWORDS: Atypical Rho GTPases, actin, cell migration, RhoD, RhoU, stress fibers
Introduction
The concept of fast-cycling small GTPases originated from studies in the early 1990s on the three-dimensional structure of H-Ras. A number of site-specific mutants were created to study the molecular interactions between the ribose ring of GTP and the key amino-acid residues in the nucleotide binding site of H-Ras. One of these mutants, with the phenylalanine at codon 28 substituted by a leucine (H-Ras/F28L), showed a higher nucleotide dissociation rate, and as a result, an elevated intrinsic GDP/GTP exchange activity.1 Expression of H-Ras/F28L in PC12 cells resulted in neurite outgrowth, which indicated that H-Ras/F28Lcan act as a constitutively active H-Ras mutant.
This concept of a small GTPases with increased intrinsic GDP/GTP exchange rates was applied to the field of the Rho GTPases by Lin et al.2 They confirmed that Cdc42/F28L has elevated nucleotide exchange activity. Moreover, NIH3T3 cells that expressed Cdc42/F28L showed oncogenic properties, through induction of anchorage-independent growth in soft agar.
The reason why elevated GDP/GTP exchange rates result in constitutively active GTPases is most likely a consequence of the differences in the cellular levels of these guanosine nucleotides. The intracellular concentration of GTP is 10-fold that of GDP, which implies that these mutant GTPases will remain predominantly in their active, GTP-bound, conformation.3
These examples demonstrate that through site-specific mutagenesis it is possible to create constitutively active mutant small GTPases that differ from the classical Ras/G12V and Ras/Q61L mutants. The classical mutants are GTPase deficient, and therefore they remain in the active GTP-bound conformation for longer. So can these Rho GTPases with high intrinsic GDP/GTP exchange activities have any impact on the world outside the test-tube? Yes, indeed, this has direct relevance, as during the quest for novel members of the Rho GTPases, the newly discovered RhoU was shown to have significantly elevated intrinsic GTP/GDP exchange activity.4,5 Moreover, it is clear that RhoD and Rif, and possibly also RhoV, have similarly elevated intrinsic GTP/GDP exchange activities.6,7 Importantly, this category of the Rho GTPases has intact GTPase activity, however, the elevated GTP/GDP exchange activity overrides the GTPase activity. These GTPases are often referred to as the ‘fast-cycling’ Rho GTPases,8 the terminology that is adopted in this review article.
The Rho GTPases
The identification of the Ras oncoproteins some 30 years ago triggered the hunt for Ras-like proteins. One of the first non-Ras proteins to be characterized was the Ras homologous protein (Rho) of the sea slug Aplysia, which was subsequently identified in mammalian cells.9 This initial observation was followed by identification of a number of additional Rho members, including Rac and Cdc42. These were shown to behave similarly to the Ras GTPases, in the sense that they are GTP-hydrolyzing enzymes that cycle between an active GTP-bound conformation and an inactive GDP-bound conformation.10 This simple scheme was considered to encompass all of the Rho GTPases, however, the first hint that things might be different came from the identification of Rnd1, Rnd2, and RhoE (also known as Rnd3).11,12 These GTPases turned out to be GTPase defective, or to show very restricted hydrolysis of GTP. This is because they have amino-acid residues in positions equivalent to 12, 59, and 61 in mutated Ras, and Ras proteins harboring mutations in these positions are GTPase-deficient and therefore constitutively GTP-bound (Fig. 1). Subsequently, RhoBTB1, RhoBTB2, and RhoH were shown to also be GTPase deficient.13
Figure 1.
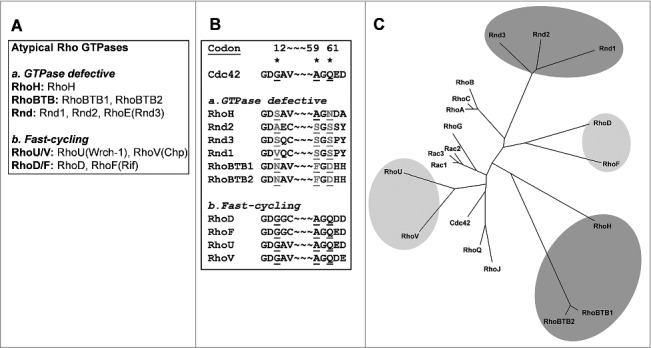
(A) The atypical Rho GTPases. (B) The amino-acid sequence over codons 12, 59, and 61 of the atypical Rho GTPases. (C) Phylogenetic tree representation of all of the human Rho GTPases. The dark grey area marks the GTPase deficient Rho GTPases and light grey are marks the fast-cycling Rho GTPases.
The Rho members that have kinetic properties that follow the archetypical cycling between the GTP-bound and GDP-bound conformations are commonly referred to as classical Rho GTPases. In addition, two atypical Rho categories can be distinguished: the GTPase-deficient Rho GTPases, and the fast-cycling Rho GTPases (Fig. 1). The fast-cycling Rho GTPases are basically identical to Cdc42 in the regions that surround codons 12 and 59/61, and therefore it is difficult to ascribe their fast-cycling to any specific amino-acid residues. The fast-cycling ability of H-Ras/F28L is a consequence of perturbed binding of the GTP purine base, its α-phosphate and β-phosphate, and the Mg2+ ion1. A similar mechanism might also apply to the fast-cycling Rho GTPases. This review focuses on these fast-cycling atypical Rho GTPases: RhoU, RhoV, RhoD, and RhoF.
RhoU and RhoV
The atypical Rho GTPases RhoU and RhoV were essentially described in a very recent review by Hodge and Ridley,14 and therefore they are mentioned briefly here, with the focus on their regulatory and kinetic properties. RhoV was cloned as Chp (Cdc42Hs homologue protein), and was shown to bind PAK2 and WASP, and to have a role in JNK activation.7 RhoU (also known as Wrch-1, Wnt-regulated Cdc42 homologous protein-1) was identified as a gene that was up-regulated in response to Wnt-1 treatment.15 RhoU was also shown to be associated with JNK activation, and ectopic expression of RhoU in Swiss 3T3 fibroblasts triggered the formation of filopodia, similar to the archetypical inducer of filopodia, Cdc42.10
Some Rho members (both classical and atypical) have N-terminal extensions, although the function of these extended regions are in most cases not clear (Fig. 2). The N-terminal extension of RhoU contains several SH3-domain-binding proline-rich motives, and RhoU has been shown to bind Nck1, Nck2, Grb2, and PLCγ via a central proline-rich APPVPPRR motif16 (Fig. 2). RhoV has a similar motif, although no proteins have so far been identified as binding to the N-terminus of RhoV. The prevailing hypothesis is that the N-terminal extension of RhoU has a regulatory role, and that SH3-domain-containing proteins that bind to the RhoU proline-rich domain, such as Grb2, are likely to contribute to this regulation. In-vitro measurements of the kinetic properties of RhoU have indicated that there are no significant differences in GDP/GTP exchange activity or GTP hydrolysis between full-length and N-terminal–truncated RhoU. However, the N-terminally truncated RhoU mutant has a stronger affinity for the effector protein PAK1, and cells that expressed this mutant showed increased anchorage-independent growth, which indicates that this truncated RhoU is more active than the intact protein.5 A similar effect was seen for RhoV, as expression of N-terminal–deleted RhoV in NIH3T3 cells resulted in a transformed phenotype in focus formation and soft agar assays.17
Figure 2.

Multiple alignments of the N-termini of the human Rho GTPases, produced using the ClustalW algorithm. *, identical amino-acid residues; :, conserved amino-acid residues. ¤, position of the amino-acid residue equivalent to codon 12 of Rac1; gray shading, APPVPPRR motif of the minimal SH3-domain-binding motif in RhoU.
To date, no Rho guanine nucleotide exchange factors (RhoGEFs) or GTPase-activating proteins (RhoGAPs) have been identified for RhoU and RhoV, which raises the question of how these Rho GTPases are regulated. Initially, Tao et al.15 showed RhoU to be under transcriptional control by Wnt-1, and therefore a positive regulatory step of RhoU at the level of transcription appeared likely. Indeed, expression of RhoU can for instance also be triggered by RANKL in osteoclasts, and by gp130 cytokines via a STAT3-dependent mechanism.18,19 To counterbalance this activation step, RhoU also needs to be negatively regulated. One mechanism for how this might be achieved was shown by studies on PAK4 and RhoU in adenocarcinoma cells.20 PAK4 is involved in the regulation of cell adhesion via RhoU. This regulation occurs through a mechanism that involves RhoU ubiquitination by the Rab40A:cullin5 complex. PAK4 protects RhoU from ubiquitination and degradation, as in the absence of PAK4, RhoU is ubiquitinated and degraded. RhoU can also be negatively regulated by tyrosine phosphorylation.21 Non-phosphorylated RhoU is localized to the plasma membrane in a GTP-bound conformation, from where it binds its downstream target protein, PAK1. However, Src-dependent phosphorylation of tyrosine 254 of RhoU results in loss of localization of RhoU at the plasma membrane, and the consequent loss of its interaction with PAK1.21
RhoU has a CCFV tetrapeptide at its C-terminus.15,22 This motif resembles the CAAX motifs seen for most Rho GTPases; however, for RhoU, this CAAX-like motif does not appear to be functional. Instead, the most C-terminal cysteine can undergo palmitoylation, and this modification is needed for the correct targeting of RhoU to the plasma membrane. Thus, mutation of this cysteine residue, or treatment of cells with the palmitoylation inhibitor 2-bromohexadecanoic acid, results in a relocalization of RhoU to cytosolic vesicles.22
The origins of RhoF and RhoD
RhoD arose very late in evolution, as it is only present in therians (e.g., marsupials, placental animals), and it appears to have originated through gene duplication of the RhoF ancestor gene in early bony vertebrates.23 RhoD is widely expressed in most tissues, and in several commonly used cell lines. Furthermore, it is expressed at high levels in mouse uterus, liver, kidney, bladder, stomach, and intestine.23,24 RhoF is also relatively widely expressed in human tissue, in particular in colon, stomach and spleen, and in HeLa cells.25 RhoF is expressed at higher levels in normal human B cells compared to other lymphocytes, which might indicate that RhoF has a role in normal functioning of B cells.26
When RhoD was first described in 1996, it was considered to fall into the category of a classical Rho GTPase.24 Similar to the archetypical Rho members, RhoA, Rac1, and Cdc42, RhoD turned out to have a strong impact on the organization of the actin filament system, and in particular on the formation of filopodia, although it was also shown to have a role in endosome trafficking.24 A few years later, RhoF was cloned and shown to have a role in the formation of dorsal filopodia (and hence originally called Rif, Rho in filopodia).25 A detailed analysis of the kinetic properties of RhoD and RhoF has revealed that they have significantly elevated intrinsic GDP/GTP exchange activities, and thus they belong to the atypical Rho GTPases, rather than the classical Rho GTPases.6
Roles in the regulation of actin dynamics
Both RhoD and RhoF are known to trigger the formation of filopodia in several cell types24,27,28 (Fig. 3). The mechanisms behind RhoD/RhoF-induced filopodia are not entirely known, although this process is independent of Cdc42, which is considered to be the bona-fide regulator of filopodia formation.25 In the case of RhoF, filopodia formation has been suggested to require Diaphanous-related formins, such as mDia1 and mDia2.29-31 In neuronal cells, filopodia-like precursors of dendritic spines elongate through a mechanism that requires RhoF and mDia2.32 Proteins that contain an inverted Bin/amphiphysin/Rvs (I-BAR) domain, such as IRTKS, are also possible mediators of filopodia formation (Fig. 3). BAR domains are known to bind lipid bilayers, and their banana-shaped three-dimensional fold can promote bending of the lipid bilayer, which is critical during initial invagination of the plasma membrane for endocytosis. In contrast, proteins with I-BAR domains can catalyze the reverse process; i.e. the formation of membrane protrusions.33 Accordingly, RhoF was shown to induce filopodia formation via a pathway that involves IRTKS, and also Eps8 and the WASP family protein WAVE2.34
Figure 3.
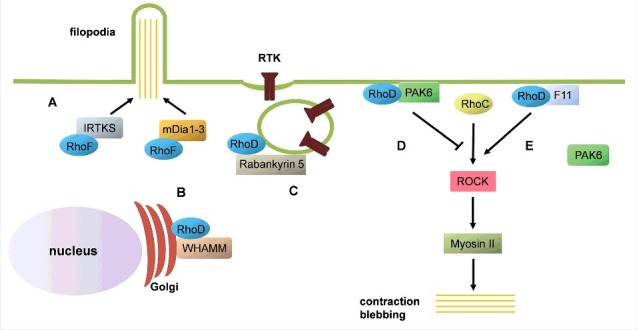
Schematic representation of RhoD and RhoF functions. (A) RhoF can trigger formation of filopodia by I-BAR-containing proteins, such as IRTKS, or Diaphanous-related formins (mDia1-3). (B) RhoD binds the Golgi complex component WHAMM, defining a role in Golgi homeostasis. (C) RhoD and its effector Rabankyrin-5 have roles in internalization and trafficking of receptor tyrosine kinanses (RTK), such as the PDGF-β receptor. (D) Under normal conditions, the RhoD:PAK6 complex inhibits ROCK activation by RhoC. (E) Under Vaccinia infection, the virus-derived protein F11 sequesters RhoD, which leads to dissociation of the RhoD:PAK6 complex. Under these conditions, RhoC can induce cell contraction and membrane blebbing in a ROCK-dependent and Myosin II-dependent manner.
The mechanisms underlying RhoD-induced filopodia formation are not so well characterized. Again, formins might be involved in this process, as RhoD was shown to interact with mDia3C (a splice variant of mDia3) in the formation of ‘cytoneme-like’ peripheral protrusions in fibroblast-like cells.31 The formation of RhoD-induced filopodia is typically accompanied by loss of stress fibers in several cell types.28,35,36 This response has sometimes been described as stress fiber dissolution, although it is important to note that the filamentous actin does not ‘disappear’; instead, it is reorganized into short bundles of actin filaments.28 The formation of these bundles of actin filaments is probably a reflection of the dispersion of the focal adhesions that can be caused by expression of wild-type RhoD or its constitutively active mutant RhoD/G26V.28,35,36 There are several potential mechanisms behind the effects of ectopic expression of RhoD on focal adhesion dynamics. One possible mediator is the Ser/Thr protein Zipper kinase (ZIPK, also known as Death-associated protein kinase 3), which is involved in the regulation of focal adhesion kinase (FAK) activity, and thereby, of cell adhesion. ZIPK is a RhoD effector, and constitutively active RhoD/G26V can modulate ZIPK activity.37 It is clear that compared to RhoA, RhoD has an opposing effect on stress fiber formation and organization. One possible mechanism for the interplay between RhoD and RhoA (or RhoC, in this case) has emerged from studies of cell contraction during Vaccinia infection. In non-infected HeLa cells, RhoD binds its effector protein PAK6, which it targets to the plasma membrane. This RhoD:PAK6 complex appears to inhibit the RhoC activation of the Ser/Thr protein kinase Rho kinase (ROCK), thus promoting normal cell spreading. However, during Vaccinia infection, the virus-derived F11 protein sequesters RhoD, which, in turn, means that the inhibitory interaction between RhoD and PAK6 is lost. Under these conditions, RhoC can bind and activate ROCK and thereby induce cell contraction38 (Fig. 3). Although the description of the relationships between RhoD and RhoA/RhoC during Vaccinia virus infection is potentially of great importance, it is still not clear how the interplay between RhoD and RhoA is regulated under normal conditions.
Compared to RhoD, RhoF appears to have different effects on stress fiber organization, as the expression of a constitutively active RhoF mutant in epithelial cells resulted in the assembly of stress fibers.39 This stress fiber response requires the concerted actions of mDia1 and ROCK. Importantly, RhoF expression does not result in increased activity of ROCK. Instead, RhoF appears to regulate the subcellular localization of ROCK, and thereby to compartmentalize the signal that regulates myosin activation and the subsequent stress fiber formation.
As RhoD and RhoF have such strong impacts on actin dynamics, it is not surprising that RhoD has important roles in the regulation of cell migration. Transfection of the constitutively active RhoD/G26V in fibroblasts resulted in decreased cell migration, as measured by the so-called ‘phagokinetic track’ assay.36 Furthermore, it has been shown that RhoD/G26V-expressing endothelial cells are more or less immotile, both in the absence and presence of chemoattractant (in this case fibroblast growth factor).40 Expression of a constitutively active RhoD mutant in glioblastoma cells resulted in less dynamic actin at the cell edges, which is in agreement with a less motile phenotype.28 Interestingly, the reverse condition, RhoD knock-down by RNA interference, resulted in the opposite effect; fibroblasts lacking RhoD moved across greater distances, although this migration was more random and hence less persistent. This indicates that the machinery for migration is not defective in cells with reduced levels of RhoD, instead, this machinery might be more active. Instead, the mechanisms that regulate cell polarization might not be fully functional. Importantly, chemotaxis towards a gradient of platelet-derived growth factor (PDGF)-BB was impaired in RhoD-depleted cells, which suggested a role for RhoD in the regulation of directed cell migration.28
Roles in membrane trafficking
From its initial characterization, RhoD was implicated in the processes that integrate early endosome motility and actin dynamics.24,40,41 RhoD in transiently transfected cells show its localization to Rab5-positive cytoplasmic vesicles, which appear to correspond to early endosomes. The perinuclear accumulation of enlarged endosomes that can be induced by overexpression of a constitutively active Rab5 (Rab/Q79L) was repressed by simultaneous expression of the constitutively active RhoD/G26V24. Moreover, RhoD has been ascribed a role in endosomal trafficking of the Src family kinases. Knock-down of RhoD has been shown to result in loss of the endosome-targeted Fyn, which suggested a role for RhoD in the regulation of Src family kinases.42
The Rab5 effector Rabankyrin-5 has also been shown to bind RhoD. RhoD and Rabankyrin-5 co-localize to Rab5-positive endosomes, which suggested a role for Rabankyrin-5 in the coordination of RhoD and Rab5 during endosomal trafficking.43 There is a relationship of mutual dependency between RhoD and Rabankyrin-5 in endosome trafficking, as knock-down of either RhoD or Rabankyrin-5 resulted in loss of a correct localization of the other protein. Moreover, RhoD and Rabankyrin-5 have been shown to interact during internalization and trafficking of receptor tyrosine kinases (Fig. 3). Knock-down of RhoD was shown to interfere with internalization of the PDGF-β receptor, and the subsequent activation of its downstream signaling cascades.43,44 This remains the possible mechanism that underlies impaired chemotaxis of RhoD-depleted cells.28 Another possible link between RhoD and cell polarization is via its binding partner WHAMM.35 A pool of endogenous RhoD localizes to the Golgi complex, and influences Golgi homeostasis.45 Therefore, cells that lack RhoD are likely to have defective Golgi reorientation, which in turn will affect cell polarization and migration in a persistent and directed manner.
Roles in cytokinesis
Expression of constitutively active RhoD/G26V in several cell types results in multinucleated cells, such as for Balb/3T3 and C3/10T1/2 fibroblasts, and N1E-115 neuroblastoma cells. This suggests a defect in cytokinesis. Defective cell cleavage has also been shown in Xenopus embryos injected with RhoD/G26V.36 Transgenic mice that express RhoD/G26V exclusively at the basal layer of the epidermis show hyperplasia and perturbed differentiation of epidermal cells.46 Apparently, RhoD has a role during entry of cells into the S phase of the cell cycle, a concept that is supported by the finding that knock-down of RhoD in endothelial cells results in accumulation of cells in the G1 phase of the cell cycle. Overexpression of RhoD/G26V results in increased cell proliferation and aberrant centrosome amplification.44 Knock-down of RhoD in fibroblasts does not interfere with cytokinesis, although the cell cycle is significantly longer in cells depleted of RhoD.28 These examples illustrate that precise RhoD activation is needed for normal cell physiology, whereby both RhoD over-activity and under-activity can have adverse effects on cell proliferation.
There is a general question regarding the role of RhoD and RhoF in vivo. There have been no studies on RhoD knock-out mice, although RhoF has been ablated in mice. RhoF−/− mice appeared healthy and breed normally.47 Moreover, the RhoF−/− mice have normal platelet, leukocyte and erythrocyte counts. Platelets isolated from these RhoF−/− mice form filopodia and spread normally on various agonist surfaces, and show normal actin dynamics. Thus, RhoF does not appear to be essential, although the phenotype might well be masked by compensatory mechanisms. Therefore, it would be interesting to analyze conditional knock-out mice, as well as mice that lack both RhoF and RhoD.
Oncogenic mutants of Rac1
We have now seen that it is possible to artificially create fast-cycling mutants of Rho GTPases, and that some of the atypical Rho GTPases are intrinsically fast cycling. However, there remains another category that needs to be considered: activated fast-cycling mutants of Rac1. The first hint of a fast-cycling variant of Rac1 came from the identification of a splice variant of Rac1 that was called Rac1b.48 This variant is caused by an alternative splicing event that resulted in 19 extra amino-acid residues after the Switch II motif in Rac1.48-50 This splice form appears to be expressed at low levels in normal cells, but importantly, its expression levels are increased in a number of human cancers.48 One possible mode of action is that Rac1b can antagonize the activity of wild-type Rac1.48,51,52 Alternatively, the constitutively active nature of Rac1b results in dysregulation of the Rac1 signal, which then simply overrides the normal and regulatable Rac1 protein. From an evolutionary perspective, Rac1b is found exclusively in amniotes.23 It is not clear why this splice form has been conserved, but it implies that Rac1b has been positively selected for a physiological function, which would be in relation to cell adhesion.
It is thus clear that both fast-cycling atypical Rho GTPases and mutations of classical Rho GTPases are associated with tumor promotion, apparently because the proteins are constitutively active in the true sense of the word. Of note, the normal so-called constitutively active mutants of the Rho GTPases, such as Rac1/G12V and Rac1/Q61L, have a separate mode of constitutive activation, as they are GTPase defective, rather than fast cycling.
Another variant on the same theme is the panel of Rac1 point mutations that have been identified, predominantly in melanoma cancer.53 Rho GTPases have been considered to be less likely to be mutated in cancers. Instead, the RhoGEFs and RhoGAPs have been implicated as targets for cancer-associated mutations. However, recent findings through the increased power of next-generation sequencing have changed this paradigm. Studies of melanoma have shown that Rac1/P29S is found in about 4% to 7% of all melanomas.53,54 Rac1/P29S is the third most recurrent mutation in melanoma, after the B-Raf/V600 and N-Ras/Q61 mutations. Rac1/P29S has a significantly increased intrinsic GDP/GTP cycling activity, and this is most likely the driver mutation that makes Rac1 an oncogene.55 The proline at codon 29 in the so-called switch I motif is conserved between all Rho GTPases and the proline to serine mutation result in direct hydrogen bonding between glycine at position 30 and the ribose hydroxyl groups. This interaction does not occur in the wild-type Rac1 since the proline at position 29 induces a conformational restraint.54 Analysis of the crystal structure of Rac1/F28L, showed that this mutant protein has a similar three dimensional fold in the Switch I motif as the wild-type Rac1, however the affinity for the nucleotide is much lower.55
Summary
The signaling networks that involve the Rho GTPases have increased in complexity more recently, and studies on the atypical Rho GTPases have significantly broadened the concept of Rho-regulated biological pathways. However, there remain many issues that need to be resolved in the future. The majority of the pioneering studies on atypical Rho GTPases have relied on ectopic expression of these proteins in various cell types, although mostly in fibroblasts. A similar approach has paved the way of our understanding of the classical Rho GTPases, RhoA, Rac1, and Cdc42. However, by applying new methods (such as CRISPR/Cas9 techniques) to ablate the atypical Rho GTPases in specific cell types, more precise information should be obtained in terms of the physiological functions of these atypical Rho GTPases. Moreover, there is the need to pin-point their binding partners, to obtain further information on the mechanisms that underlie the signaling of the atypical Rho GTPases. We have already seen that disturbances in their function can result in oncogenic transformation. It is likely that dysfunctional atypical Rho GTPases result in other disease conditions, which makes it important to increase the focus of our attention on these atypical Rho GTPases and the biological processes that they are involved in.
Funding Statement
Swedish cancer society, 2014/0644.
References
- 1.Reinstein J, Schlichting I, Frech M, et al. P21 with a phenylalanine 28→leucine mutation reacts normally with the GTPase-activating protein GAP, but nevertheless has transforming properties. J Biol Chem. 1991;266(26):17700–17706. [PubMed] [Google Scholar]
- 2.Lin R, Bagrodia S, Cerione R, et al. A novel Cdc42Hs mutant induces cellular transformation. Curr Biol. 1997;7(10):794–797. doi: 10.1016/S0960-9822(06)00338-1. [DOI] [PubMed] [Google Scholar]
- 3.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140(1):1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 4.Saras J, Wollberg P, Aspenström P. Wrch1 is a GTPase-deficient Cdc42-like protein with unusual binding characteristics and cellular effects. Exp Cell Res. 2004;299(2):356–369. doi: 10.1016/j.yexcr.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 5.Shutes A, Berzat AC, Cox AD, et al. Atypical mechanism of regulation of the Wrch-1 Rho family small GTPase. Curr Biol. 2004;14(22):2052–2056. doi: 10.1016/j.cub.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Jaiswal M, Fansa EK, Dvorsky R, et al. New insight into the molecular switch mechanism of human Rho family proteins: shifting a paradigm. Biol Chem. 2013;394(1):89–95. doi: 10.1515/hsz-2012-0207. [DOI] [PubMed] [Google Scholar]
- 7.Aronheim A, Broder YC, Cohen A, et al. Chp, a homologue of the GTPase Cdc42Hs, activates the JNK pathway and is implicated in reorganizing the actin cytoskeleton. Curr Biol. 1998;8(20):1125–1128. doi: 10.1016/S0960-9822(98)70468-3. [DOI] [PubMed] [Google Scholar]
- 8.Fidyk N, Wang JB, Cerione RA. Influencing cellular transformation by modulating the rates of GTP hydrolysis by Cdc42. Biochemistry. 2006;45(25):7750–7762. doi: 10.1021/bi060365h. [DOI] [PubMed] [Google Scholar]
- 9.Madaule P, Axel R. A novel Ras-related gene family. Cell. 1985;41(1):31–40. doi: 10.1016/0092-8674(85)90058-3. [DOI] [PubMed] [Google Scholar]
- 10.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–2469. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 11.Foster R, Hu KQ, Lu Y, et al. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in-vivo farnesylation. Mol Cell Biol. 1996;16(6):2689–2699. doi: 10.1128/MCB.16.6.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nobes CD, Lauritzen I, Mattei MG, et al. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141(1):187–197. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aspenström P, Ruusala A, Pacholsky D. Taking Rho GTPases to the next level: the cellular functions of atypical Rho GTPases. Exp Cell Res. 2007;313(17):3673–3679. doi: 10.1016/j.yexcr.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 14.Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496–510. doi: 10.1038/nrm.2016.67. [DOI] [PubMed] [Google Scholar]
- 15.Tao W, Pennica D, Xu L, et al. Wrch-1, a novel member of the Rho gene family that is regulated by Wnt-1. Genes Dev. 2001;15(14):1796–1807. doi: 10.1101/gad.894301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Risse SL, Vaz B, Burton MF, et al. SH3-mediated targeting of Wrch1/RhoU by multiple adaptor proteins. Biol Chem. 2013;394(3):421–432. doi: 10.1515/hsz-2012-0246. [DOI] [PubMed] [Google Scholar]
- 17.Chenette EJ, Abo A, Der CJ. Critical and distinct roles of amino- and carboxyl-terminal sequences in regulation of the biological activity of the Chp atypical Rho GTPase. J Biol Chem. 2005;280(14):13784–13792. doi: 10.1074/jbc.M411300200. [DOI] [PubMed] [Google Scholar]
- 18.Brazier H, Stephens S, Ory S, et al. Expression profile of RhoGTPases and RhoGEFs during RANKL-stimulated osteoclastogenesis: identification of essential genes in osteoclasts. J Bone Miner Res. 2006;21(9):1387–1398. doi: 10.1359/jbmr.060613. [DOI] [PubMed] [Google Scholar]
- 19.Schiavone D, Dewilde S, Vallania F, et al. The RhoU/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem J. 2009;421(2):283–292. doi: 10.1042/BJ20090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dart AE, Box GM, Court W, et al. PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J Cell Biol. 2015;211(4):863–879. doi: 10.1083/jcb.201501072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alan JK, Berzat AC, Dewar BJ, et al. Regulation of the Rho family small GTPase Wrch-1/RhoU by C-terminal tyrosine phosphorylation requires Src. Mol Cell Biol. 2010;30(17):4324–4338. doi: 10.1128/MCB.01646-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berzat AC, Buss JE, Chenette EJ, et al. Transforming activity of the Rho family GTPase, Wrch-1, a Wnt-regulated Cdc42 homolog, is dependent on a novel carboxyl-terminal palmitoylation motif. J Biol Chem. 2005;280(38):33055–33065. doi: 10.1074/jbc.M507362200. [DOI] [PubMed] [Google Scholar]
- 23.Boureux A, Vignal E, Faure S, et al. Evolution of the Rho family of Ras-like GTPases in eukaryotes. Mol Biol Evol. 2007;24(1):203–216. doi: 10.1093/molbev/msl145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy C, Saffrich R, Grummt M, et al. Endosome dynamics regulated by a Rho protein. Nature. 1996;384(6608):427–432. doi: 10.1038/384427a0. [DOI] [PubMed] [Google Scholar]
- 25.Ellis S, Mellor H. The novel Rho-family GTPase Rif regulates coordinated actin-based membrane rearrangements. Curr Biol. 2000;10(21):1387–1390. doi: 10.1016/S0960-9822(00)00777-6. [DOI] [PubMed] [Google Scholar]
- 26.Gouw LG, Reading NS, Jenson SD, et al. Expression of the Rho-family GTPase gene RhoF in lymphocyte subsets and malignant lymphomas. Br J Haematol. 2005;129(4):531–533. doi: 10.1111/j.1365-2141.2005.05481.x. [DOI] [PubMed] [Google Scholar]
- 27.Aspenström P, Fransson A, Saras J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J. 2004;377(2):327–337. doi: 10.1042/bj20031041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blom M, Reis K, Heldin J, et al. The atypical Rho GTPase RhoD is a regulator of actin cytoskeleton dynamics and directed cell migration. Exp Cell Res. 2017;352(2):255–264. doi: 10.1016/j.yexcr.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Pellegrin S, Mellor H. The Rho family GTPase Rif induces filopodia through mDia2. Curr Biol. 2005;15(2):129–133. doi: 10.1016/j.cub.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Goh WI, Sudhaharan T, Lim KB, et al. Rif-mDia1 interaction is involved in filopodium formation independent of Cdc42 and Rac effectors. J Biol Chem. 2011;286(15):13681–13694. doi: 10.1074/jbc.M110.182683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.31. Koizumi K, Takano K, Kaneyasu A, et al. RhoD activated by fibroblast growth factor induces cytoneme-like cellular protrusions through mDia3C. Mol Biol Cell. 2012;23(23):4647–4661. doi: 10.1091/mbc.E12-04-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hotulainen P, Llano O, Smirnov S, et al. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185(2):323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salzer U, Kostan J, Djinović-Carugo K. Deciphering the BAR code of membrane modulators. Cell Mol Life Sci. 2017;74(13):2413–2438. doi: 10.1007/s00018-017-2478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sudhaharan T, Sem KP, Liew HF, et al. The Rho GTPase Rif signals through IRTKS, Eps8 and WAVE2 to generate dorsal membrane ruffles and filopodia. J Cell Sci. 2016;129(14):2829–2840. doi: 10.1242/jcs.179655. [DOI] [PubMed] [Google Scholar]
- 35.Gad AK, Nehru V, Ruusala A, et al. RhoD regulates cytoskeletal dynamics via the actin nucleation-promoting factor WASp homologue associated with actin Golgi membranes and microtubules. Mol Biol Cell. 2012;23(24):4807–4819. doi: 10.1091/mbc.E12-07-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsubakimoto K, Matsumoto K, Abe H, et al. Small GTPase RhoD suppresses cell migration and cytokinesis. Oncogene. 1999;18(15):2431–2440. doi: 10.1038/sj.onc.1202604. [DOI] [PubMed] [Google Scholar]
- 37.Nehru V, Almeida FN, Aspenström P. Interaction of RhoD and ZIP kinase modulates actin filament assembly and focal adhesion dynamics. Biochem Biophys Res Commun. 2013;433(2):163–169. doi: 10.1016/j.bbrc.2013.02.046. [DOI] [PubMed] [Google Scholar]
- 38.Durkin CH, Leite F, Cordeiro JV, et al. RhoD inhibits RhoC-ROCK-dependent cell contraction via PAK6. Dev Cell. 2017;41(3):315–329.e7. doi: 10.1016/j.devcel.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan L, Pellegrin S, Scott A, Mellor H. The small GTPase Rif is an alternative trigger for the formation of actin stress fibers in epithelial cells. J Cell Sci. 2010;123(8):1247–1252. doi: 10.1242/jcs.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy C, Saffrich R, Olivo-Marin JC, et al. Dual function of RhoD in vesicular movement and cell motility. Eur J Cell Biol. 2001;80(6):391–398. doi: 10.1078/0171-9335-00173. [DOI] [PubMed] [Google Scholar]
- 41.Gasman S, Kalaidzidis Y, Zerial M. RhoD regulates endosome dynamics through Diaphanous-related Formin and Src tyrosine kinase. Nat Cell Biol. 2003;5(3):195–204. Erratum in: Nat Cell Biol.2003;5(7):680. doi: 10.1038/ncb935. [DOI] [PubMed] [Google Scholar]
- 42.Sandilands E, Brunton VG, Frame MC. The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J Cell Sci. 2007;120(15):2555–2564. doi: 10.1242/jcs.003657. [DOI] [PubMed] [Google Scholar]
- 43.Nehru V, Voytyuk O, Lennartsson J, et al. RhoD binds the Rab5 effector Rabankyrin-5 and has a role in trafficking of the platelet-derived growth factor receptor. Traffic. 2013;14(12):1242–1254. doi: 10.1111/tra.12121. [DOI] [PubMed] [Google Scholar]
- 44.Schnatwinkel C, Christoforidis S, Lindsay MR, et al. The Rab5 effector Rabankyrin-5 regulates and coordinates different endocytic mechanisms. PLoS Biol. 2004;2(9):E261. doi: 10.1371/journal.pbio.0020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blom M, Reis K, Nehru V, et al. RhoD is a Golgi component with a role in anterograde protein transport from the ER to the plasma membrane. Exp Cell Res. 2015;333(2):208–219. doi: 10.1016/j.yexcr.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 46.Kyrkou A, Soufi M, Bahtz R, et al. RhoD participates in the regulation of cell-cycle progression and centrosome duplication. Oncogene. 2013;32(14):1831–1842. doi: 10.1038/onc.2012.195. [DOI] [PubMed] [Google Scholar]
- 47.Goggs R, Savage JS, Mellor H, et al. The small GTPase Rif is dispensable for platelet filopodia generation in mice. PLoS One. 2013;8(1):e54663. doi: 10.1371/journal.pone.0054663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan P, Brazåo R, Boavida MG, et al. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999;18(48):6835–6839. doi: 10.1038/sj.onc.1203233. [DOI] [PubMed] [Google Scholar]
- 49.Fiegen D, Haeusler LC, Blumenstein L, et al. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004;279(6):4743–4749. doi: 10.1074/jbc.M310281200. [DOI] [PubMed] [Google Scholar]
- 50.Singh A, Karnoub AE, Palmby TR, et al. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23(58):9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- 51.Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem. 2003;278(50):50442–50448. doi: 10.1074/jbc.M308215200. [DOI] [PubMed] [Google Scholar]
- 52.Nimnual AS, Taylor LJ, Nyako M, et al. Perturbation of cytoskeleton dynamics by the opposing effects of Rac1 and Rac1b. Small GTPases. 2010;1(2):89–97. doi: 10.4161/sgtp.1.2.14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halaban R. RAC1 and melanoma. Clin Ther. 2015;37(3):682–685. doi: 10.1016/j.clinthera.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis MJ, Ha BH, Holman EC, et al. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc Natl Acad Sci USA. 2013;110(3):912–917. doi: 10.1073/pnas.1220895110. [DOI] [PMC free article] [PubMed] [Google Scholar]