

ABSTRACT
Extracellular vesicles (EVs) are cell-derived nanoparticles that act as natural carriers of nucleic acids between cells. They offer advantages as delivery vehicles for therapeutic nucleic acids such as small RNAs. Loading of desired nucleic acids into EVs can be achieved by electroporation or transfection once purified. An attractive alternative is to transfect cells with the desired small RNAs and harness the cellular machinery for RNA sorting into the EVs. This possibility has been less explored because cells are believed to secrete only specific RNAs. However, we hypothesized that, even in the presence of selective secretion, concentration-driven RNA sorting to EVs would still be feasible. To show this, we transfected cells with glycine 5ʹ tRNA halves, which we have previously shown to better resist RNases. We then measured their levels in EVs and in recipient cells and found that, in contrast to unstable RNAs of random sequence, these tRNA halves were present in vesicles and in recipient cells in amounts proportional to the concentration of RNA used for transfection. Similar efficiencies were obtained with other stable oligonucleotides of random sequence. Our results demonstrate that RNA stability is a key factor needed to maintain high intracellular concentrations, a prerequisite for efficient non-selective RNA sorting to EVs and delivery to cells. Given that glycine 5ʹ tRNA halves belong to the group of stress-induced tRNA fragments frequently detected in extracellular space and biofluids, we propose that upregulation of extracellular tRNA fragments is consequential to cellular stress and might be involved in intercellular signalling.
KEYWORDS: tiRNAs, vesicles, exosomes, secretion, intercellular communication
Introduction
Synthetic oligonucleotides have emerged as promising novel therapeutics. Their potential has been reinforced by the recent approval of oligonucleotide-based therapies for several diseases such as spinal muscle atrophy and hyperlipidemias [1]. Single-stranded DNA oligonucleotides can bind specific mRNAs by anti-sense base pairing interactions, mediating RNase H-dependent cleavage, or modulating their translation or splicing [2]. Double-stranded small RNAs (siRNAs) can also induce cleavage of specific transcripts by exploiting the RNA interference pathway. Indeed, the first siRNA-based therapy intended for human use was approved by the FDA in 2018. Ongoing clinical trials are evaluating short single-stranded RNAs as microRNA mimics to achieve translational repression or target degradation [1] and locked nucleic acids are being studied as anti-sense microRNA inhibitors [3]. Together, oligonucleotide therapies may become an innovative technique for silencing or correcting the expression of virtually any gene, many of them previously thought as ‘undruggable’ by conventional pharmacological strategies that work at the protein level [4].
A key aspect and major limiting factor in oligonucleotide therapy is the delivery of the oligonucleotide into the internal cellular compartments where they can exert their functions. Nucleic acids are hydrophilic and have a limited ability to spontaneously penetrate cells [1]. However, some sequences can adopt cell-penetrating structures [5]. Approaches used in experimental or approved drugs include oligonucleotide conjugation to achieve tissue-specific receptor-mediated uptake or encapsulation in lipidic nanoparticles. Extracellular vesicles (EVs) are attracting great attention as delivery systems as they are natural carriers of regulatory RNAs between cells [6–9]. They also have several advantages over other delivery vectors, for example, biocompatibility, protection against RNase degradation, decreased immunogenicity (when derived from proper cell sources) and their capacity to cross the blood-brain barrier [10]. EVs also contain a set of surface proteins that can confer selectivity towards specific tissues or cell types [11], a property that can be exploited for targeted delivery. Compared to liposomes, EVs contain surface proteins that inhibit clearance by phagocytes and enhance their retention in circulation [12]. Moreover, EVs can be produced under clinical-grade good manufacturing practices and in large quantities [13].
Strategies to enrich EVs with therapeutic nucleic acids can be divided into two main categories. Vesicles can be loaded with the nucleic acids after their purification from parental cells [12,14], or the parental cells can be transfected or electroporated with the oligonucleotides before collecting the EVs [15]. However, the feasibility of the latter method has been questioned given that some reports suggest that cells have sorting mechanisms that are selective when incorporating small RNAs into EVs [16,17]. Were this true, it is possible that RNAs that differ in their nucleotide arrangements would be sorted differently into the EVs and in turn differentially or selectively secreted.
A selective RNA sorting mechanism is supported by the description of different RNA-binding proteins responsible for loading specific RNAs inside the vesicles [18–20]. Also, microRNAs harbour different non-template nucleotide additions in cells and vesicles [21], suggesting that certain sequence motifs might label RNAs for their secretion. In addition, there are reports describing microRNAs expressed at very low levels, but highly enriched in the EVs [22]. Specific EV microRNA signatures have been described by many groups, with cells and EVs from different sources tending to cluster together when profiled by their microRNA content [23].
The assumption that only specific RNAs are selected and secreted in extracellular vesicles was challenged by our observation that extracellular and intracellular microRNA profiles are highly correlated in MCF-7 cells grown under serum-free conditions [24]. Other studies have also failed to find multiple microRNAs enriched in EVs with respect to their parental cells [25]. Discrepancies among studies can be cell type-dependent, but it should be noted that many of the microRNAs usually observed as selectively enriched in human EVs are likely remnants from foetal bovine serum or other additives used in cell culture [26–28]. In contrast, other small RNA classes, such as YRNA- and tRNA-derived fragments, showed patterns consistent with selective export in studies performed by several research groups, including ours [24,25,29]. However, we have recently shown that some of the most extracellularly enriched tRNA-derived fragments can form RNA dimers which confer resistance against RNase degradation [30]. Other tRNA-derived fragments can also fold into highly stable intermolecular tetramers stabilized by G-quadruplex structures [31]. Furthermore, angiogenin (ANG), the nuclease responsible for the generation of some tRNA-derived fragments (namely stress-induced tRNA halves [32]) is abundant in extracellular vesicles [29], together with its full-length tRNA substrates [19]. Thus, it is at least feasible that the lack of correlation between extracellular and intracellular small RNA profiles can be explained by a combination of factors, including contamination, extracellular biogenesis (as already described for microRNAs [33];), and differential extracellular stability rather than selective RNA export.
Studying tRNA-derived fragments by sequencing or RT-qPCR is intrinsically challenging because these small RNAs harbour a variety of modified nucleobases [34] that can greatly interfere with reverse transcription and sequencing [35,36]. Thus, small RNA-seq does not give a representative image of all small RNAs present in a sample. Even in the absence of modified bases, different RNAs can be amplified and sequenced with very different efficiencies, also affecting quantitative estimates of relative abundance [37]. Despite these caveats, the profiles of tRNA-derived fragments that we have obtained inside cells were highly heterogeneous, while the same sequencing techniques applied to the extracellular space showed a decrease in variability and a remarkable enrichment of glycine and glutamic acid 5ʹ tRNA halves [24]. We have also identified these tRNA halves in the extracellular, non-vesicular fraction by Northern blot (Tosar et al. unpublished), while others have done so in serum [38]. Thus, the presence and enrichment of these specific tRNA-derived fragments in the extracellular space, particularly in the non-EV fraction, does not seem to be an artefact of RNA-seq.
The precise mechanisms of small RNA sorting into the extracellular space remain poorly understood, with both selective and non-selective secretion mechanisms being recognized as possible pathways in two recent position papers published by the International Society for Extracellular Vesicles [39,40]. However, it would stand to reason that overexpressing a therapeutic RNA in a cell would tend to enhance the likelihood of incorporation into EVs.
To study nucleic acid secretion and to address whether RNA overexpression in a cell can serve as a method to obtain EVs enriched in the overexpressed RNAs, we transfected human cells with synthetic 5ʹ tRNAGlyGCC halves and then measured their levels in EVs as well as in recipient cells that accepted those EVs. Because these sequences are identical to previously described Angiogenin-generated tRNA-derived stress-induced RNAs (termed tiRNAs [32], we will refer to them as ‘5ʹ tiRNAGly ’. We relied on 5ʹ tiRNAGly for this study because: i) it is stable inside the cell for at least 9 hours after transfection, ii) it is also stable in the extracellular space [30], and iii) we hypothesized it would also be stable in recipient cells which could internalize the EVs. If the RNA is degraded inside EV-producing cells or inside the EVs after their release onto the extracellular space, it would be difficult to distinguish between selective and non-selective secretion, since the RNA would not be detectable extracellularly. Additionally, 5ʹ tiRNAGly are amongst the most abundant small RNA entities in MCF-7 EVs, being amenable to standard small RNA sequencing or ligation-independent stem-loop RT-droplet digital PCR [24]. Moreover, recent data supports a specific role for these sequences in vesicle-mediated intercellular communication pathways [41,42]. Thus, beyond the utilitarian use of 5ʹ tiRNAGly as a degradation-resistant RNA, we proposed to gain insights into a possible connection between cellular stress, RNA release and intercellular communication.
In the present work, we provide experimental evidence indicating that, when cells are transfected in vitro with synthetic stable oligonucleotides, these are loaded into released vesicles in a concentration-dependent manner regardless of their sequence. This process was demonstrated for 5ʹ tiRNAGly, a stable mutant retaining its dimer-forming ability and irrelevant DNA sequences. Under the experimental conditions described herein, synthetic stable oligonucleotides loaded in secreted vesicles can be easily transferred to other cells in culture. Unstable RNAs could still be transferred between cells; however, the efficiency of the process was much lower and their levels in acceptor cells could only be assessed using highly sensitive techniques such as qPCR. In light of these results, we speculate that any small RNA can be loaded into intact EVs, provided that the transfected RNA be relatively stable against RNases or rendered stable by standard chemical modifications. Also, our results point out at 5ʹ tiRNAGly as strong candidates to mediate intercellular communication during acute cell stress.
Methods
Synthetic oligonucleotides
Synthetic RNA oligonucleotides were purchased from Integrated DNA Technologies (USA).
-
tRNAGlyGCC 5ʹ halves (5ʹ tiRNAGly):
5ʹ GCAUUGGUGGUUCAGUGGUAGAAUUCUCGC 3ʹ
-
Mutated tRNAGlyGCC 5ʹ halves (5ʹ tiRNA9GG/AA):
5ʹ GCAUUGGUAAUUCAGUGGUAGAAUUCUCGC 3ʹ
-
Mutated tRNAGlyGCC 5ʹ halves (5ʹ tiRNA25U/C):
5ʹ GCAUUGGUGGUUCAGUGGUAGAAUCCUCGC 3ʹ
-
Scrambled tRNAGlyGCC 5ʹ halves (SCR):
5ʹ GUAUAGGUGUGUCGGUAGUAGUAUCCUCGC 3ʹ
Nucleotides that are in bold are different from the wild-type sequence. A modified version of this sequence (5ʹ tiDNAGly) containing a DNA instead of an RNA backbone (alternatively viewed as an RNA oligonucleotide bearing a 2ʹ-deoxy modification in every base) was also used.
5ʹ biotinylated versions of all oligonucleotides were used for confocal microscopy-based assays. In addition, an irrelevant biotinylated DNA oligonucleotide sequence was used:
DNA_16mer:
5ʹ/Bio/CCCTTTTGCTAAATCC 3ʹ
Primer sequences
SL-RT primer (‘X’ denotes assay-specific 3ʹ overhangs): GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACXXXXXX;
miR-21-5p [23nt] SL-RT 3ʹ overhang: GTCAAC;
tRNAGlyGCC [WT] SL-RT 3ʹ overhang: GCGAGA;
tRNAGlyGCC [SCR] SL-RT 3ʹ overhang: GCGAGG;
miR-21-5p [23nt] forward primer: gccccgTAGCTTATCAGACTGATGT;
tRNAGlyGCC [WT] forward primer: ccGCATTGGTGGTTCAGTGGTA;
tRNAGlyGCC [SCR] forward primer: ggGTATAGGTGTGTCGGTAGTA;
tRNAGlyGCC [9GG/AA] forward primer: gctcgGCATTGGTAATTCAGTGGTA;
Universal reverse primer: GTGCAGGGTCCGAGGT;
Lowercase letters show 5ʹ overhangs included to increase the Tm of the forward primers after the first PCR cycle.
Cell culture
MCF-7 cells were purchased from ATCC and ΔYBX-1 MCF-7 cells were generated using CRISPR/Cas9 technology as described in [43]. Both cell lines were tested in-house for Mycoplasma. Cells were cultured in DMEM medium plus 10% foetal bovine serum (FBS) in a humidified chamber at 37°C with CO2 at 5% and without any antibiotics. The day prior to transfection, cells were washed with PBS, dissociated using a solution of Trypsin-EDTA, and 4 × 104 or 9 × 103 cells were plated in 24 or 96 well plates, respectively. All reagents used for cell culture were purchased from Gibco (Thermo Fisher Scientific) unless otherwise stated.
Transfection with synthetic small RNAs
Synthetic RNAs and Lipofectamine 2000 (L2K, Invitrogen, Thermo Fisher Scientific) were mixed in a desired volume (depending on the area of the culture wells, but typically 250 µL for 24-well plates containing cells grown at 70% confluency) of serum-free Mammary Epithelial Growth Medium (MEGM, Lonza Clonetics, without addition of any antibiotics or bovine pituitary extract included in the kit) for 15 min at room temperature. A fixed ratio of 2 µL L2K per 50 pmol RNA was used in all cases. Cells were washed with PBS and incubated with the L2K/RNA mixture in MEGM for 30 min at 37°C. Then, cells were washed three times with PBS to remove as much non-internalized RNA as possible. Fresh MEGM media was added and cells were incubated for variable amounts of time (typically 6 or 9 hours) before collecting the cell conditioned medium. 40 units of RNase inhibitor (murine, New England Biolabs) was added immediately, and the cell-conditioned medium was centrifuged at 800 x g for 5 minutes at 4°C before storing the supernatants at −20°C for later use. In some experiments, an aliquot of 3 µL of the cell-conditioned medium was directly used for stem-loop reverse-transcription quantitative PCR (SL-RT-qPCR), detailed later.
Purification of extracellular vesicles
Extracellular vesicles were purified as described in previous work from our group [24]. However, there were minor modifications (e.g., we did not intend to separate ‘microvesicles’ from ‘exosomes’ considering the limitations explained in MISEV2018 [40],). In brief, the harvested cell-conditioned medium was centrifuged at 2,000 x g at 4ºC to remove debris and apoptotic blebs. Then, the supernatant was centrifuged at 100,000 x g for 2.5 hrs at 4ºC (SW40Ti rotor; Optima XPN-90 ultracentrifuge, Beckman Coulter Instruments) to collect a pellet enriched in extracellular vesicles (‘EV’ fraction) and a supernatant presumably enriched in non-EV extracellular ribonucleoprotein complexes (‘non EV’ fraction). The 100,000 x g pellet was resuspended in 250 µl of PBS (for NTA) or MEGM media (for quantitative PCR). Resuspension was performed by repetitive pipetting back and forth (20 times) and by gently scraping ultracentrifuge tube walls with a p200 micropipette tip. The supernatant was concentrated to 250 µl with an ultrafiltration membrane with a 10,000 kDa cut-off (Vivaspin 20, Sartorious Stedim Biotech). Characterization of the vesicular fraction was performed by nanoparticle tracking analysis (NTA; ZetaView, Particle Metrix, Germany), microfluidic resistive pulse sensing (Spectradyne, USA) and solid-phase tetraspanin capture and profiling using an ExoView R100 (NanoView Biosciences, USA). For Spectradyne analysis, TS300 cartridges were used. Samples were run until at least 6,000 events were obtained through resistive pulse sensing, where size and concentrations were analysed. For ExoView analysis, control and transfected biological duplicates were diluted 1:1 in incubation buffer and incubated on chips coated with canonical tetraspanin markers CD81, CD63, and CD9 for 16 hours. Chips were then washed extensively with both wash and rinse solutions, allowed to fully dry, and then run on the ExoView for EV capture using label-free interferometric imaging. Each biological replicate was analysed in triplicate.
For EV quantitation and characterization experiments, two S75 flasks were seeded with 2.3 × 106 cells and incubated overnight at 37°C in DMEM + 10% FBS. Cells were then washed with PBS and transfected with 100 nM 5ʹ tiRNAGly as previously described. In parallel, cells were treated with transfection reagent in the absence of RNA and were used as controls ‘(NT)’. After washing three times with PBS, cells were incubated in MEGM for another two hours at 37°C. Cell conditioned medium was centrifuged at 2,000 x g at 4°C and then at 100,000 x g for 2.5 hours to collect EVs as previously described. The pellet was suspended in 250 µL of 0.22 µm-filtered ice-cold PBS, stored at −80°C and shipped in dry ice for off-site analysis.
RNA extraction and stem-loop RT quantitative PCR
RNA purification was performed with Trizol (Invitrogen, Thermo Fisher Scientific) according to manufacturer’s instructions. For the detection of both endogenous and synthetic 5ʹ tRNA halves, we performed an adaptation of the stem-loop RT microRNA quantification method [44] as described in our previous work [24,30]. SL-RT-qPCR was performed in Trizol-purified RNA extracted from cells, which was first diluted to 10 ng/µL and 2 µL were used as input. Alternatively, the assay was performed directly from the concentrated cell-conditioned medium (without RNA extraction). To do this, 3 µL of RNA-containing samples were initially heated at 95ºC for 5 minutes and immediately placed on ice. From this point onwards, the protocol was the same as with purified RNA.
The 1X reverse transcription reaction (final volume = 7 µL) contained the RNA plus 1X first strand buffer, 1mM dNTP mix, 3.8 U murine ribonuclease inhibitor, 0.01 M DTT, 18 U of Superscript II reverse transcriptase and 10.7 nM of the stem-loop primer folded as described in previous work [24]. All reagents were purchased from Invitrogen with the exception of the SL primer which was purchased from IDT. Thermal programme: 30 min at 16°C, 30 min at 42°C, 5 min at 85°C. The cDNA was immediately used or stored at −20°C. Quantitative real time PCR was performed with SYBR Green PCR Master Mix (2X) from Applied Biosystems, in a reaction volume of 10 µL containing 2 µL of cDNA (diluted 1/3), 1.5 µM of Forward primer and 0.7 µM of the universal reverse primer (complementary to a sequence present in the SL primer).
Fluorescence microscopy
MCF-7 cells (1x104) were plated onto microscope slides containing a culture area equivalent to that of a well of a 96-well plate. 24 hours later, cells were transfected with 100 nM biotinylated synthetic oligonucleotides as described above. After the desired experiment duration, cells were fixed using 4% paraformaldehyde for 15 min at RT and permeabilized with PBS-tween 0.3% for 3 minutes. Then, cells were incubated with blocking buffer (5% BSA in PBS) for 1 hour at 37°C. The cells were washed three times with PBS and incubated in streptavidin–Allophycocyanin (APC) (BioLegend, 0.2 mg/mL) diluted 1:500 in blocking buffer containing 300 µM DAPI (Santa Cruz Biotechnologies) for 30 minutes at room temperature in the dark. After washing with PBS, coverslips were mounted using Prolong Gold Antifade Reagent (Thermo Fisher Scientific). Cells were photographed with a Zeiss confocal microscope (Axio Observer Z1, LSM 800) using a 63X oil immersion objective and analysed using Fiji-ImageJ software. Acquisition parameters were the same for non-transfected cells (negative control) and for cells that were transfected with different biotinylated RNAs.
EV uptake experiments
EVs from transfected and non-transfected MCF-7 cells were purified as described above and incubated with 1 × 104 non-transfected ‘acceptor’ cells in a ratio of 4:1 (donor to acceptor ratio), either in 96 well plates (for RT-qPCR) or microscope slides. Treated cells were incubated for 4 hours at 37°C prior to analysis.
Microarray experiments
(Acceptor) MCF-7 cells were incubated with EVs from (donor) MCF-7 cells transfected with 1 µM tiRNAGly or SCR oligonucleotide in a donor to acceptor cell ratio of 10:1 (i.e., EVs from 4 × 105 donor cells plated in two 6-well plates and collected over a period of 2 hours at 37°C in MEGM medium were added to 4 × 104 MCF-7 cells plated in one 24-well plate). As a control, we followed the same purification protocol starting from non-conditioned MEGM medium and added the fraction which would otherwise correspond to EVs to an equivalent number of acceptor MCF-7 cells (-EVs). After an incubation of 4 hours in the presence of EVs, acceptor cells were washed down three times with PBS and RNA extraction was performed using Trizol. Purified RNA was reprecipitated overnight at −20°C in ethanol + 0.3M sodium acetate buffer pH = 5 and RNase-free glycogen to remove traces of phenol and other contaminants and resuspended in RNase-free water. The extracted RNA was assessed for purity and concentration in a Nanodrop 1000 Spectrophotometer (Thermo Scientific) and an Agilent 2100 Bioanalyzer.
Microarray analysis was performed by using a Human Gene Expression 4x44K v2 Microarray (Agilent Design ID 026652) in a one-colour design. Briefly, 150 ng of total RNA was reverse-transcribed into cDNA, and this was transcribed into cRNA and Cy-5 labelled using the Low Input Quick Amp Labelling Kit, One-colour (Agilent Technologies). The labelled cRNA was purified with Illustra RNAspin Mini Isolation kit (GE Healthcare, United States). The quality of each cRNA sample was verified by total yield and specificity calculated based on NanoDrop ND-1000 spectrophotometer measurements (NanoDrop Technologies, United States). Hybridization, washing and scanning were performed according to the protocol specified by Agilent. The slides were scanned using an Agilent microarray scanner G2505C. Feature Extraction software (version 9.5.1) was used to extract data, quality control, filtering, and data normalization. Three biological replicates were performed for each condition. Differential gene expression analysis was performed with Agi4x44PreProces R package. Genes significantly up- and down-regulated were identified by t-test with a p-value of 0.01 and a Benjamini–Hochberg false discovery rate correction for multiple testing using limma R package.
Results
Overexpression of 5ʹ tiRNAGly increases their extracellular abundance
To study if intracellular levels of tRNA halves correlate with their extracellular abundance, we transfected MCF-7 cells with synthetic 5ʹ-biotinylated oligonucleotides bearing the sequence of wild-type tRNAGlyGCC 5ʹ halves (5ʹ tiRNAGly), or a scrambled oligonucleotide sequence (SCR) with the same length and base composition. Previous work from our group [30] has shown that the 5ʹ tiRNAGly are capable of forming RNase-resistant homodimers, while SCR oligonucleotides are not. Thus, this experimental approach could serve to study the influence of RNA stability on the secretion process.
Transfection with 250 µL of 5ʹ tiRNAGly at 100 nM increased their intracellular levels by a factor of 213, reverting the miR-21-5p (used as reference) to 5ʹ tiRNAGly ratio as measured by fragment-specific SL-RT-qPCR (Fig. 1A). As a control, cells were exposed to the same concentration of oligonucleotide, but in the absence of transfection reagent (-L2K). This did not cause any measurable changes in the intracellular levels of 5ʹtiRNAGly with respect to non-transfected cells. As a consequence, in our experimental conditions, we conclude that there is no measurable spontaneous uptake of these oligonucleotides by MCF-7 cells.
Figure 1.
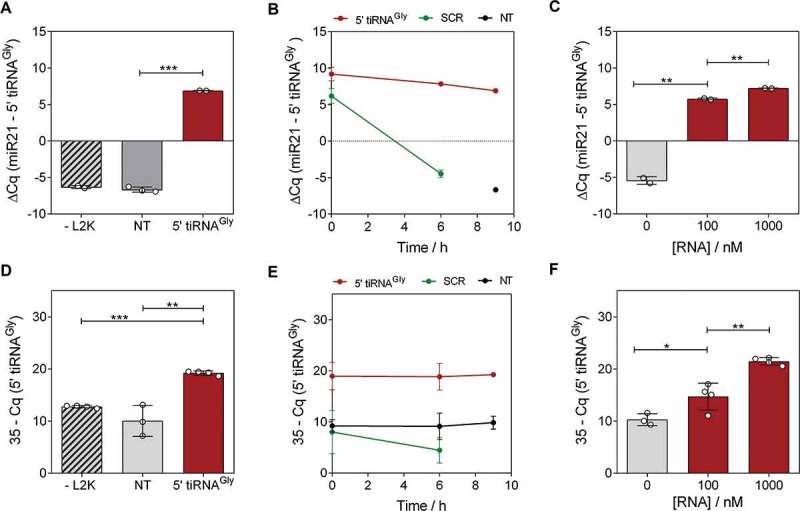
Transfection of 5ʹtiRNAGly in MCF-7 increases their intracellular and extracellular abundance. (A) Relative quantification by stem-loop RT-qPCR of tRNAGly 5ʹ halves in cells exposed to synthetic RNAs in the absence of lipofectamine (-L2K), to lipofectamine in the absence of RNA (NT, for not transfected) and in transfected cells (5ʹ tiRNAGly). Levels of miR-21-5p in the same cells were used for normalization. RNA concentration was 100 nM and cells were lysed at 9 hours post-transfection. (B) Stability of transfected RNAs in a period of time comprising 9 hours post-transfection. SCR: transfected with a scrambled oligonucleotide at 100 nM. Primers were changed accordingly. (C) Intracellular levels as a function of the RNA concentration used for transfection. (D) Same as (A), but aliquots of 3 µL of the conditioned medium were directly analysed without RNA extraction at the conclusion of the experiment. Normalization was performed based on volume for the theoretical inability of finding an RNA not affected by the variable under study (secretion). (E, F) same as (B, D), but in the conditioned medium. For this and subsequent figures: error bars represent the standard error of the mean for experimental replicates and asterisks correspond to results of two-tailed unpaired t tests on the corresponding datasets. *p < 0.05; **p < 0.01; ***p < 0.001.
Reproducing our previous observations [30], the intracellular levels of the transfected RNAs remained stable for at least 9 hours (Fig. 1B). In contrast, transfection with the SCR oligonucleotide showed a 210 drop (10 PCR cycles) in intracellular abundance at the 6 hour post-transfection mark. When transfecting with escalating concentrations of 5ʹ tiRNAGly from 100 to 400 nM, their intracellular levels increased by 1.3 PCR cycles (expected: 2; p < 0.01), plateauing at 400 nM (Fig. 1C and Fig. S1A).
We then asked whether intracellular overexpression of 5ʹtiRNAGly by transfection with synthetic RNA mimics was followed by an increase in their extracellular levels. Indeed, direct SL-RT-qPCR analysis of an aliquot of the cell-conditioned medium (at 9 hours post transfection) showed a gain of approximately 9 PCR cycles in transfected vs not transfected cells (p < 0.01) (Fig. 1D), comparable to the increase observed inside the cells (Fig. 1A). In contrast, extracellular levels of the SCR oligonucleotide were 210 times lower despite being transfected at an identical concentration (Fig. 1E). Interestingly, extracellular levels of 5ʹ tiRNAGly grew exponentially with the increase in the RNA concentration used for transfection (Fig. 1F and S1B), suggesting secretion efficiency is concentration-driven, at least for this sequence. This is probably also the case for the unstable SCR oligonucleotides. Being rapidly degraded inside cells, their effective intracellular concentration drops, and this is reflected by low extracellular levels. However, at least a fraction of this RNA is detectable in the media. This implies that there is a temporal window during which transfected RNAs can be captured by the secretion machinery of the cell and released to the extracellular space while it is still abundant intracellularly.
We adjusted our washing protocol to keep remnant levels at a point where they no longer interfere such that the observed extracellular increase in transfected RNA levels was not due to remnants of the RNA used for transfection. Incubating cells with RNA but no transfection reagent (-L2K control; Fig. 1D), or the RNA/L2K complexes in the absence of cells or with cells incubated at 4°C (which limits overall cell metabolism and vesicular trafficking) (Fig. S2), collectively establish a confident measurement of RNA remnants which cannot explain the extracellular increase in 5ʹ tiRNAGly levels. Thus, these results strongly support the concept that variation of extracellular levels is the consequence of the secretion of cell-internalized RNA, rather than simple carry-over.
Transfected 5ʹ tiRNAGly are packaged and secreted through extracellular vesicles
Next, we analysed whether the overexpressed tRNA halves were present in the cell-conditioned medium as either vesicle-associated or extra-vesicular RNA. Although previous work from our lab showed that 5ʹ tiRNAGly are stable in the non-vesicular fraction as a consequence of their homodimer forming ability [30], we added RNase inhibitor to the cell conditioned medium to further limit degradation. After low-speed centrifugation to remove debris and the largest vesicles/apoptotic blebs, we fractionated the cell conditioned medium into 100,000 x g pellets (EV fraction) and 100,000 x g supernatants (non-EV fraction). The latter was concentrated by ultrafiltration to a volume equal to that used for pellet resuspension.
In contrast with our previous study using the same cell line, culture conditions, and both vesicular and non-vesicular fractions [24], we now omitted the 16,000 x g centrifugation, collecting a mixture of large and small vesicles as a total ‘EV’ fraction. Nanoparticle tracking analysis (NTA) showed that the majority of the vesicles had a diameter below 150 nm, and tetraspanin profiling by ExoView showed these vesicles were positive for CD9, CD63 and CD81 (Fig. S3). However, following the recommendations of the International Society for Extracellular Vesicles [40] we avoided discriminating between exosomes and microvesicles since we cannot unambiguously trace the biogenesis of different vesicular populations. Furthermore, we observed that the RNA present in the 100,000 x g pellets partially pelleted also at 16,000 x g (data not shown), without specific association to neither large nor small EVs.
Reproducing what was observed for the intact cell-conditioned medium, an increase of 10 PCR cycles was obtained when assessing 5ʹ tiRNAGly in the EV fraction of transfected vs. not transfected or – L2K control cells (Fig. 2B, C) (p < 0.001). In contrast, the increase in the non-EV fraction was rather small. As we have previously shown [24], glycine tRNA halves were more concentrated in the non-vesicular fraction in not transfected cells (Fig. 2B; compare columns 2 and 5; p < 0.05). However, this was reverted when cells were transfected with synthetic 5ʹ tiRNAGly (p < 0.01; Fig. 2B; compare columns 1 and 4 and Fig. 2C).
Figure 2.
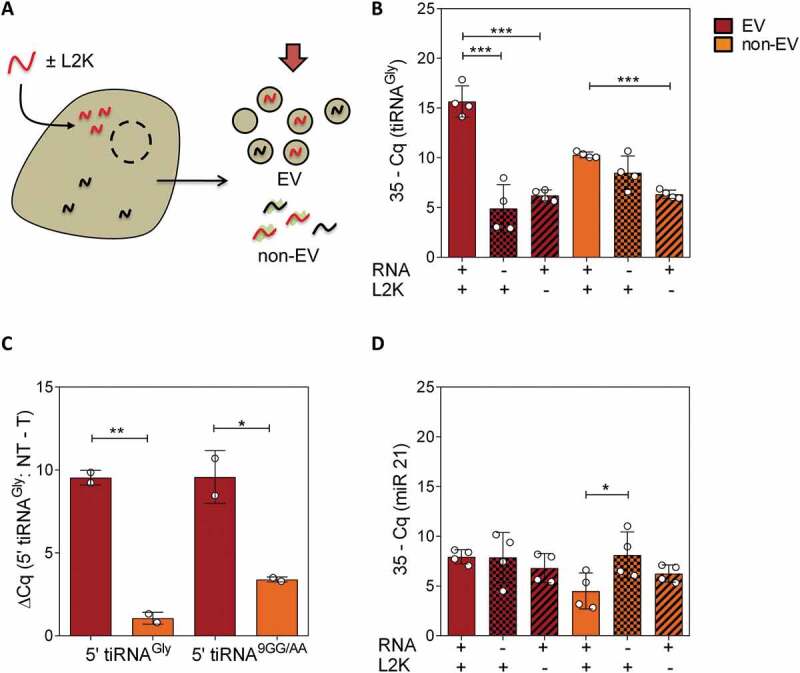
Transfected RNAs are secreted in extracellular vesicles. (A) Schematic representation of a cell being transfected with synthetic RNAs (red). Ultracentrifugation at 100,000 x g was performed at 6 hours post-transfection to separate EV and non-EV fractions, which were concentrated to the same volume and analysed by SL-RT-qPCR. (B) qPCR results in EV (red) and non-EV (orange) fractions, in transfected, NT and – L2K cells (from left to right). (C) Difference between Cq values of not transfected (NT) vs transfected cells, in both extracellular fractions. A mutated version of glycine tRNA halves (called 5ʹ tiRNA9GG/AA) was also tested. Primers were changed accordingly. (D) Same as (B), but assaying miR-21-5p.
Importantly, there was no increase of miR-21-5p (internal reference) in the EVs of transfected vs. non-transfected cells (Fig. 2D), suggesting that vesicles were efficiently loaded with the transfected RNA rather than transfected cells producing a higher number of vesicles with a constant RNA-per-vesicle ratio. This possibility was ultimately eliminated by vesicular quantitation by either NTA or microfluidic resistive pulse sensing (Spectradyne), which did not show significantly increased vesicle production between transfected and control (only lipofectamine) cells (Fig. S3).
Lastly, we repeated the same assays, but transfecting cells with a mutated version of glycine 5ʹ halves (9GG/AA mutant) which we have previously demonstrated to also be RNase-resistant [30]. This enabled us to quantify the exogenous RNAs with less interference of endogenous glycine tRNA halves, given the presence of two consecutive mismatches in the middle of the SL-RT-qPCR forward primer. Once again, a difference of 10 PCR cycles was obtained in the EV fraction of transfected vs not transfected cells, being significantly higher than the difference obtained in the non-EV fraction (Fig. 2C).
All in all, considering no differences in the number of purified EVs between transfected and not transfected cells, we conclude that transfection-mediated overexpression of 5ʹ tiRNAGly increased their concentration in EVs by approximately 1,000 (210) fold. In other words, from 0.06 copies of the endogenous small RNA per small EV [24] to 62 copies of the synthetic RNA mimic per vesicle.
YBX1 independent sorting of overexpressed RNAs in extracellular vesicles
It is tempting to speculate that the mechanism for sorting a > 1,000-fold overexpressed RNA to the intraluminal space during EV biogenesis needs to follow a concentration-driven sequence-independent mechanism. Although many research groups have reported the action of specific RNA-binding proteins to selectively deliver certain RNAs into the sites of EV biogenesis (reviewed in [39]), saturation of this machinery is reasonably expected in our experimental setup. Nevertheless, we decided to test the effect of loss-of-function mutants in at least one of the reported selective RNA sorting pathways.
YBX1 (YB-1) is an RNA-binding protein involved in the stress response [5] that shows binding affinity towards certain tRNA-derived fragments [43,45,46]. Moreover, it has been suggested as the protein responsible for the selective delivery of microRNAs [18] and full-length tRNAs [19] to the EV lumen. However, when we repeated transfection and EV/non-EV fractionation of cell conditioned medium from CRISPR/Cas9 functional YBX1 knock-out MCF-7 cells, we obtained comparable results to MCF-7 wild-type cells (Fig. S4). The difference between the ΔΔCq values (transfected – non transfected; miR21-normalized) in intracellular and EV fractions was comparable between wild-type and YBX1 knock-out cells (2.3 vs 2.6, respectively), implying that there is no difference in secretion efficiencies between the cell lines. In conclusion, and concerning synthetic RNA molecules transfected at 100 nM, YBX1 is not essential for their sorting into EVs.
Glycine tRNA halves can be transferred from transfected to recipient cells via EVs
Next, we studied whether the vesicles containing a higher cargo of glycine tRNA halves (i.e., vesicles derived from transfected cells) were able to deliver these RNAs to untransfected MCF-7 cells. We used oligonucleotides labelled with biotin in 5ʹ, which we had previously shown to retain stability and homodimer forming capacity [30].
Vesicles were purified from transfected, untransfected and – L2K control cells, and added in batch to separate flasks of untransfected MCF-7 cells. Unless otherwise stated, the ratio between donor and acceptor cells was 4:1 (e.g. EVs purified from 8,000 cells were added to 2,000 cells), which is probably higher than the steady-state levels of EVs in cell culture, but considerably lower than the number of EVs used in other previous functional studies [23]. After incubation with the EVs, cells were fixed and observed by confocal microscopy using APC-coupled streptavidin for RNA visualization.
Foci corresponding to APC-streptavidin/biotin-RNA (Red) accumulation were only observable in cells incubated with EVs coming from transfected cells, but neither in cells incubated with the non-EV fraction of transfected cells nor the EV fraction of untransfected nor – L2K cells (Fig. 3B). This was confirmed by quantitative analysis of at least 50 cells present in at least 10 randomly-picked fields (Fig. 3C). To confirm that the full sequence was transferred, rather than just labelled degraded fragments, SL-RT-qPCR (which is specific for RNAs containing an intact 3ʹ end) gave a significant increase (p < 0.05) in 5ʹ tiRNAGly of 3.2 PCR cycles in cells exposed to EVs of transfected vs. untransfected cells (vs. 1.8 cycles in the non-EV fraction, which was not significant; Fig. 3D).
Figure 3.
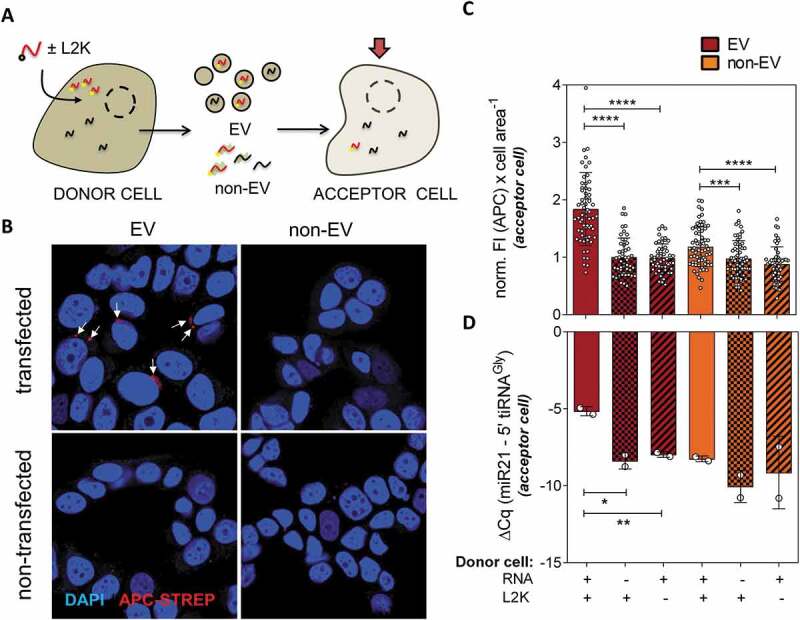
Extracellular vesicles containing transfected RNA can deliver their cargo to recipient cells. (A) Schematic representation of MCF-7 cells being transfected with biotinylated RNAs ‘(donor cells)’. The EV and non-EV extracellular fractions were purified and incubated with not transfected MCF-7 cells, which we will call ‘acceptor cells’. Biotinylated RNAs were then measured in acceptor cells by fluorescence microscopy and SL-RT-qPCR. (B) Representative confocal microscopy images of cells exposed to either EVs or the non-EV fraction of either transfected or not transfected cells and stained with APC-coupled streptavidin (red). Nuclei were stained with DAPI. (C) Quantitation of cell area-normalized total APC fluorescence in at least 50 cells from 10 random fields for each experimental condition. Values were normalized to the average of column 2 (EVs from non-transfected cells). (D) Relative quantitation by SL-RT-qPCR in two independent replicates of the same experiment. miR-21-5p was used for normalization.
The impact of RNA stability in EV-mediated intercellular RNA transfer
Although the previous results strongly suggest that synthetic tRNA halves can be transferred between cells, our experimental readout is based on an analysis of biotin rather than RNA itself. Thus, we wondered whether RNA could be hydrolysed in donor cells, releasing the biotin group, and whether free biotin could then be loaded into EVs and internalized by recipient cells. It can be argued that SL-RT-qPCR depends on RNA integrity, and this method also showed an increase in intracellular 5ʹ tiRNAGly levels in acceptor cells (Fig. 3D). Still, we cannot distinguish between endogenous and synthetic tRNA halves using this technique, and cells can rapidly upregulate 5ʹ tiRNA levels in response to stimuli [32,47]. Therefore, we decided to repeat our biotin-streptavidin fluorescence-based uptake assay (Fig. 3A–C) but now using a combination of biotinylated stable and unstable RNAs. We reasoned that unstable RNAs would not be efficiently transferred between cells, and that this would indicate whether or not the biotin group could be transferred independently of the RNA.
MCF-7 cells were transfected with biotinylated 5ʹ tiRNAGly, a biotinylated version of the SCR RNA, and a third biotinylated RNA where the uracil at position 25 of 5ʹ tiRNAGly was substituted for a cytosine (25U/C mutant). We have previously shown that the 25(U/C) substitution impairs the capacity of 5ʹ tiRNAGly to form homodimers, and that this single change is sufficient to abrogate the high resistance of 5ʹ tiRNAGly to intra- and extracellular RNases [30]. As expected, the normalized fluorescence intensities when using EVs derived from cells transfected with SCR or 25U/C were undistinguishable from background at 4 hours post treatment. On the contrary, the increase in Streptavidin-APC fluorescence in acceptor cells exposed to EVs containing the wild-type 5ʹ tiRNAGly was highly significant (Fig. S5A–C). This result also shows that the accumulation of the fluorescence signal as discrete foci in Fig. 3B is probably an artefact caused by setting the microscope’s acquisition parameters to the high end of the intensity histogram, because we now obtained a more uniform cytoplasmic distribution of 5ʹ tiRNAGly in acceptor cells.
This experiment demonstrates that the fluorescence signal we are monitoring is a reliable surrogate measurement of the levels of synthetic RNAs in acceptor cells and discards the possibility that the biotin group was transferred independently of the RNA. Interestingly, at least some SCR oligonucleotide is loaded inside EVs derived from SCR-transfected cells, as evidenced by SL-RT-qPCR performed directly on the EVs (Fig. S5D). This suggests that the lack of fluorescence signal in acceptor cells exposed to SCR-EVs is not only a consequence of reduced SCR secretion/release (Fig. 1E), but also of increased degradation of the RNA in acceptor cells in comparison with the highly stable 5ʹ tiRNAGly. Consequently, we reasoned that if the SCR oligonucleotide was originally internalized by acceptor cells and later degraded, we should still be able to amplify a remaining fraction by SL-RT-qPCR, which has a much higher sensitivity than fluorescence microscopy. Additionally, and unlike qPCR interrogation of 5ʹ tiRNAGly, there is no endogenous SCR that could interfere with the measurement and cells cannot upregulate this sequence in response to stress. As expected, there was no amplification of SCR in untreated cells, while the sequence could be amplified consistently in MCF-7 cells exposed to SCR-EVs (Fig. S5E). The specificity of our SL-RT- assay for SCR and 5ʹ tiRNAGly was considered high (Fig. S5D, E).
In summary, different RNA sequences can be loaded into EVs and transferred between cells, but RNA stability seems to be a fundamental determinant of the efficiency of the overall process.
Overexpression of tiRNAs is correlated with EV loading and post-uptake intracellular levels
Dose-response studies were performed in order to evaluate if intracellular levels of RNA in transfected cells correlate with their respective levels in secreted vesicles. As depicted in Fig. 4A, the levels of transfected RNA in EVs strongly correlated (Spearman r = 0.976; p < 0.001) with the concentration of transfected RNA. In contrast, levels of the endogenous control miR-21-5p did not vary to a significant extent (Spearman r = 0.39; p = 0.327), suggesting that the previous effect was neither due to cell lysis nor increased EV production (Fig. S3). Variations in miR-21-5p levels between 0 and 100 nM probably reflect alterations induced by the addition of cationic lipids. However, they were in any case not significant, compared with a significant (p < 0.005) difference of 8 PCR cycles for 5ʹ tiRNAGly within the same concentration range. These results suggest that RNA levels in EVs can be modified according to the intracellular RNA levels in EV-releasing cells.
Figure 4.
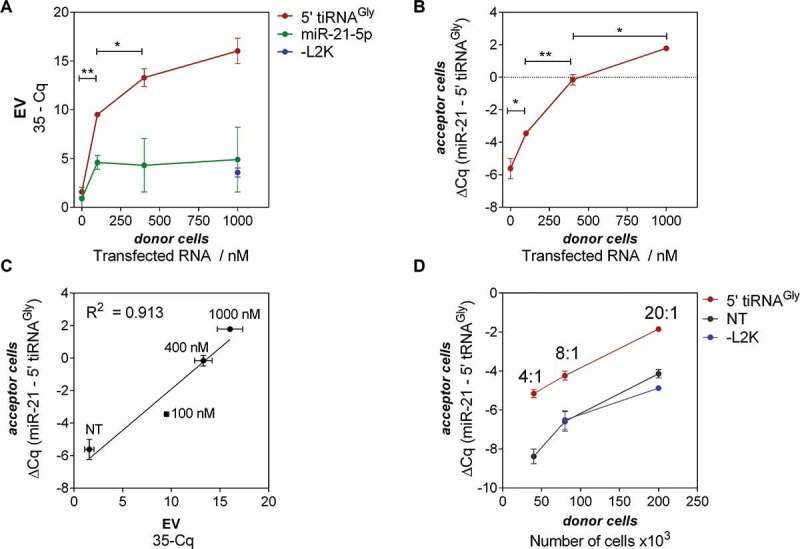
Transfected RNAs are secreted in EVs and delivered to acceptor cells in a donor cell concentration-dependent manner. (A) Quantitation of tRNAGly 5ʹ halves (red; used for transfection) and miR-21-5p (green; not transfected) in EVs, as a function of the concentration of RNA used for transfection. (B) Relative quantitation of tRNAGly 5ʹ halves (normalized to miR-21-5p levels) in acceptor cells, as a function of the concentration of RNA used for donor cell transfection. (C) Correlation between RNA levels in EVs and in acceptor cells. For all these experiments, a fixed donor to acceptor cell ratio of 4:1 was used. (D) Same as (B) but varying the donor to acceptor cell ratio at a fixed RNA concentration of 100 nM (red). EVs were also purified from not transfected (black) or – L2K cells (blue).
We then added a constant number of EVs containing different payloads of biotin-labelled 5ʹ tiRNAGly by exposing untransfected MCF-7 cells (acceptor cells) to a constant number of EVs purified from cells transfected with increased RNA concentrations (donor cells). Post-uptake intracellular levels in the acceptor cells were measured by SL-RT-qPCR and again were highly correlated to the RNA concentration used for transfection (Spearman r = 0.976; p < 0.001) (Fig. 4B). Moreover, the miR-21-5p/5ʹ tiRNAGly ratio in acceptor cells was inverted when donor cells were transfected with RNA concentrations above 400 nM. This increase was directly proportional (in Log2 qPCR scale) to the EV-RNA cargo (Fig. 4C).
Similar results were obtained when performing the reverse experiment (i.e. incubating acceptor cells with EVs obtained from increasing numbers of donor cells transfected at a fixed RNA concentration) (Fig. 4D). Here, the number of EVs varied while the RNA-per-EV content was presumably constant. Again, the levels of 5ʹ tiRNAGly in the EV-treated cells were proportional to the levels in the corresponding EV preparation (R2 = 0.882, data not shown). Although we routinely express intracellular RNA levels normalized to miR-21-5p, it is important to note that the difference between cells exposed to EVs from transfected and untransfected (or – L2K) donor cells was only observable in the Cq values of 5ʹ tiRNAGly (and not for miR-21).
In agreement with the transferred RNA being encapsulated inside EVs, RNase A treatment of the EV preparation did not affect the ability of transfected donor cell-derived EVs to increase the concentration of glycine tRNA halves in acceptor cells. This was observed even with transfected RNA concentrations as low as 40 nM and a 4:1 donor to acceptor cell ratio (Fig. S5).
Overall, these results show that by manipulating the levels of 5ʹ tiRNAGly in donor cells, we can also manipulate RNA levels in acceptor cells treated with donor cell-derived EVs. This property can be exploited for targeted RNA enrichment in intact EVs and EV-mediated delivery of specific oligonucleotides to cells. Depending on the donor cells, selective EV uptake by specific cell types might be also achieved, but this is beyond the scope of this study.
Gene expression changes induced by transfected cell-derived EVs
Although we have provided evidence for EV-mediated cell-to-cell transfer of labelled RNAs, we do not know the fate of these tRNA halves in acceptor cells and whether EV-mediated transfer is able to induce RNA-dependent gene expression changes. To study the bioactivity of the transferred 5ʹ tiRNAGly in acceptor cells, we performed transcriptomic profiling of cells treated with transfected cell-derived EVs. 5ʹ tiRNAGly was recently reported to interact with other ncRNAs which in turn modify chromatin accessibility [48]. As a consequence, we expected to find at least some transcriptomic changes upon EV-mediated 5ʹ tiRNAGly uptake.
To investigate this, we transfected cells with identical concentrations of 5ʹ tiRNAGly and the scrambled (SCR) control RNA oligonucleotide. Then, EVs were purified as previously described and incubated for 2 hours with untransfected (acceptor) MCF-7 cells, keeping a donor-to-acceptor ratio of 10:1. RNA was extracted from cells subjected to three experimental conditions: i) cells not exposed to purified EVs, but containing any possible lab-derived contaminant ‘(-EV)’, ii) exposed to EVs from SCR-transfected cells ‘(+EV(SCR))’ and iii) exposed to EVs from 5ʹ tiRNAGly-transfected cells ‘(+EV(GLY))’.
We hypothesized that by using the same cell line for both donor and acceptor cells, effects arising from exposure to EV cargo other than transfected RNA would be minimal. Indeed, gene expression microarrays showed almost two hundred genes consistently affected (2-fold; adjusted p-value < 0.001) between cells exposed to EVs containing synthetic glycine tRNA halves vs. untreated cells (Fig. 5A). In contrast, no changes were detected at that level of significance when using EVs from cells transfected with the SCR oligonucleotide (Fig. 5B). Thus, obtained hits are RNA-specific rather than constituting an unspecific response to an increase in the EV concentration of the media. Furthermore, significantly altered genes were strongly biased towards downregulation (180 vs. 15), suggesting that we are registering early responses at the level of mRNA stability. Although these genes are not necessarily direct interactors of 5ʹ tiRNAGly, it is clear that transferred RNA is exerting measurable effects in the acceptor cells.
Figure 5.
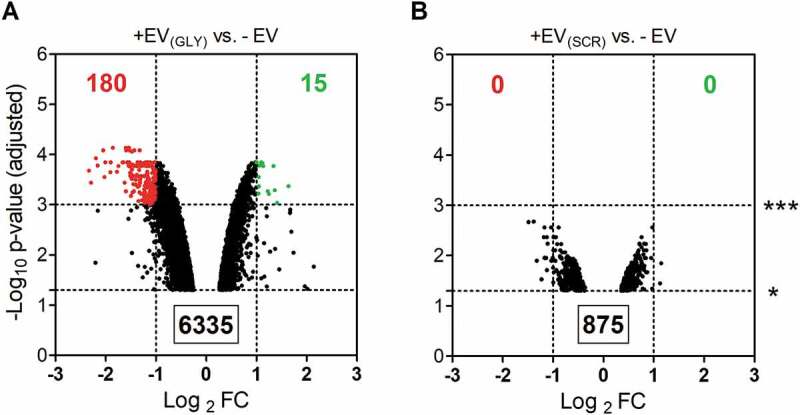
Transfected RNAs secreted in EVs are bioactive in acceptor MCF-7 cells. Volcano plots correspond to genes significantly (p < 0.05) affected by exposure to EVs from cells transfected with glycine tRNA halves (A) or a scrambled oligonucleotide (B) vs. untreated cells. Genes which were downregulated (red) and upregulated (green) more than two-fold with Banjamini-Hochberg adjusted p-value < 0.01 are highlighted. Data was obtained with Agilent gene expression 4x 44K microarrays.
It should be considered that we did not expect EVs from SCR-transfected cells to contain comparable amounts of the SCR oligonucleotide as EVs from cells transfected with 5ʹ tiRNAGly (Fig. 1E). However, this control enables us to rule out transfection-induced changes in donor cells at the level of EV production or composition. Moreover, we have previously shown that transfection with glycine tRNA halves neither altered tetraspanin profiles at the surface of the EVs nor increased their concentration in the media (Fig. S3).
Stable oligonucleotides can be packed in EVs and transferred to other cells
We speculated that at high intracellular concentrations any hypothetical selective RNA sorting machinery would be saturated. In this scenario, one could propose that any excess of free RNA could passively enter the EV lumen by the aid of a concentration gradient. We hypothesized that synthetic 5ʹ tiRNAGly are being loaded in the EVs because they are abundant inside cells irrespective of their sequence or structure being specifically recognized by a putative EV sorting protein. Conversely, scrambled oligonucleotides (SCR) are not enriched in EVs because they are rapidly degraded so that their effective intracellular concentration drops soon after transfection and/or uptake. Nevertheless, we still showed that SCR are not per se excluded from EV encapsulation (Fig. S5D).
To investigate this hypothesis, we transfected cells with biotinylated oligonucleotides which, being relatively stable against RNase degradation, are never found in a human cell (e.g, DNA_16mer; or stable mutants of 5ʹ tiRNAGly). As a consequence, we can rule out the existence of a specific RNA sorting mechanism that evolved to aid secretion of these specific sequences. We tested synthetic 5ʹ tiRNAGly and a mutant where guanines in positions 9 and 10 were changed to adenines (5ʹ tiRNA9GG/AA). We have previously shown that this mutant retains stability and the capacity of forming homodimers [30]. We also tested a modified version of 5ʹ tiRNAGly where the 2ʹ-OH ribose was changed to a deoxyribose (5ʹ tiDNAGly), but keeping the same sequence (including uracil). This oligonucleotide does not form homodimers [30] but is obviously stable against RNase action as it harbours a 2ʹ-deoxy modification in the backbone. Lastly, we tested a biotinylated DNA oligonucleotide of 16 random bases (DNA_16mer) that is not related to 5ʹ tiRNAGly in sequence, structure or length, and is not related to any known human small RNA as judged by in silico predictions.
A constant number of cells was transfected with each oligonucleotide at 100 nM, and the EVs were purified and incubated with untransfected MCF-7 cells. Fluorescence microscopy with APC-streptavidin was used to assay the capacity of these oligonucleotides to be packed into EVs and transferred to the acceptor cells. Strikingly, the four oligonucleotides tested were not only detectable in acceptor cells, but present at comparable levels (as estimated from an average and relatively constant 20% increase in cell area-normalized fluorescence intensity; Fig. 6). It is unreasonable to assume that these oligonucleotides were secreted because of the presence of a yet-to-be-discovered molecular machinery shaped by evolution to detect and selectively export these specific sequences or structures. In contrast, these results strongly suggest that any small DNA or RNA fragment, as long as it is stable enough to be accumulated in the cytosol, will be loaded into EVs in a concentration-dependent manner, though we cannot exclude other variables. Furthermore, increasing the concentration of an oligonucleotide in a given cell will likely increase not only its extracellular abundance, but also its concentration in other cells capable of accepting the EVs. The biotechnological and physiological implications of these findings will be discussed in the following section.
Figure 6.
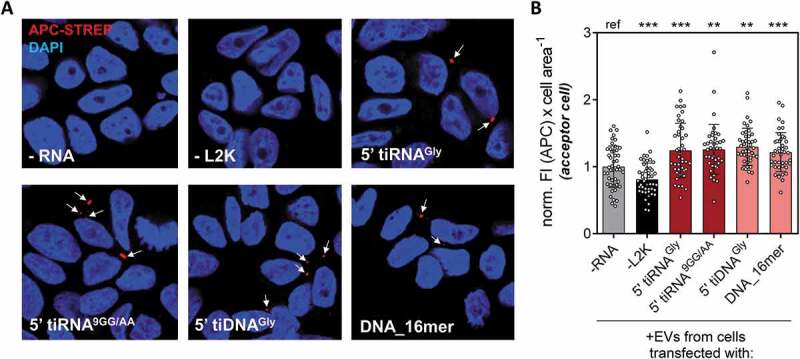
Similar efficiencies for EV-mediated intercellular oligonucleotide transference with different stable RNA (red) and DNA (pink) oligonucleotides. (A) Donor cells were transfected with different biotinylated oligonucleotides (controls: not transfected cells, -RNA, and cells exposed to biotinylated glycine tRNA halves in the absence of lipofectamine, -L2K). EVs were purified from each condition and added to non-transfected acceptor MCF-7 cells, which were stained with APC-streptavidin (red) and DAPI (blue) and analysed by confocal microscopy. (B) Cell area-normalized APC fluorescence intensity, after counting at least 10 random fields from a representative experiment. Values were normalized to the average in column 1 (-RNA). One way analysis of variance was performed to assess the statistical significance of every sample against the ‘–RNA’ control.
Discussion
In this work, we afford experimental evidence that stable non-degradable oligonucleotides can be loaded in EV by transfection-mediated overexpression of producing cells. Stability is key to this process as single-stranded RNAs incapable of forming RNase-protecting dimers were not enriched in the extracellular space even when transfected at the same concentration. Furthermore, these oligonucleotides can be transferred to other cells, implying that the method can be generalized for EV-mediated delivery of target oligonucleotides as long as RNase stability is conferred.
There is compelling evidence that mammalian cells use EVs for intercellular exchange of lipids, proteins and nucleic acids in a manner that is independent of cell-to-cell contact. For example, glioma cells can transmit a transformed phenotype by sharing a truncated oncogenic form of the epidermal growth factor receptor via EVs [49]. Vesicle-mediated intercellular communication can also work systemically through EVs released into the bloodstream or lymphatic circulation. There is evidence that EVs are key players for pre-conditioning the metastatic niche [11,50]. An elegant demonstration that cells can also share nucleic acids in vivo comes from the injection of tumour cells expressing Cre recombinase and cells expressing a fluorescent reporter flanked by LoxP sequences in contralateral mouse mammary glands [51,52] . The authors showed that Cre+ cells released EVs containing Cre mRNA rather than the protein, implying mRNA translation is possible after vesicular uptake, as suggested in previous [6,7,9,53] and later studies [54]. The EVs showed enrichment in specific mRNAs in several studies [9,52], though long mRNAs may be sterically hindered from their loading into EVs, with the consequent enrichment in smaller messengers [29].
In addition to mRNAs, functional transfer of regulatory small RNAs has also attracted great attention. Vesicles containing microRNAs are released and exchanged between innate immune cells. The effects of transferred microRNAs in acceptor cells can be shown by using reporting constructs containing microRNA target sites in cloned 3ʹ UTRs or by silencing/overexpressing specific microRNAs in EVs and analysing downstream phenotypic effects in acceptor cells [23,55,56]. The adipose tissue is also an important source of extracellular EV-associated microRNAs in the bloodstream, which can regulate gene expression distally [8].
If EVs are natural carriers of regulatory RNAs between cells, EVs enriched in therapeutic RNAs emerge as promising biomedicines. A phase I clinical trial with EVs electroporated with siRNAs against oncogenic KRAS for the treatment of metastatic pancreatic cancer is beginning patient recruitment (ClinicalTrials.gov identifier: NCT03608631). Overexpressing the therapeutic RNA in parental cells before EV purification might have some drawbacks. However, upon extensive characterization of the EVs released by transfected cells, it has the advantage that EVs are not themselves manipulated. On the other hand, theoretical feasibility of this method strongly depends on the physiological mechanisms of small RNA sorting to EVs. We will leave mRNA/lncRNA secretion beyond the scope of this discussion, since it imposes additional constraints owing to a much larger size [29].
We can envision two mutually-exclusive mechanisms of small RNA sorting into EVs with different consequences regarding a prediction of which RNAs would be secreted and which would not. Either (a), small RNAs cannot get internalized in the EV lumen during EV biogenesis unless targeted by an active vesicle packaging mechanism, or (b), there is no gatekeeper for small RNAs at the site of EV biogenesis, and their probability of encapsulation strongly relies on their effective concentration in a free, unbound state at the site of EV biogenesis. The former model can be regarded as ‘purely selective secretion’ and the consequence would be a poor correlation between small RNA profiles in cells and EVs. This was observed in several studies [23,57], but contamination with RNA-containing cell culture additives [26–28] or cross-contamination between samples containing low RNA inputs during library preparation [58] can induce bias in interpretation of experimental results. Furthermore, extracellular RNA processing [33] and differential RNA stabilities in the extracellular space [30] can also confound with the effects of selective secretion. As a consequence, lack of significant positive correlation between intracellular and extracellular small RNA profiles is necessary but not sufficient to claim selective secretion as a general mechanism. On the other hand, the latter model (b) does not exclude the possibility that some RNAs are subject to preferential or selective secretion, but does imply that the most abundant species will get into the vesicles unless specifically retained by the cell. This mixture of selective secretion on top of a generalized non-selective secretion model is in agreement with data from a recent study showing EV-mediated silencing of fungal pathogen virulence genes by host plant-derived small RNAs [59].
With the previous considerations in mind, we were interested in determining whether a barrier for the secretion of abundant RNAs exists (i.e., to discriminate between model ‘a’ and model ‘b’). Our results clearly showed that 5ʹ halves from glycine tRNAs can be secreted in EVs in a concentration-dependent manner. Importantly, this was not a singularity of these specific RNA sequences, because other oligonucleotides showing point mutations (i.e., 5ʹ tiRNA9GG/AA), different backbones and structure (5ʹ tiDNAGly) and completely different sequences and size (DNA_16mer) could be secreted and transferred to recipient cells with almost identical efficiencies. In contrast, the efficiency of the process was greatly diminished when using unstable RNAs, although low levels of extracellular release and intercellular transfer could still be measured.
By no means does the previous exclude the possibility of facilitated transport of specific RNAs. It simply shows that, at high concentrations, RNAs might not need to harbour specific sequence motifs to be secreted out onto EVs. If the goal is to obtain EVs enriched in a specific RNA for therapeutic or biotechnological applications, our results suggest that increasing their effective concentration in parental cells might be a suitable method. However, this is not the same as simply expressing or transfecting desired RNAs inside the cell: our study also points out at intracellular and extracellular stability as key variables which can have a profound effect on the outcome. In this regard, transfection of cells and EV-mediated delivery to recipient cells of the transfected RNA has already been shown for stable dsRNAs such as siRNAs [15].
Although we have obtained evidence for transcriptomic changes in MCF-7 cells after exposure to EVs containing high payloads of 5ʹtiRNAGly, these results were intended to show bioactivity of transferred RNAs rather than the identification of putative physiological targets of tRNA-derived fragments, what would require further exploration. In mice, sperm acquires 5ʹtiRNAGly from seminal vesicles (epididymosomes) and these tRNA-derived fragments regulate gene expression in preimplantation embryos [41]. However, later work from the same group showed that snRNAs are the direct interactors of these tRNA-derived fragments, and the affected mRNAs are probably a downstream effect arising from Cajal body alterations and global changes in chromatin state [48]. Alterations in snRNAs and other ncRNAs would not be detected in our gene expression microarray analysis. It is also implausible that EVs showing the degree of enrichment achieved for 5ʹtiRNAGly in our study would be found in nature, and endogenous 5ʹ tiRNAGly probably bares a yet-to-be-characterized set of modified bases. However, it is well known that several cellular stressors such as sodium arsenite increase Angiogenin-mediated tRNA cleavage and the generation of certain tRNA halves, which are termed stress-induced tRNA-derived fragments or tiRNAs (reviewed in [60,61]). It now seems clear that the role of Angiogenin in stress-induced tRNA cleavage is at least partially redundant with the action of other RNases [62]. Regardless of the enzyme(s) responsible, stress-induced tRNA cleavage is a conserved gene regulatory mechanism from mammals [5,32,46,47] to protozoa [63,64]. Considering that some of these fragments are highly stable, it is reasonable to think that their increase in intracellular concentration will be followed by a consequent increase in their EV-dependent secretion. We are currently studying the gene expression changes induced by EVs from cells exposed to different cell stressors, hypothesizing that tRNA-derived fragments, and 5ʹ tiRNAGly in particular, might work as paracrine signals transmitting stress-related information between neighbouring cells.
The above stated hypothesis is sustained by three key observations: i) tRNA halves are produced at higher rates in stressed cells [32,47], ii) some 5ʹ tRNA halves are both thermodynamically [31] and enzymatically stable [30], and iii) overexpressed RNAs are likely secreted at higher rates (this study). We hypothesize that stressed cells secreting higher-than-normal levels of EV-associated tiRNAs might be sensed by cells accepting these EVs as an alarm signal. A similar mechanism, with an evident analogy to the role of interferon during viral infection, has been proposed for Angiogenin [32,61], and the idea that the RNase and its product may act synergistically is attractive. Future studies will tell to what extent tiRNAs are the actual mediators of an interferon-like intercellular signalling pathway during acute cell stress.
Supplementary Material
Acknowledgments
JPT received funding from Comisión Sectorial de Investigación Científica (CSIC, Universidad de la República, Uruguay) and Agencia Nacional de Investigación e Innovación (ANII, Uruguay; FCE-148745). This work was supported by the National Institutes of Health [RO1 GM126150 to P.I., K99 GM124458 to S.L.]. JPT, AC, and KW also received support from UG3CA241694, supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director. GG, AC and JPT are members and receive funding from Sistema Nacional de Investigadores (ANII, Uruguay).
Funding Statement
This work was supported by the Agencia Nacional de Investigación e Innovación [FCE-148745]; NIH Office of the Director [UG3CA241694]; National Institutes of Health [K99GM124458-01A1 and RO1GM126150].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary materials
Supplemental data for this article can be accessed here.
References
- [1].Levin AA. Treating disease at the RNA Level with oligonucleotides. N Engl J Med. 2019;380:57–70. [DOI] [PubMed] [Google Scholar]
- [2].Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14:9–21. [DOI] [PubMed] [Google Scholar]
- [3].Chakraborty C, Sharma AR, Sharma G, et al. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 2017;8:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dang CV, Reddy EP, Shokat KM, et al. Drugging the “undruggable” cancer targets. Nat Rev Cancer. 2017;17:502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ivanov P, O’Day E, Emara MM, et al. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc Natl Acad Sci U S A. 2014;111:18201–18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ratajczak J, Miekus K, Kucia M, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20:847–856. [DOI] [PubMed] [Google Scholar]
- [7].Skog J, Wurdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Thomou T, Mori MA, Dreyfuss JM, et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valadi H, Ekstrom K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- [10].EL Andaloussi S, Mager I, Breakefield XO, et al. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. [DOI] [PubMed] [Google Scholar]
- [11].Hoshino A, Costa-Silva B, Shen T-L, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mendt M, Kamerkar S, Sugimoto H, et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight. 2018;3:99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alvarez-Erviti L, Seow Y, Yin H, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. [DOI] [PubMed] [Google Scholar]
- [15].Zhang Y, Li L, Yu J, et al. Microvesicle-mediated delivery of transforming growth factor beta1 siRNA for the suppression of tumor growth in mice. Biomaterials. 2014;35:4390–4400. [DOI] [PubMed] [Google Scholar]
- [16].Lener T, Gimona M, Aigner L, et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles. 2015;4:30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang D, Lee H, Zhu Z, et al. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2017;312:L110–L121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, et al. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife. 2016;5:e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shurtleff MJ, Yao J, Qin Y, et al. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc Natl Acad Sci U S A. 2017;114:E8987–E8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koppers-Lalic D, Hackenberg M, Bijnsdorp IV, et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014;8:1649–1658. [DOI] [PubMed] [Google Scholar]
- [22].Fong MY, Zhou W, Liu L, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tosar JP, Gámbaro F, Sanguinetti J, et al. Assessment of small RNA sorting into different extracellular fractions revealed by high-throughput sequencing of breast cell lines. Nucleic Acids Res. 2015;43:5601–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chiou N-T, Kageyama R, Ansel KM. Selective export into extracellular vesicles and function of tRNA fragments during T cell activation. Cell Rep. 2018;25:3356–3370.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Auber M, Fröhlich D, Drechsel O, et al. Serum-free media supplements carry miRNAs that co-purify with extracellular vesicles. J Extracell Vesicles. 2019;8:1656042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tosar JP, Cayota A, Eitan E, et al. Ribonucleic artefacts: are some extracellular RNA discoveries driven by cell culture medium components? J Extracell Vesicles. 2017;6:1272832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wei Z, Batagov AO, Carter DRF, et al. Fetal bovine serum RNA interferes with the cell culture derived extracellular RNA. Sci Rep. 2016;6:31175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wei Z, Batagov AO, Schinelli S, et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun. 2017;8:1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tosar JP, Gambaro F, Darre L, et al. Dimerization confers increased stability to nucleases in 5ʹ halves from glycine and glutamic acid tRNAs. Nucleic Acids Res. 2018;46:9081–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lyons SM, Gudanis D, Coyne SM, et al. Identification of functional tetramolecular RNA G-quadruplexes derived from transfer RNAs. Nat Commun. 2017a;8:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yamasaki S, Ivanov P, Hu G-F, et al. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Melo SA, Sugimoto H, O’Connell JT, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang X, Cozen AE, Liu Y, et al. Small RNA modifications: integral to function and disease. Trends Mol Med. 2016;12:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cozen AE, Quartley E, Holmes AD, et al. ARM-seq: alkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods. 2015;12:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zheng G, Qin Y, Clark WC, et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods. 2015;12:835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Giraldez MD, Spengler RM, Etheridge A, et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat Biotechnol. 2018;36:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dhahbi JM, Spindler SR, Atamna H, et al. 5ʹ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genomics. 2013;14:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mateescu B, Kowal EJK, van Balkom BWM, et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - An ISEV position paper. J Extracell Vesicles. 2017;6:1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sharma U, Sun F, Conine CC, et al. Small RNAs are trafficked from the epididymis to developing mammalian sperm. Dev Cell. 2018;46:481–494.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lyons SM, Achorn C, Kedersha NL, et al. YB-1 regulates tiRNA-induced stress granule formation but not translational repression. Nucleic Acids Res. 2016;44:6949–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goodarzi H, Liu X, Nguyen HCB, et al. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161:790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ivanov P, Emara MM, Villen J, et al. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fu H, Feng J, Liu Q, et al. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583:437–442. [DOI] [PubMed] [Google Scholar]
- [48].Boskovic A, Bing XY, Kaymak E, et al. Control of noncoding RNA production and histone levels by a 5ʹ tRNA fragment. BioRxiv. 2018. doi: 10.1101/498949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624. [DOI] [PubMed] [Google Scholar]
- [50].Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ridder K, Sevko A, Heide J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015;4:e1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zomer A, Maynard C, Verweij FJ, et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015;161:1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aliotta JM, Sanchez-Guijo FM, Dooner GJ, et al. Alteration of marrow cell gene expression, protein production, and engraftment into lung by lung-derived microvesicles: a novel mechanism for phenotype modulation. Stem Cells. 2007;25:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kojima R, Bojar D, Rizzi G, et al. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat Commun. 2018;9:1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li J, Zhang Y, Liu Y, et al. Microvesicle-mediated transfer of microRNA-150 from monocytes to endothelial cells promotes angiogenesis. J Biol Chem. 2013;288:23586–23596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Montecalvo A, Larregina AT, Shufesky WJ, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guduric-Fuchs J, O’Connor A, Camp B, et al. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genomics. 2012;13:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tosar JP, Rovira C, Naya H, et al. Mining of public sequencing databases supports a non-dietary origin for putative foreign miRNAs: underestimated effects of contamination in NGS. RNA. 2014;20:754–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cai Q, Qiao L, Wang M, et al. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science. 2018;360:1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gebetsberger J, Polacek N. Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 2013;10:1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lyons SM, Fay MM, Akiyama Y, et al. RNA biology of angiogenin: current state and perspectives. RNA Biol. 2017b;14:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Su Z, Kuscu C, Malik A, et al. Angiogenin generates specific stress-induced tRNA halves and is not involved in tRF-3-mediated gene silencing. J Biol Chem. 2019;294:16930–16941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fricker R, Brogli R, Luidalepp H, et al. A tRNA half modulates translation as stress response in Trypanosoma brucei. Nat Commun. 2019;10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Garcia-Silva MR, Frugier M, Tosar JP, et al. A population of tRNA-derived small RNAs is actively produced in Trypanosoma cruzi and recruited to specific cytoplasmic granules. Mol Biochem Parasitol. 2010;171:64–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.