

ABSTRACT
Recent work has associated point mutations in both zinc fingers (ZnF) of the spliceosome component U2AF35 with malignant transformation. However, surprisingly little is known about the functionality of the U2AF35 ZnF domains in general. Here we have analysed key functionalities of the ZnF domains of mammalian U2AF35 and its paralog U2AF26. Both ZnFs are required for splicing regulation, whereas only ZnF2 controls protein stability and contributes to the interaction with U2AF65. These features are confirmed in a naturally occurring splice variant of U2AF26 lacking ZnF2, that is strongly induced upon activation of primary mouse T cells and localized in the cytoplasm. Using Ribo-Seq in a model T cell line we provide evidence for a role of U2AF26 in activating cytoplasmic steps in gene expression, notably translation. Consistently, an MS2 tethering assay shows that cytoplasmic U2AF26/35 increase translation when localized to the 5ʹUTR of a model mRNA. This regulation is partially dependent on ZnF1 thus providing a connection between a core splicing factor, the ZnF domains and the regulation of translation. Altogether, our work reveals unexpected functions of U2AF26/35 and their ZnF domains, thereby contributing to a better understanding of their role and regulation in mammalian cells.
KEYWORDS: U2AF, zinc finger, protein stability, translation, splicing, T cell activation
Introduction
Splicing is an essential step during pre-mRNA processing of most genes that removes introns and joins exons to yield the mature mRNA with a continuous open reading frame. Splicing is catalysed by the spliceosome that assembles de novo on every intron through binding to several sequence motives on the pre-mRNA [1]. The 3ʹ splices site (3’ss) is marked by a polypyrimidine tract followed by an AG-dinucleotide directly at the intron-exon boundary. The 3’ss is recognized by the U2 auxiliary factor (U2AF) heterodimer composed of the large subunit U2AF65 binding to the polypyrimidine tract and the small subunit U2AF35 (U2AF1) binding to the AG-dinucleotide [2–6]. Structural analysis of a minimal U2AF heterodimer identified a 28-residue segment in U2AF65, directly upstream of its RRMs, and two short 13- and 8-residue fragments in U2AF35 as interacting regions between both proteins [7]. The recent structure of full-length yeast U2AF23 (the U2AF35 homolog) with a short fragment of yeast U2AF59 (the U2AF65 homolog) revealed an additional interaction surface, a short alpha-helix (α6), in the C-terminus of U2AF23 directly downstream of its second zinc finger [8], which was shown to play a role in the expression of mammalian U2AF35 as well [9].
U2AF35 consists of an U2AF homology motif (UHM) flanked by two zinc finger domains (ZnFs) and a C-terminal RS-domain important for subcellular localization (see Fig. 1A) [10–14]. This architecture is highly conserved across species and several paralogs are found in mouse and human [15]. One example is the smaller paralog U2AF26 (U2AF1L4) that lacks parts of the C-terminal RS-domain but shares 89% of the amino acids in the N-terminal 190 amino acids with U2AF35 [16–18]. U2AF26 is itself alternatively spliced and gives rise to at least two different isoforms in addition to the full-length protein [18]. The alternative U2AF26 splice variants lack either exon 7 or exons 6 and 7 and localize to the cytoplasm in contrast to the nuclear full-length U2AF26 [18,19]. Exclusion of exon 6 and 7 changes the reading frame of the mRNA and leads to a new prolonged C-terminus in U2AF26 that influences circadian rhythms in mice [19]. The function of cytoplasmic U2AF26 lacking only exon 7 is so far unknown.
Figure 1.
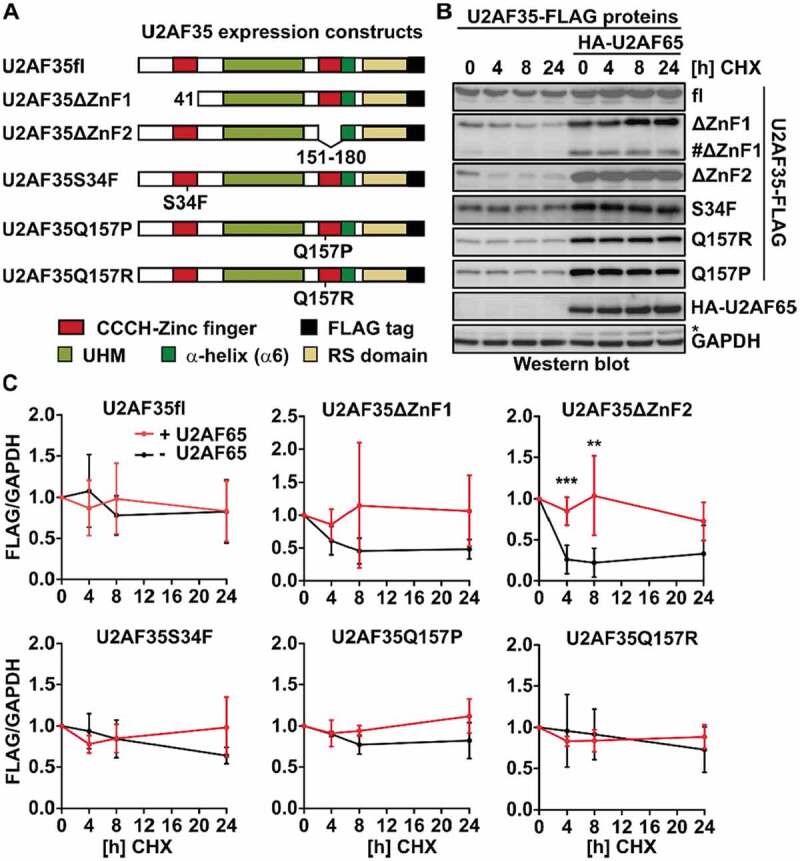
The second zinc finger of U2AF35 is required for protein stability.
Although the role of U2AF35 in directly binding the AG at the 3ʹ splice site has been discovered 20 years ago, the protein domain that makes direct contact to the RNA remained elusive. In early studies, it was reported that a minimal U2AF heterodimer composed out of the UHM of U2AF35 and a short N-terminal fragment of U2AF65 is sufficient to bind an RNA containing a 3ʹ splice site consensus sequence [7]. A comprehensive mutational analysis provided first indications that, besides the UHM, both zinc fingers of yeast U2AF23 are important for viability and that all three domains contribute to RNA binding [11]. Recently, the crystal structure of the full length yeast U2AF35 homolog U2AF23 was solved showing that RNA-binding of U2AF23 is mediated through its ZnF domains. The UHM however serves as a scaffold for the ZnF domains and as a protein–protein interaction surface [8]. As this work was based on in vitro assays and a structure of the yeast homolog U2AF23, with a conservation of 60% to mouse U2AF35 in the N-terminus, functional implications of this structure remain to be confirmed in the mammalian system. The ZnF domains in human U2AF35 have attracted further attention since the identification of cancer-associated mutations that exclusively occur in either ZnF1 (S34F/Y) or ZnF2 (Q157R/P) of U2AF35 and ZnF2 of U2AF26 (C116X) and not in the other domains [20–22]. Although several studies have shown that these mutations affect the binding specificity of U2AF35 to the 3’ss [23–28], additional roles of the ZnF domains in controlling U2AF35 function remain largely unknown.
In the present work, we have analysed several key functions of U2AF26 and U2AF35 using ZnF deletions or cancer-associated point mutations in mammalian cells. We show that ZnF2 is required for basal expression, protein stability and stable interaction with U2AF65, whereas cancer-associated point mutations only display slightly reduced basal expression. We have furthermore quantified the effect of the ZnF deletion mutants on splicing of several U2AF35-dependent exons and show loss of function for ZnF-deleted U2AF35; similar results were obtained with the paralog U2AF26. We further validate our observation with a naturally occurring alternative splice variant of U2AF26 that lacks exon 7, which leads to the deletion of ZnF2. This variant localizes to the cytoplasm and is upregulated upon activation of primary mouse T cells. Ribo-Seq in a T cell line and an MS2-based reporter assay suggest that cytoplasmic U2AF26 activates translation, in part through ZnF1. Our work provides evidence for a new function of cytoplasmic U2AF26 and demonstrates the importance of the ZnF domains for U2AF35 and U2AF26 function beyond RNA binding.
Results
ZnF2 stabilizes U2AF35 and U2AF26
Despite the identification of U2AF35 as the AG-binding protein around 20 years ago and the recent discovery of several cancer-associated mutations within U2AF35, surprisingly little is known about the contribution of the different protein domains to U2AF35 function. The ZnF domains have been suggested to mediate RNA-protein contacts and they also represent hotspots for somatic mutations associated with malignant transformation. We therefore cloned U2AF35 constructs that lack either the first ZnF (ΔZnF1, this construct also lacks the N-terminal amino acids 1–17 just upstream of ZnF1; results are confirmed with a single ZnF-destroying point mutation, see below) or the second ZnF (ΔZnF2) domain. In addition, we introduced the cancer-associated mutation S34F in ZnF1 and Q157R and Q157P in ZnF2 of U2AF35 (Fig. 1A). The sequences correspond to the mouse ortholog, which is basically identical to human U2AF35 at amino acid level; the only difference is the length of a Glycine tract in the C-terminus, 11 G-residues in mouse, 12 in human. We expressed these U2AF35 constructs as Flag-tagged proteins in HEK293T cells and addressed a role in regulating protein stability, interaction with U2AF65 and in controlling alternative splicing.
To analyse stability of the different U2AF35 mutants we treated transfected cells with the translation inhibitor Cycloheximide (CHX) for different time points and analysed protein expression by western blot. The full length U2AF35 protein (fl) has a protein half-life above 24 h that is not affected by co-expression of U2AF65 (Fig. 1B, C). Removal of the first zinc finger leads to a slightly reduced basal expression level and a reduction of the half-life to less than 8 h. Deletion of the second zinc finger has a strong impact on the basal expression (Supplementary Figure S1B) and the stability is reduced to a half-life below 4 h (Fig. 1B, C). This is not due to reduced solubility of the ZnF2-deleted U2AF35, as we do not observe an enrichment of this variant in the pellets of cell debris after whole cell extract preparation compared to the other U2AF35 mutants (Supplementary Figure S1C). Furthermore, reduced basal expression is not due to altered mRNA levels, as we find similar amounts of overexpressed mRNAs (Supplementary Figure S1D). These results suggest the second zinc finger of U2AF35 to play a role in controlling protein abundance and stability. Interestingly, both, reduced basal expression and decreased stability, are rescued by co-expression of U2AF65, suggesting that the formation of a U2AF heterodimer stabilizes the structure (Fig. 1B, C). Similar results were observed for its shorter paralog U2AF26 that, apart from its RS-rich, C-terminal domain, has 89% sequence conservation to U2AF35 (Supplementary Figure S1A and S1E). This is consistent with in vitro data showing that the UHM of U2AF35 adapts an ordered structure only upon binding of U2AF65 [29] and suggests that the second ZnF contributes to the formation of a stabilized protein fold in the absence of U2AF65 interaction. In the ΔZnF1 construct, we consistently observe a faster migrating protein species that, like the ΔZnF2 mutant, shows decreased basal expression and stability that is partially rescued by co-expression of U2AF65. This protein likely is the result of the usage of an internal ATG; based on size, it could be the methionine at position 74. This finding supports the idea that several parts of the protein are required, i.e., the UHM and the second ZnF, to allow a stable protein fold in cells in the absence of U2AF65. Finally, to rule out that deletion of the ZnF domains globally alters protein conformation, we repeated the experiments with point mutants in which one of the Zn-coordinating Cysteine-residues was mutated to Alanine (ZnF1: C18A; ZnF2: C155A). We also included a double point mutant (L181P/L185P) that interferes with the formation of the helix α6 downstream of ZnF2, that was suggested to contribute to the U2AF65 interaction (Supplementary Figure S1A). The single point mutants recapitulate the effect of whole ZnF deletions, with strongly reduced basal expression and stability in the C155A mutant (Supplementary Figure S1F). A similar effect was observed in the mutant preventing α6 to form (Supplementary Figure S1F), further validating a crucial role of these domains for U2AF26/35 expression.
Given the prominent role of the ZnF domains in controlling expression and stability of U2AF35, we further analysed cancer-associated mutations falling within these domains, namely S34F (c.101 C > T) in ZnF1 and Q157R (c.470A>G) and Q157P (c.470A>C) in ZnF2. Cancer-associated U2AF35 mutations have been primarily analysed with respect to splicing defects and RNA-binding specificity but additional features conferred by these mutations have not been characterized in detail. We therefore tested the stability of the S34F, Q157R and Q157P mutants in the presence or absence of U2AF65. In these assays, we observe slightly reduced basal expression for all three point mutants similar to the complete ZnF1 deletion and a protein half-life above 24 h (Fig. 1C and Supplementary Figure S1B). Again, the reduced basal expression of the S34F, Q157R and Q157P mutations are completely rescued by co-expression of U2AF65 suggesting that the cancer-associated mutations can interfere with proper expression of the U2AF35 monomer in conditions where U2AF65 is limiting for the formation of the heterodimeric U2AF complex.
ZnF2 and α6 are required for stable U2AF heterodimer formation
Although the co-expression experiments suggested that all U2AF35 mutants were able to interact with U2AF65, we analysed this interaction in more detail, as it is crucial for U2AF function. To this end, we performed CoIP experiments with U2AF65 and the different U2AF35 mutations and ZnF deletions. All U2AF35 constructs show a similar interaction with U2AF65 under standard conditions (400 mM, Fig. 2A), which is RNA independent (Fig. 2B). Interestingly, this interaction is almost completely lost in more stringent (high salt: 1200 mM) conditions for the mutant lacking ZnF2 (Fig. 2A, bottom). Again we observe the same pattern for the paralog U2AF26 (Fig. 2C). These data are in agreement with the U2AF35 crystal structure from yeast, which shows the presence of a U2AF35-U2AF65 helical interaction surface in the C-terminal region of U2AF35, which is independent of the interaction mediated by the UHM [8]. Although the C-terminus of mouse and yeast U2AF35 is not well conserved, a helix in the C-terminus of mouse U2AF35 is predicted directly downstream of the second zinc finger that may function as additional U2AF65 interaction surface (Supplementary Figure S2A). Deletion of the second ZnF in U2AF35 likely alters the location, orientation or fold of this interaction surface, which reduces the interaction strength and results in loss of heterodimer formation under stringent conditions. To directly test this prediction, we used the ZnF2 (C155A) or helix-disrupting (L181P/L185P) point mutants and tested their interaction with U2AF65 under stringent conditions. In both cases, heterodimer formation is completely lost (Fig. 2D), confirming an essential role for α6 in the interaction with U2AF65, which also requires an intact ZnF2. The disease-associated mutations S34F in the first and Q157R/P in the second ZnF do not affect the interaction with U2AF65 (Fig. 2A), suggesting that they do not interfere with the C-terminal interaction domain. It should be noted that we observe strong differences in heterodimer formation between the different mutants only under stringent, high salt, conditions. While this is above the physiological salt concentration, it is difficult to predict which IP condition most accurately reflects the densely packed nucleoplasm. Furthermore, our IP assay likely is not sensitive enough to monitor subtler differences that may be present at lower, physiological salt concentration. We therefore suggest that the ZnF2 and α6 domains will likely impact U2AF heterodimer formation also in living cells.
Figure 2.
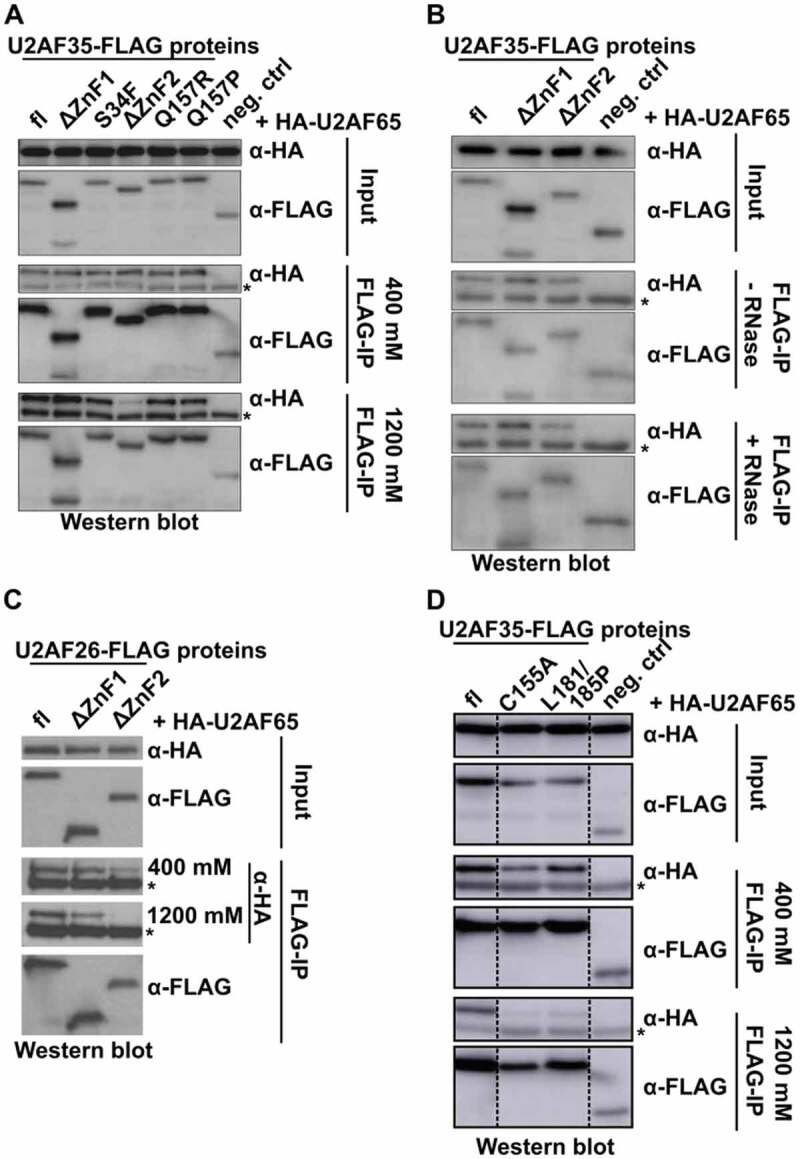
The second zinc finger of U2AF35 mediates stable interaction with U2AF65.
Both ZnFs but not α6 are essential for splicing regulation
The structure of yeast U2AF35 showed that both ZnF domains of U2AF35 are positioned on its UHM and cooperatively bind to the target RNA sequence [8]. To directly test a role of the ZnF domains in splicing regulation we used an established knock-down complementation assay with quantitative read-out, in which missplicing of exons sensitive to loss of endogenous U2AF35 can be rescued by expression of U2AF35 variants [19,27]. Endogenous U2AF35 mRNA was knocked down to around 10% and replaced by the respective ectopically expressed mutants (Fig. 3A, B). For our analysis, we have chosen eight exons that, based on previous studies, show a substantial change in exon inclusion upon U2AF35 knock down [27,30]. For seven out of eight exons we observe decreased exon inclusion upon knock-down of U2AF35 and one exon shows the opposite effect (Fig. 3C and Supplementary Figure S3), which is in agreement with previously published work [27,30]. Overexpression of U2AF35fl reverses this effect and leads to an almost complete rescue of the splicing defect in all cases thus validating our experimental system. In contrast, mutants with deletion of either the first or the second ZnF domain completely lose the ability to rescue splicing defects induced by knock-down of endogenous U2AF35 (Fig. 3C). For the effect of cancer-associated point mutants on alternative splicing, we refer to previously published work [23–27]. Our data, showing that ZnF deletions completely lose the ability to rescue splicing defects, are in agreement with the recent yeast U2AF23 structure, showing that both ZnF domains are required to make contact with the RNA [8]. Stabilization of unstable ZnF-deleted U2AF35 mutants by co-expressing U2AF65 has no influence on the rescue abilities of U2AF35 (Fig. 3D) ruling out that reduced protein levels of the mutants are causing the inability to rescue. U2AF35 knock-down is also rescued through the full-length paralog U2AF26 but not by U2AF26 lacking either the first or the second zinc finger (Fig. 3C). This is in agreement with in vitro splicing assays for two model RNAs were U2AF26 was shown to fully substitute for U2AF35 function in splicing regulation [16]. To further substantiate the role of the ZnF and α6 domains in splicing regulation, we tested the rescue ability of the respective point mutants. While single point mutants deleting a Zn-coordinating Cysteine-residue in ZnF1 or ZnF2 are sufficient to completely abolish the rescue ability, the α6 mutant is able to rescue like the wt protein (Fig. 3E). These data suggest that α6 is required for the formation of a stable U2AF heterodimer, but that, in this overexpression setting, this is not required to control alternative splicing of U2AF35-responsive exons.
Figure 3.
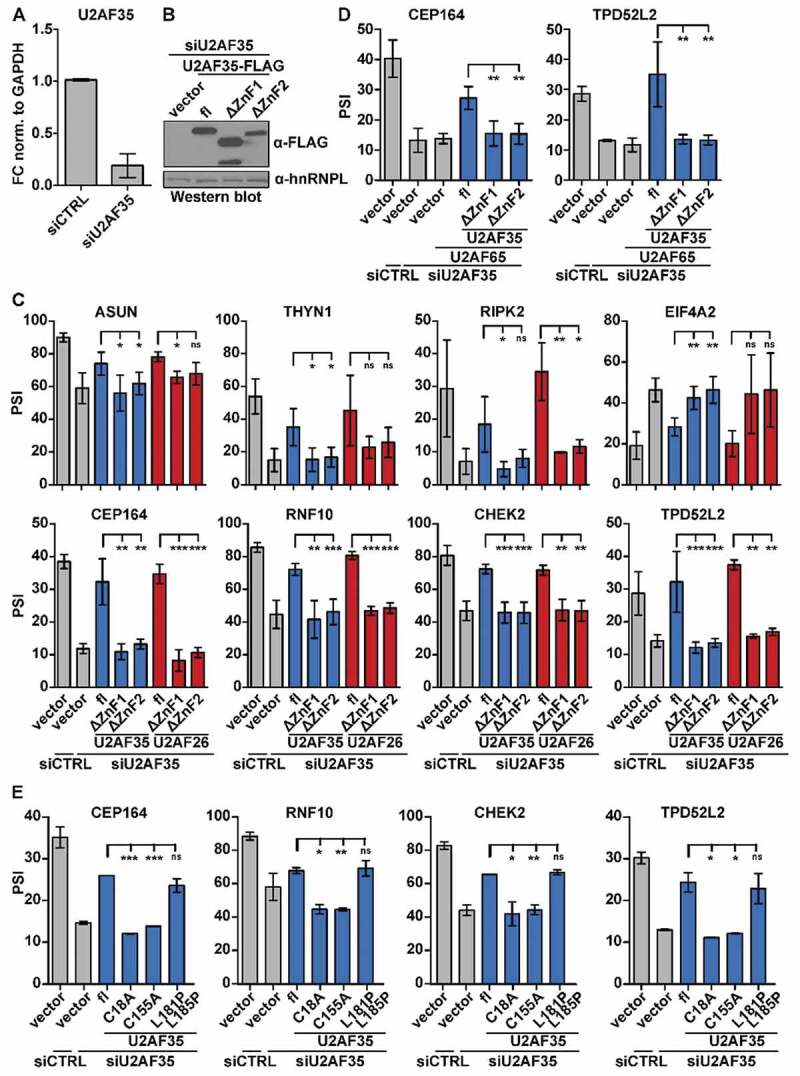
Both ZnFs are indispensable for U2AF35-dependant regulation of alternative splicing.
A U2AF26 variant lacking ZnF2 and α6 is strongly induced upon activation of primary mouse T cells
Previous studies have shown that U2AF26 itself is alternatively spliced [18,19]. Interestingly, exclusion of U2AF26 exon 7 (U2AF26ΔE7) leads to the removal of three of the four zinc complexing residues of ZnF2 and 20 additional downstream amino acids, including α6 (Fig. 4A, Supplementary Figure S2B, bottom). Based on previous work [17] we purified and stimulated mouse primary T cells and quantified U2AF26 isoform expression by splicing sensitive RT-PCR. For three different stimuli, we observe a strong increase of the U2AF26ΔE7 variant, whereas the second known variant, U2AF26ΔE67, is only mildly affected (Fig. 4B, C). In the following, we have therefore used U2AF26ΔE7 to investigate the functionality of ZnF2 and α6 in an endogenously occurring U2AF26 isoform. Loss of exon 7 leads to a slightly reduced basal expression and reduced stability of the U2AF26ΔE7 protein, but the effects are milder than for the full ZnF2 deletion (Fig. 4D and Supplementary Figure 1E). Both, basal protein expression and stability are rescued by co-expression of U2AF65 similar as seen for the full ZnF2 deletion. Binding of U2AF26ΔE7 to U2AF65 is similar as for full-length U2AF26 in low salt conditions, but abolished in the presence of high salt (Fig. 4E), thus resembling behaviour of the ZnF2 deletion and α6 mutation, which is consistent with α6 being absent in U2AF26ΔE7 (Supplementary Figure S2B, bottom). U2AF26ΔE7 is also not able to rescue miss-splicing upon U2AF35 knock-down (Fig. 4F), which is likely a combined effect of the lack of ZnF2 and predominant localization of this variant in the cytoplasm [18]. These results confirm the functional studies of artificial deletion and point mutants in an endogenously occurring U2AF26 variant.
Figure 4.
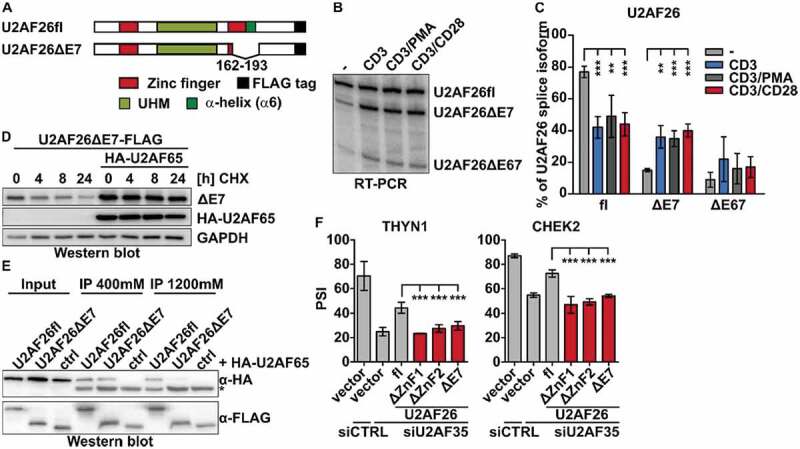
The naturally occurring U2AF26ΔE7 behaves similar as the ZnF2 deletion mutant.
U2AF26ΔE7 controls gene expression in the cytoplasm
Based on the above results we further characterized the functionality conferred by the lack of ZnF2 and α6 in U2AF26ΔE7. As this isoform is strongly induced in activated T cells, we used a mouse T cell line, EL4, and generated independent cell lines stably overexpressing U2AF26ΔE7 (Fig. 5A). To investigate a potential gene regulatory function of the U2AF26ΔE7, we performed ribosome profiling (Ribo-Seq) experiments using triplicate samples of U2AF26ΔE7-expressing (clone #7) and control cells (clone #2), yielding approximately 20 million uniquely mapped reads per sample (Supplementary table 2). As expected for ribosome protected fragments (RPF) these aligned reads have an average length of ~29 nucleotides (Supplementary table 2). In parallel we sequenced total RNA from the same clones in duplicates to normalize changes in ribosome occupancy to changes in total transcript levels, yielding approximately 15 million uniquely mapped reads per sample (Supplementary table 2). After normalization and stringent filtering, we obtained 269 genes that generate significantly different numbers in input-normalized RPFs (fold change more than 1.5-fold, p-value below 0.05) in the U2AF26ΔE7-expressing cells (Fig. 5B, Supplementary table 3). Changes in RPF levels are highly consistent in the triplicate samples (Fig. 5C). Notably, over two-thirds of the genes show an increase in input-normalized RPFs upon overexpression of U2AF26ΔE7, indicating a translational upregulation (Fig. 5B,C). GO term analysis of the genes that produce increased numbers of RPFs suggest an involvement in cell cycle (p = 1.11E-06) and metabolic processes (p = 4.18E-06) whereas no GO term is enriched in the set of genes with a decrease in RPF numbers upon forced U2AF26ΔE7 expression. Upon the genes that show an increase in normalized RPFs in U2AF26ΔE7-overexpressing cells are two related genes, Nr4a1 and Nr4a2. In RT-qPCR validations, we included an additional independently generated U2AF26ΔE7 expressing clone (#3) and confirm that Nr4a1 mRNA levels are not affected by U2AF26ΔE7 expression (Fig. 5D). Unchanged mRNA levels but increased ribosome association after overexpression of U2AF26ΔE7 indicates that U2AF26ΔE7 can affect ribosome loading, and possibly translation of this specific and other select mRNAs. To provide further evidence in favour of this hypothesis we analysed protein levels of Nr4a1 by western blot (Fig. 5F). Here we observe an increase in protein expression in the U2AF26ΔE7 cell line, which, together with the unchanged mRNA levels points to U2AF26ΔE7-mediated translational upregulation. This finding is in agreement with recent work suggesting a role of cytoplasmic full-length U2AF35 in controlling translation [28, also see discussion].
Figure 5.
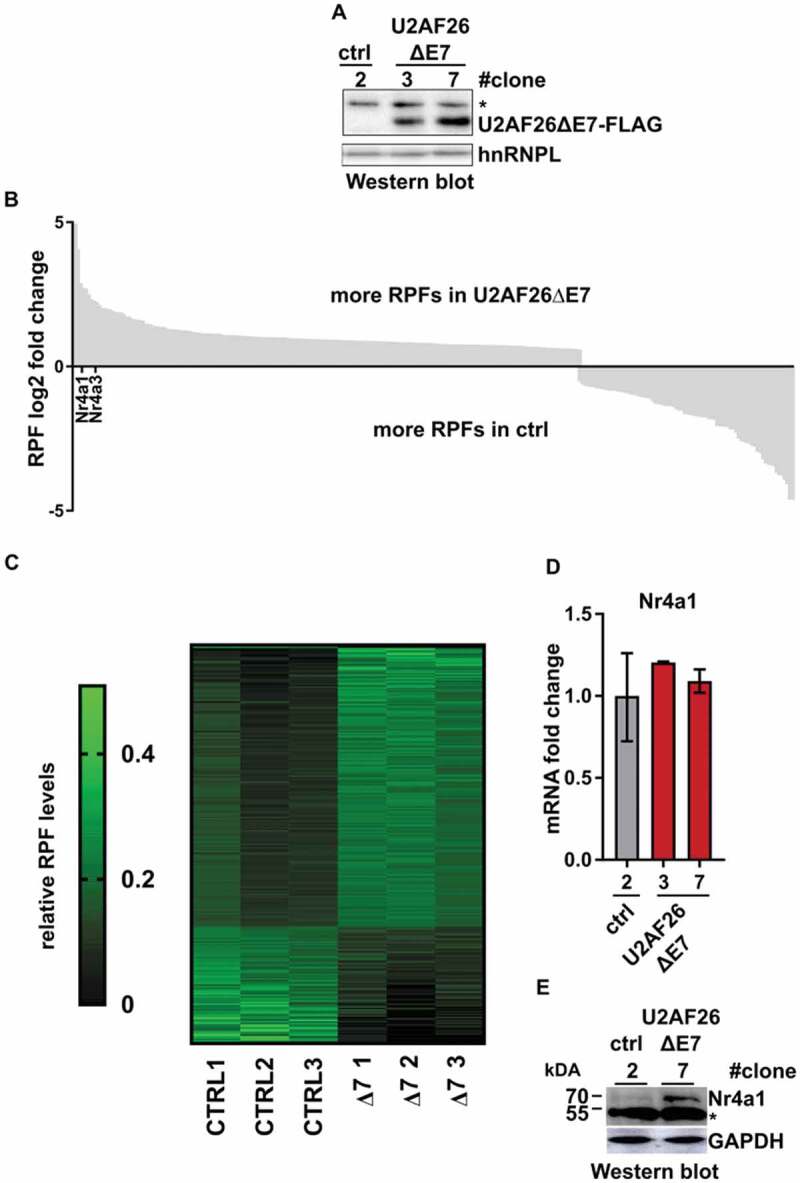
U2AF26ΔE7 activates translation in T cells.
U2AF26ΔE7 regulates translation of a reporter when tethered to the 5ʹUTR
To further substantiate a direct role of U2AF26ΔE7 in activating translation, we set up a luciferase reporter assay where we cloned three MS2-stem loops upstream of the firefly-luciferase gene and recruited MS2-tagged U2AF26ΔE7 to the reporter (Fig. 6A). The MS2-protein alone and the MS2-tagged U2AF26ΔE7 are both localized in the cytoplasm (Supplementary Figure S4A), in contrast to MS2-tagged U2AF26 localizing to the nucleus (Supplementary Figure S4B). Binding of MS2-U2AF26ΔE7 to the forward orientated MS2-loops in the 5ʹ UTR of the reporter leads to a substantial and significant increased luciferase expression in HEK293T (Fig. 6A) and HeLa cells (Supplementary Figure S4 C). Consistent, with a specific effect of cytoplasmic U2AF26ΔE7, MS2-U2AF26 barely increased luciferase expression (Supplementary Figure S4 C). A construct with the reverse complementary sequences in the MS2 loops, which cannot be bound by the MS2-protein is unaffected by the expression of MS2-U2AF26ΔE7 (Fig. 6A and Supplementary Figure S4B). Furthermore, reporter constructs with insertions of forward and reverse orientated MS2-loops in the 3ʹ UTR are unaffected by the expression of MS2-U2AF26ΔE7 (Supplementary Figure S4D), indicating a strongly site-specific effect of U2AF26ΔE7. The increase in luciferase expression is mediated by binding of U2AF26ΔE7 through the fused MS2-domain as we do not detect a change in luciferase expression when we overexpressed a GFP-tagged U2AF26ΔE7 construct (Supplementary Figure S4E). Furthermore, the increased luciferase expression is not due to increased RNA turnover or altered mRNA export, as we observe similar levels of total and cytoplasmic luciferase mRNA in the presence of MS2-U2AF26ΔE7 compared to MS2 alone (Fig. 6B). As an additional read out we used western blot analysis that shows a strong increase in Luciferase protein expression for the forward orientated MS2-loops when MS2-U2AF26ΔE7 is present (Fig. 6C). As mRNA abundance and localization are not affected by the expression of MS2-tagged U2AF26ΔE7, increased expression of the reporter is most likely mediated by stimulation of translation, thus linking U2AF26ΔE7 to translational control. To identify the domain within U2AF26ΔE7 that is involved in the control of translation, we cloned MS2-tagged deletions of U2AF26ΔE7 and analysed their activity in the luciferase reporter assay (Fig. 6D). Deletion of ZnF1 reduces the reporter activity and additional deletion of the RNP2 fully abolishes the activation of the reporter gene (Fig. 6E). Expression of the MS2-ZnF1 alone partially activates the reporter which is even stronger when expressing MS2-RNP2 alone or in combination with ZnF1 (Fig. 6E). These results are confirmed by western blot analysis where a strong increase in luciferase expression is only observed in constructs including the ZnF1 and RNP2 domains (Supplementary Figure S4 F). For the reporter with the reverse MS2-loop sequence no such regulation is detected, confirming specificity of the assay (Fig. 6E and Supplementary Figure S4F). These results indicate that binding of U2AF26ΔE7 to the 5ʹ UTR of target mRNAs increases translation efficiency of the bound mRNA through the N-terminal ZnF1 and RNP2 domains of U2AF26ΔE7, thus providing evidence for a cytoplasmic function of U2AF26. Given the high sequence similarity of U2AF26 and U2AF35 in the N-terminal region, including ZnF1 and RNP2, we were interested in investigating a similar function of U2AF35. MS2-U2AF35, wt or with cancer-associated point mutants, which are mainly located in the nucleus, has an only modest effect in our reporter assays. However, deleting the C-terminal RS domain which harbours the NLS of U2AF35 [13], leads to a strong increase in reporter gene expression (Supplementary Figure S4G), providing evidence that both, U2AF26 and U2AF35, are able to control translation when localized in the cytoplasm.
Figure 6.
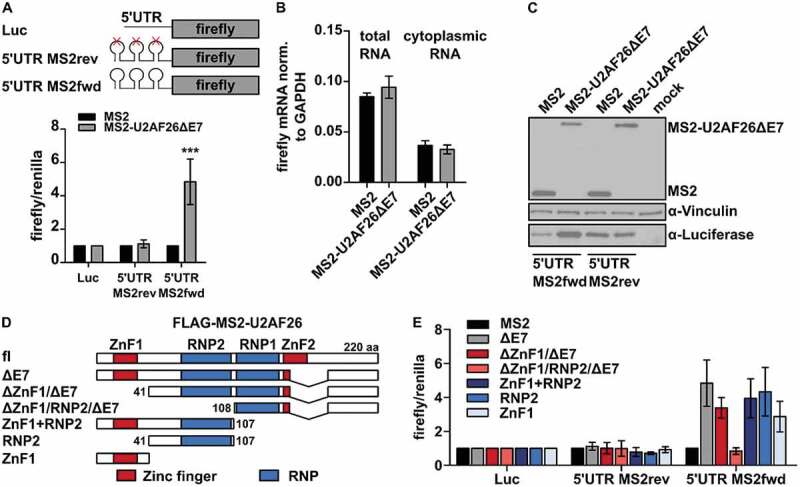
Cytoplasmic MS2-U2AF26ΔE7 regulates translation in a MS2 tethering assay.
Discussion
In our work, we address the impact of both ZnF domains of U2AF26 and U2AF35 on their key functionalities in mammalian cells. Our data show that ZnF2 plays a crucial role in stabilizing U2AF26 and U2AF35, probably by allowing formation of a more stable protein structure, which in the absence of the zinc finger requires interaction with U2AF65 to form. Furthermore, we suggest ZnF2 to properly position α6, which acts as an additional interaction surface with U2AF65. Previous structural work with the yeast homolog U2AF23 suggested the presence of this interaction surface and showed reduced expression of recombinant U2AF23 lacking the very C-terminal helix directly downstream of the second ZnF, whereas deletion of the first ZnF had no effect on protein expression and stability [8]. In the present work, we have observed similar results for mouse U2AF35 in living cells. It was also shown that binding of the AG dinucleotide at the 3`ss is accomplished through both ZnFs and not through the UHM. In line with this, ZnF-deleted U2AF35 were unable to rescue splicing of U2AF35-dependent alternative exons in cells. With our study we thus confirm structure- and in vitro-based conclusions from yeast U2AF23 in cell culture for mouse U2AF26 and U2AF35.
We also note that U2AF26 can functionally substitute U2AF35 in alternative splicing regulation. This confirms in vitro data showing that recombinant U2AF26 interacts with U2AF65 and that U2AF26 can substitute U2AF35 in in vitro splicing assays [16]. Based on SELEX experiments a previous study suggested that U2AF26 and U2AF35 possess different RNA-binding specificities that differ at the +1 position located directly downstream of the AG-dinucleotide [31]. However, in our knockdown and complementation assay U2AF26 rescues splicing of all eight U2AF35-dependent exons independent of the base at the +1 position (Guanine for ASUN, THYN1, EIF4A2; Cytosine for CHEK2, TPD52L2; Adenine for CEP164, RNF10; Thymine for RIPK2). Our results thus indicate that U2AF26 and U3AF35s bind various 3ʹ splice sites with similar affinity, although rescue efficiencies were slightly different between the two proteins, which could indicate subtle binding preferences (e.g., RIPK2 in Fig. 3C).
Interestingly, we have validated our results with an alternative splice variant of U2AF26 (U2AF26ΔE7) that lacks the largest part of the second zinc finger and α6 and is endogenously expressed in different mouse cell lines and tissues, especially after T cell activation [18,19,32]. We further focused our study on the function of this cytoplasmic U2AF26 variant. There are several examples of splicing factors that localize to the cytoplasm upon alternative splicing and have functions in cytoplasmic mRNA processing. The alternative splicing regulator muscleblind-like 1 (MBNL1) plays a crucial role in the development of skeletal and heart muscles and is itself alternatively spliced during development [33,34]. MBNL1 lacking exon 5 localizes to both the nucleus and the cytoplasm in contrast to nuclear MBNL1 full-length protein [33,34]. So far the function of cytoplasmic MBNL1ΔE5 is unknown. However, its paralog MBNL3 that lacks the region encoded by exon 5 is also localized into the nucleus and the cytoplasm and cytoplasmic MBNL3 was shown to bind 3ʹ UTRs of genes involved in cell growth and proliferation [35]. HuR is another RBP with altered functionality depending on nuclear or cytoplasmic localization. Here, an autoregulatory feedback mechanism is dependent on intracellular distribution and tightly controls HuR protein levels, with a misregulation contributing to malignant transformation [36]. Another example is the RNA-binding protein Quaking (Qk) in which alternative splicing of the C-terminus localizes the Qk6 and Qk7 splice variants in both the cytoplasm and the nucleus whereas the Qk5 splice variant is localized into the nucleus [37]. The authors show that the cytoplasmic Qk6 promotes its own translation and represses Qk5 translation through 3ʹUTR binding, although mechanistic details are still missing. In our study, ribosome profiling and MS2 tethering assays suggest that U2AF26ΔE7 controls translation when bound to the 5ʹ UTR of a model mRNA. The strong positional effect, U2AF26ΔE7 has no effect in our reporter assays when tethered to the 3ʹUTR, may indicate a role in translation initiation, for example, through contributing to the recruitment of the small ribosomal subunit or through altering RNA secondary structures in the 5ʹUTR. In addition, cytoplasmic U2AF26ΔE7 alters the abundance of many mRNAs, which could indicate a role in controlling mRNA stability. As U2AF26ΔE7 lacks the largest part of the ZnF2, RNA binding might be accomplished by the ZnF1 alone. RNA binding of a single ZnF was shown for one ZnF domain of the tristetraprolin protein [38], which, interestingly, has two CCCH-type ZnFs similar to U2AF26. However, binding U2AF26ΔE7 to cytoplasmic target mRNAs might also be mediated indirectly through recruitment by other RNA-binding proteins. In this case, sequence specificity would be mediated by other RBPs and U2AF26ΔE7 would be recruited through protein–protein interactions to then contribute to translational regulation.
The best described RBP family that fulfils nuclear as well as cytoplasmic functions is the SR-protein family. SR-proteins contain one or two RNA recognition motives and a C-terminal Arginine/Serine-rich domain which functions as a protein interaction platform and is important for shuttling between cytoplasm and nucleus. Cytoplasmic SR-proteins where shown to regulate mRNA stability and translation (reviewed in [39]). For example, SRSF1 was shown to bind to the 3ʹUTR of protein-kinase-C-interacting protein mRNA and to induce its degradation [40]. Furthermore, an association of SRSF1 and SRSF7 with monosomes or polysomes was shown that appeared to be regulated by phosphorylation [41]. In SRSF1 the RRM2 seems to play an important role in controlling translation, probably through protein–protein interactions mediated by this domain [41]. We obtained similar results for U2AF26ΔE7, as deletion of RNP2 and ZnF1 leads to the loss of translational activation (Fig. 5E). Interestingly, both domains alone alter translation, suggesting that this regulation is controlled through protein–protein interactions that are mediated at least in part independently by ZnF1 and RNP2.
Interestingly, a recent study provides further evidence for a role of U2AF35 in controlling translation [28]. In this work, a substantial fraction of U2AF35 was localized to the cytoplasm, where it was found to bind mRNAs with a 5ʹUTR containing an oligopyrimidine stretch followed by an AG. Polysome-profiling suggested an involvement of U2AF35 in regulating translation by an unknown mechanism and the authors followed this further for a group of genes whose translation is decreased upon U2AF35 binding. Binding of these transcripts was reduced by the S34F mutation, leading to translation-derepression and suggesting a contribution of U2AF35-controlled translation to malignant transformation [28]. Our work complements this study, by showing that a cytoplasmic variant of U2AF26 is also able to increase translation; the precise binding position in the 5ʹUTR of target mRNAs as well as available cofactors and/or posttranslational modifications are variables that likely determine the direction of translational regulation. Of note, and in agreement with [28], we also find some mRNAs that show reduced ribosome association upon U2AF26ΔE7 expression, which may indicate translational repression. As discussed above, U2AF26ΔE7 lacks the second ZnF and will therefore have an altered mode of RNA recruitment, which may explain a different set of targets and no overlap with the mRNAs regulated by full-length U2AF35. Along similar lines, we did not observe an effect of cancer-associated point mutations in our MS2 tethering assay (Supplementary Figure S4F), which is consistent with the translational derepression depending on reduced mRNA binding of the S34F mutant [28].
As expression of U2AF26ΔE7 is strongly increased during activation of primary mouse T cells [42,43], we used a mouse T cell line stably overexpressing U2AF26ΔE7 to address functional consequences. We identified Nr4a1 expression to be upregulated in U2AF26ΔE7 expressing cells, likely through increased translation. Nr4a1 belongs to the family of nuclear receptors that acts as transcription factors. Its mRNA is upregulated within hours after T cell activation and the protein mediates a metabolic switch that is essential to control activated T cells and prevent hyperactivation [44]. Further increasing Nr4a1 expression at the translational level through the formation of U2AF26ΔE7 may be part of a feedback mechanism to restrict T cell activation and prevent autoimmunity. In addition, Nr4a1 has been characterized as an essential factor controlling T cell tolerance during chronic infection or in the tumour microenvironment with implications for tumour immunotherapy [45,46]. Of note, translational upregulation of transcription factors such as Nr4a1, Nr4a2 or Nr1d1 may account for some of the differences in gene expression we observed in the U2AF26ΔE7 cell line; e.g. Gata3 was identified as a direct target of Nr4a1 [45,46]. Interestingly, we have previously shown that U2AF26fl promotes formation of a less active CD45 protein isoform through alternative splicing regulation to restrict T cell activation [17]. Together with the current manuscript, these data point to the control of T cell activation as a common functionality of U2AF26. This is achieved at different levels and mechanisms, translation and alternative splicing, and is potentially even more wide spread, when considering cytoplasmic regulation of mRNA abundance as an additional regulatory layer. With respect to the current work, it will be especially interesting to functionally connect U2AF26ΔE7 expression in activated T cells with Nr4a1 and a role in inducing T cell tolerance.
Taken together, our data show that the ZnF domains control key functionalities of U2AF35 and U2AF26 and, consistent with recent work [28] connect this core splicing factor to the cytoplasmic regulation of gene expression. The identification of RRM2/ZnF1 interacting proteins will be important to unravel the mechanism by which cytoplasmic U2AF26ΔE7 or U2AF35 regulate translation when bound to the 5ʹUTR of target mRNAs. This will further help to elucidate the link between this cytoplasmic function of U2AF26/35 in physiological processes such as T cell activation and pathophysiological conditions such as cancer.
Materials and methods
Cell culture, transfection and treatments
HEK293 T and HeLa cells were cultured in DMEM High glucose (Biowest) containing 10% FCS (Biochrom) and 1% Penicillin/Streptomycin (Biowest). EL4 cells were cultivated in RPMI medium (Biowest) with the same additives. Transfections of plasmid and siRNA were performed using RotiFect (Carl Roth) following manufacturer’s instructions. For protein stability assays, 1.5 × 105 HEK293T cells in a 12-well plate were transfected with 0.4 µg expression vectors for Flag-tagged U2AF35 and U2AF26 in the presence or absence of 0.4 µg HA-U2AF65. Twenty-four hours after transfection, cells were treated with 30 ng/mL Cycloheximide (Sigma) for 4, 8 or 24 h and lysed as described below. For rescue experiments, knockdown of U2AF35 was performed by transfecting 0.5 × 105 HEK293 T cells in a 12-well plate with 20 pmol siRNAs (siU2AF35: GAAAGUGUUGUAGUUGAUUGA; siCTRL: UUCUCCGAACGUGUCACGU). After 24 h, knockdown was rescued by transfection of 0.8 µg expression vectors for Flag-tagged U2AF35 and U2AF26 expression constructs for an additional 48 h. For mRNA stability assays, cells were treated with 5 µg/mL Actinomycin D (Sigma).
Constructs
U2AF35-Flag, U2AF35-Q157R-Flag, U2AF35-Q157P-Flag, U2AF26-Flag, U2AF26ΔE7-Flag and U2AF65-HA protein expression constructs have been described previously [18,27]; these plasmids served as template for insertions of deletions and point mutations using standard cloning procedures (Supplementary Figure S1A). For MS2 tethering assays U2AF26 deletions were cloned into pEF-MS2 plasmid, encoding an N-terminal MS2 tag. To introduce the MS2 binding loop, we designed oligonucleotides encoding the MS2 binding loop and overhangs for HindIII (5ʹUTR) or XbaI (3ʹUTR), annealed the oligonucleotides (5ʹUTR: fwd: agcttgcgatcaccatcagggatcgca, rev: agcttgcgatccctgatggtgatcgca; 3ʹUTR: fwd: ctagagcgatcaccatcagggatcgct, rev: ctagagcgatccctgatggtgatcgct) and cloned them into pGL3 luciferase (Promega) using either HindIII (5ʹUTR) or XbaI (3ʹUTR). We obtained several constructs with different amounts and orientations of the MS2 binding loop. For this study, we used constructs containing either three loops in forward or in reverse orientation for the 5ʹUTR, and one loop in forward or reverse orientation for the 3ʹUTR. All constructs were confirmed by sequencing. Primer sequences are provided in the Supplementary table 1.
RNA, RT-PCR and RT-qPCR
RNA extraction, radioactive RT-PCR, Phosphor imager quantification and RT-qPCR were done as previously described [47]. Results of endogenous alternative splicing represent the mean value of at least three independent experiments with the according to standard deviation. Significance, if not differently labelled, was calculated by Student’s unpaired t-Test: *p < 0.05, **p < 0.01, ***p < 0.001. Primer sequences are provided in the Supplementary table 1.
Immunoblotting, immunoprecipitation (IP) and antibodies
Cells were lysed with a buffer containing 60 mM Tris pH 7.5, 30 mM NaCl, 1 mM EDTA, 1% TritionX-100 and protease inhibitors. SDS-page and immunoblotting were performed according to standard protocols. For isolation of nuclear and cytoplasmic proteins, cells were incubated in low salt buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl and protease inhibitors) for cytoplasmic proteins followed by incubation in high salt buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% glycerol and protease inhibitors) to isolate nuclear proteins. For IPs, Flag- and HA-tagged proteins were overexpressed in HEK293T cells for 48 h. One hundred-microgram lysate was pre-incubated with protein A/G-plus agarose (Santa Cruz) in 400 µL RIPA buffer containing 400 mM or 1200 mM NaCl, 2% BSA and protease inhibitors in the presence or absence of 40 µg RNase A (Roth) and 100 units RNase T1 (Thermo Scientific). Afterwards, IP was performed with the pre-cleaned lysates using anti-Flag® M2 affinity gel (Sigma). After protein binding, beads were washed four times with RIPA buffer (10 mM Tris HCl pH 8, 400 mM/1200 mM NaCl, 2 mM EDTA, 1% NP-40, 5 mg/mL Sodium deoxycholate) and then boiled in 2x SDS loading dye. Protein expression and IPs were analysed by SDS-page, blotting to a PVDF membrane and detection with the following antibodies: α-Flag (Cell Signalling, 2368), α-HA (Santa Cruz, sc-7392), α-hnRNPL (Santa Cruz, sc-32317), α-GAPDH (Epitope Biotech Inc., L001), α-Firefly Luciferase HRP (GeneTex, GTX20635), α-Nr4a1 (Invitrogen, 12.14), α-Vinculin (Santa Cruz, sc-5573).
Secondary structure prediction
Secondary structure prediction was performed with the PSIPRED online tool [48].
Luciferase assays
For luciferase assays 1.5 × 105 HEK293T or 1.0 × 105 HeLa cells were seeded into a 12-well plate using DMEM medium containing 10% FBS. Cells were transfected with Lipofectamin 2000 according to the manufacturer’s instructions, using 0.6 µg of the different pEF-MS2 constructs, 0.2 µg of the different pGL3 luciferase reporter constructs and 0.01 µg of a Renilla control vector (Promega). As a control, the pEF-MS2 constructs were replaced by GFP or an C-terminal GFP-tagged variant of U2AF26ΔE7 [18]. Forty-eight hours after transfection cells were harvested using the passive lysis buffer from p.j.k. according to the manufacturer’s instructions. Luciferase activity (beetle juice, p.j.k.) and renilla activity (renilla juice, p.j.k) were measured in technical duplicates by an Orion L microplate luminometer (Berthold Detection Systems). Average luciferase expression was normalized to averaged renilla expression. Data were normalized to the pGL3 control luciferase and pEF-MS2 expressing cells. Depicted data represent the average and standard deviation of several independent transfections.
Primary T cell preparation
Spleens of C57Bl6 mice where squeezed through a 70 µm cell strainer into complete medium (RPMI, 10% FCS, 1% Penicillin/Streptomycin, 1x NEAA, 0,1 mM ß-Mercaptoethanol, 1 mM Pyruvat, 2 mM Glutamine) to get a single cell suspension. Cells were washed with PBS++ (1x PBS, 2% FCS, 2 mM EDTA) followed by incubation in 1 mL red blood cell lysis buffer (Sigma) for 5 min at room temperature. After washing with 5 mL 1x PBS, 2% FCS, 2 mM EDTA, cells were counted and 5 × 107 cells were used to purify T cells with the Dynabeads™ Untouched™ Mouse T Cells Kit (Thermo Fischer) following the manufacturer’s protocol. For T cell stimulation, 12-well plates were coated with 1 µg mouse α-CD3 antibody (BD Pharmingen) in 500 µL 1xPBS for 2 h at 37°C followed by washing with 1 mL 1xPBS. 1 × 106 purified T cells were seeded in 1 mL complete medium in the coated 12-well plates and were additionally treated with either 20 ng/µL PMA or 1 µg mouse α-CD28 antibody (BD Pharmingen) or were treated with 1 µL DMSO as control. Forty-eight hours after the stimulation total RNA was isolated.
Stable EL4 cells and ribosome profiling
For generation of stable EL4 cells, 5 µg of linearized pCMV-U2AF26ΔE7-Flag plasmid was transfected into 2 × 106 EL4 cells by electroporation with the Nucleofector II (Amaxa Biosytems) electroporation system following the manufacturer’s protocol. Forty-eight hours after transfection, clones were selected with 4 mg/ml G418 (Sigma) in RPMI and clones were expanded. For ribosome profiling, harvested cells were lysed for 15 min on ice in a buffer containing 20 mM Tris pH7.4, 150 mM NaCl, 5 mM MgCl2, 1% Triton-X, 1 mM DTT, 0.1 mg/ml cycloheximide, RNasin and 1x cOmplete protease inhibitor cocktail (Roche). The cell lysate was cleared by centrifugation and digested with 0.25 U RNaseI (Ambion) per 1 µg RNA for 15 min at 4°C. To stop the reaction, 1 U SUPERaseIn RNase Inhibitor (Invitrogen) was added per unit of RNaseI. Ribosomal complexes were separated on 12 ml 10-50% sucrose gradients (in a buffer containing 20 mM Tris pH7.4, 150 mM NaCl, 5 mM MgCl2, 0.1 mg/ml cycloheximide, and 1 mM DTT) by centrifugation at 4°C and 35,000 rpm for 3 h in a SW41 rotor. Fractions were collected at 0.5 ml/min with continuous monitoring of conductivity and UV absorption at 254 nm. Fractions containing 80 S monosomes were diluted with an equal amount of RNase-free water followed by organic extraction with Phenol:Chloroform:Isoamylalkohol 25:24:1 (Roth) and ethanol precipitation. RNAs were then separated by denaturing Urea-PAGE and visualized by SYBR Gold staining. RNAs with a length of 27–33 nt were excised from the gel and eluted overnight in a buffer containing 300 mM NaOAc pH 5.5, 1 mM EDTA, and 0.25% SDS followed by ethanol precipitation. After dephosphorylation for 1 h at 37°C with 20 U of T4 Polynucleotide Kinase (NEB), RNAs were ligated to a universal miRNA cloning linker (NEB) using truncated T4 RNA ligase 2. After gel purification reverse transcription was performed using SuperscriptIII reverse transcriptase and a primer complementary to the ligated adapter, followed by alkaline hydrolysis of the RNA strand. The resulting cDNAs were gel purified and contaminating sequences originating from rRNA were depleted by substractive hybridization to a mixture of biotinylated DNA oligonucleotides and Streptavidin magnetic beads (NEB). The recovered RNAs were then circularized with CircLigase II ssDNA Ligase (Epicentre) according to the manufacturer’s instructions and subjected to PCR amplification to generate amplicons suitable for Illumina sequencing. After gel purification, DNA concentration was determined using a Qubit Fluorometer (Qubit 2.0, Thermo Fisher Scientific). Bioanalyzer or TapeStation (Agilent) analyses were performed to assess the quality of the samples followed by deep sequencing on an Illumina HiSeq platform essentially as described [27]. We have sequenced three independently generated samples from clone #2 (ctrl) and #7 U2AF26ΔE7 (see Fig. 5A). Sequencing reads from both input and riboseq were aligned to the mouse reference genome (mm10) using STAR (2.7.3a) [49] with standard settings for the input data. For the riboseq data outFilterScoreMinOverLread and outFilterMatchNminOverLread were both set to 0.3 to allow mapping of short fragments. RNASeQC [50] was used for calculating gene-wise TPM values for each sequenced replicate using the aligned reads. GENCODE release M21 (GRCm38.p6) was supplied as annotation for gene counting. Read counts were transformed to a count matrix using standard python code. Triplicate RPFs were normalized to the average of the respective input samples. To identify genes with altered translation efficiency in response to U2AF26ΔE7 overexpression we filtered for genes with adjusted p-values ≤ 0.05 and a fold change above 1.5. To increase confidence, we additionally filtered out genes with a mean expression level < 0.5 in input samples and with a fold change in input samples >1.5. For the heat map, the sum of the six RPF samples is set to one for each gene.
Supplementary Material
Acknowledgements
We would like to thank members of the Heyd lab for discussion and comments on the manuscript.
Funding Statement
This work was supported by the German Research Foundation (DFG) [SFB960/2, B11 to J.M. and an Emmy-Noether-Fellowship He5398/3 and 278001972 - TRR 186, A15 to F.H.] and the German Federal Ministry of Education and Research [BMBF, 01ZX1401D to J.M.]. MP is funded by a post-doc stipend of the Peter and Traudl Engelhorn Foundation.
Author contributions
O.H. and M.P. performed most experiments. S.R and J.M. prepared samples and libraries for Ribo-Seq. RNA-Seq was performed by B.T., D.O. and M.P. performed bioinformatics analysis. F.H., O.H. and M.P. designed the study, planned the experiments and analyzed the data. F.H., O.H. and M.P. wrote the manuscript. F.H. supervised the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Wahl MC, Will CL, Luhrmann R.. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. [DOI] [PubMed] [Google Scholar]
- [2].Zamore PD, Patton JG, Green MR. Cloning and domain structure of the mammalian splicing factor U2AF. Nature. 1992;355:609–614. [DOI] [PubMed] [Google Scholar]
- [3].Zorio DA, Blumenthal T. Both subunits of U2AF recognize the 3ʹ splice site in Caenorhabditis elegans. Nature. 1999;402:835–838. [DOI] [PubMed] [Google Scholar]
- [4].Merendino L, Guth S, Bilbao D, et al. Inhibition of msl-2 splicing by sex-lethal reveals interaction between U2AF35 and the 3ʹ splice site AG. Nature. 1999;402:838–841. [DOI] [PubMed] [Google Scholar]
- [5].Wu S, Romfo CM, Nilsen TW, et al. Functional recognition of the 3ʹ splice site AG by the splicing factor U2AF35. Nature. 1999;402:832–835. [DOI] [PubMed] [Google Scholar]
- [6].Mackereth CD, Madl T, Bonnal S, et al. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature. 2011;475:408–411. [DOI] [PubMed] [Google Scholar]
- [7].Kielkopf CL, Rodionova NA, Green MR, et al. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. [DOI] [PubMed] [Google Scholar]
- [8].Yoshida H, Park S-Y, Oda T, et al. A novel 3ʹ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev. 2015;29:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kralovicova J, Vorechovsky I. Alternative splicing of U2AF1 reveals a shared repression mechanism for duplicated exons. Nucleic Acids Res. 2017;45:417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang M, Zamore PD, Carmo-Fonseca M, et al. Cloning and intracellular localization of the U2 small nuclear ribonucleoprotein auxiliary factor small subunit. Proc Natl Acad Sci U S A. 1992;89:8769–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Webb CJ, Wise JA. The splicing factor U2AF small subunit is functionally conserved between fission yeast and humans. Mol Cell Biol. 2004;24:4229–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu T, Fu XD. Genomic functions of U2AF in constitutive and regulated splicing. RNA Biol. 2015;12:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gama-Carvalho M, Carvalho MP, Kehlenbach A, et al. Nucleocytoplasmic shuttling of heterodimeric splicing factor U2AF. J Biol Chem. 2001;276:13104–13112. [DOI] [PubMed] [Google Scholar]
- [14].Kielkopf CL, Lucke S, Green MR. U2AF homology motifs: protein recognition in the RRM world. Genes Dev. 2004;18:1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mollet I, Barbosa-Morais NL, Andrade J, et al. Diversity of human U2AF splicing factors. Febs J. 2006;273:4807–4816. [DOI] [PubMed] [Google Scholar]
- [16].Shepard J, Reick M, Olson S, et al. Characterization of U2AF26, a splicing factor related to U2AF35. Mol Cell Biol. 2002;22:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heyd F, Ten Dam G, Moroy T. Auxiliary splice factor U2AF26 and transcription factor Gfi1 cooperate directly in regulating CD45 alternative splicing. Nat Immunol. 2006;7:859–867. [DOI] [PubMed] [Google Scholar]
- [18].Heyd F, Carmo-Fonseca M, Moroy T. Differential isoform expression and interaction with the P32 regulatory protein controls the subcellular localization of the splicing factor U2AF26. J Biol Chem. 2008;283:19636–19645. [DOI] [PubMed] [Google Scholar]
- [19].Preussner M, Wilhelmi I, Schultz A-S, et al. Rhythmic U2af26 alternative splicing controls PERIOD1 stability and the circadian clock in mice. Mol Cell. 2014;54:651–662. [DOI] [PubMed] [Google Scholar]
- [20].Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. [DOI] [PubMed] [Google Scholar]
- [21].Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Przychodzen B, Jerez A, Guinta K, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ilagan JO, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Okeyo-Owuor T, White BS, Chatrikhi R, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shirai CL, Ley J, White B, et al. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell. 2015;27:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Herdt O, Neumann A, Timmermann B, et al. The cancer-associated U2AF35 470A>G (Q157R) mutation creates an in-frame alternative 5ʹ splice site that impacts splicing regulation in Q157R patients. RNA. 2017;23:1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Palangat M, Anastasakis DG, Fei DL, et al. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 2019;33:482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kellenberger E, Stier G, Sattler M. Induced folding of the U2AF35 RRM upon binding to U2AF65. FEBS Lett. 2002;528:171–176. [DOI] [PubMed] [Google Scholar]
- [30].Shao C, Yang B, Wu T, et al. Mechanisms for U2AF to define 3ʹ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol. 2014;21:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shepard JB. Characterization of U2AF26, a paralog of the splicing factor U2AF35 [Dissertation]. Univ. Texas Southwest. Med. Cent; 2004. [Google Scholar]
- [32].Preussner M, Goldammer G, Neumann A, et al. Body temperature cycles control rhythmic alternative splicing in mammals. Mol Cell. 2017;67:433–446.e434. [DOI] [PubMed] [Google Scholar]
- [33].Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. [DOI] [PubMed] [Google Scholar]
- [34].Terenzi F, Ladd AN. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol. 2010;7:43–55. [DOI] [PubMed] [Google Scholar]
- [35].Poulos MG, Batra R, Li M, et al. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet. 2013;22:3547–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kotta-Loizou I, Giaginis C, Theocharis S. Clinical significance of HuR expression in human malignancy. Med Oncol. 2014;31:161. [DOI] [PubMed] [Google Scholar]
- [37].Fagg WS, Liu N, Fair JH, et al. Autogenous cross-regulation of Quaking mRNA processing and translation balances Quaking functions in splicing and translation. Genes Dev. 2017;31:1894–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Michel SL, Guerrerio AL, Berg JM. Selective RNA binding by a single CCCH zinc-binding domain from Nup475 (Tristetraprolin). Biochemistry. 2003;42:4626–4630. [DOI] [PubMed] [Google Scholar]
- [39].Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: more than splicing factors. Febs J. 2011;278:3246–3255. [DOI] [PubMed] [Google Scholar]
- [40].Lemaire R, Prasad J, Kashima T, et al. Stability of a PKCI-1-related mRNA is controlled by the splicing factor ASF/SF2: a novel function for SR proteins. Genes Dev. 2002;16:594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sanford JR, Ellis JD, Cazalla D, et al. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc Natl Acad Sci U S A. 2005;102:15042–15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martinez NM, Pan Q, Cole BS, et al. Alternative splicing networks regulated by signaling in human T cells. RNA. 2012;18:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schultz AS, Preussner M, Bunse M, et al. Activation-dependent TRAF3 exon 8 alternative splicing is controlled by CELF2 and hnRNP C binding to an upstream intronic element. Mol Cell Biol. 2017;37. DOI: 10.1128/mcb.00488-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liebmann M, Hucke S, Koch K, et al. Nur77 serves as a molecular brake of the metabolic switch during T cell activation to restrict autoimmunity. Proc Natl Acad Sci U S A. 2018;115:E8017–e8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu X, Wang Y, Lu H, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019. DOI: 10.1038/s41586-019-0979-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen J, López-Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019. DOI: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wilhelmi I, Kanski R, Neumann A, et al. Sec16 alternative splicing dynamically controls COPII transport efficiency. Nat Commun. 2016;7:12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Buchan DW, Minneci F, Nugent TC, et al. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41:W349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. DOI:10.1093/bioinformatics/bts635%J Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].DeLuca DS, Levin JZ, Sivachenko A, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.