

ABSTRACT
Transfer RNA fragments (tRFs) are an emerging class of small RNA molecules derived from mature or precursor tRNAs. They are found across a wide range of organisms and tissues, in small RNA fraction or loaded to Argonaute in numbers comparable to microRNAs. Their functions and mechanisms of action are largely unknown, and results obtained on individual tRFs are often hard to generalize. Here we predicted binding mechanisms and specific target interaction sites of 26 human Argonaute-loaded tRFs of different types using large-scale meta-analyses of available experimental data. Strikingly, our findings matched all interaction sites detected in a recent experimental screen, confirming the validity of our computational approach. Such sites are primarily located on the 5ʹ end of tRFs and often involve additional binding along the tRF length, similar to microRNAs. Indicative of multiple layers of regulation, diverse regulatory non-coding RNAs comprised a third of the tRF targets, with the rest being protein-coding transcripts. In the latter, coding sequence and 3ʹ UTRs were the likely primary target regions, although we observed interactions of tRFs with 5ʹ UTRs. Another novel phenomenon we report, a large number of putative interactions between tRFs and introns, is compatible with the roles of Argonaute in the nucleus. Further, observed tRF-intron binding modes suggest a mechanism of interaction of tRFs with Argonaute-dependent introns, and we predict here >20 candidate introns of this type. Taken together, these results present tRFs as regulatory molecules with a rich functional spectrum.
KEYWORDS: Argonaute, small RNA, non-coding RNA, tRNA, tRNA fragments
Introduction
Recent advances in RNA sequencing technologies have contributed substantially towards the discovery of novel non-coding RNAs (ncRNAs). Based on their size, ncRNAs can be grouped into three categories: long (>200 nts), medium (>40 nts) and small RNAs (<40 nts). In this study we focused on a particular type of small RNAs that originate from transfer RNA (tRNA) genes and are called tRNA-derived fragments (tRFs).
Mature tRNAs are usually less than 90 nts long and their secondary structure resembles a cloverleaf. Properly folded tRNAs contain four distinct arms: D arm, anticodon loop, T arm and a variable loop. Transfer RNAs are crucial components of the cell’s translational machinery. They facilitate translation of mRNA codons into amino-acids through base pairing between the mRNA codon and the anti-codon trinucleotide located in the middle of the tRNA molecule. Despite the fact that there are only 64 codons encoding for 20 amino acids, the number of tRNA genes ranges in hundreds for multiple species. For example, the human genome contains more than 600 tRNA genes, ranking them among the most abundant RNA molecules in the human transcriptome [1,2]. Such abundance of tRNA genes suggests that these molecules may have additional functions and properties. Hence, it is not surprising that recently there has been an explosion of reports describing abundant levels and potential novel functions of their fragments (tRFs) in different species.
tRFs have been posited to arise from directed cleavage of cellular tRNAs, including both tRNA precursors and mature tRNA molecules. They are categorized into two groups based on the length of the small RNA: tRNA halves (28 ~ 40 nts) and tRNA-derived fragments (16 ~ 24 nts). tRNA halves are considered to be a product of cleavage of mature tRNAs under stress conditions [3–5].
On the other hand, tRFs can be classified into four distinct subgroups based on their location: tRF-5, tRF-i, tRF-3 and tRF-1 (following the general notation of the first tRF report [6]). tRF-5 are derived from the 5ʹ end of the mature tRNA molecule through endonucleolytic cleavage near the D loop/arm [7]. tRF-i (internal tRFs) is the most recently identified type of tRFs, which span a variety of contiguous regions across tRNA molecules other than the very 5ʹ and 3ʹ ends [8,9]. The last two subgroups originate from the 3ʹ end of the transfer RNA molecule. 3ʹ CCA tRFs (tRF-3) contain the post-transcriptional CCA trinucleotide addition and they are products of direct cleavage of the mature tRNA molecule (most frequently at the T arm/loop), while tRF-1 (also known as 3 ‘U tRFs) derive from the uracil-rich sequence on the 3ʹ end of the precursor tRNA molecule [10].
tRFs have often been excluded from small-RNA studies and considered to be non-functional degradation products of their parental molecules. However, there is both biochemical and computational evidence for the role of tRFs as functional molecules in multiple biological processes [11–14]. tRFs have been shown to bind to Argonaute complexes in multiple species [10,12] and they have been proposed to function similarly to microRNAs (miRNAs) by regulating mRNAs or by affecting miRNA loading and processing [15–17]. Supporting these similarities between miRNAs and tRFs, a recent study has shown two miRNAs being, in fact, tRF-1 species derived from the trailer sequences of tRNA genes [18].
Most of the experimental studies of tRFs have focused on a one or two of molecules; there is no overlap and the findings are hard to generalize. The mode of action for tRFs with regards to RISC mediated post transcriptional RNA silencing still remains vague. It is unclear if tRFs act as plant miRNAs (which are almost fully complementary to their target RNAs) or as animal miRNAs (which recognize their targets based on complementarity of a short ‘seed’ region located on the 5ʹ end of the small RNA molecule). Different results have been reported for such seed regions in tRFs. One group has demonstrated a silencing mechanism similar to miRNAs and based on the complementarity of the 5ʹ seed sequence of a tRF to a 3ʹ UTR of a reporter gene [19]. Another study has shown that such a seed region can be on the 3ʹ end of a tRF molecule and can induce mRNA repression [20]. In our previous work in fruit fly and rat we have found that a seed region can be located on either end of a tRF molecule based on matches with conserved target sequences primarily found in 3ʹ UTRs of mRNAs [12,13].
The seed-driven target identification is a standard practice for miRNAs, and it has been simply adopted for tRFs with limited support from computational and experimental data [10,12,19]. On the other hand, there is also an increasing amount of evidence for non-canonical hybridization modes for both miRNAs and tRFs [19–21]. Additionally, we have shown earlier that potentials seed sequences with exact match to conserved 3ʹ UTRs are much more frequent in tRF-3 compared to tRF-5 in rat brain suggesting possible differences in binding for different tRF types [13]. Thus in order to effectively study tRFs, there is a need to identify and understand their targeting/hybridizing modes.
Here we investigated targeting sites of tRFs and their putative binding mechanisms using computational analysis of large-scale datasets produced by several experimental projects. In our ab initio analysis of tRF targeting modes we used the Crosslinking, Ligation, and Sequencing of Hybrids (CLASH) data series from Ago1 pulldowns in HEK293 cells [22], which has generated a valuable resource of chimeric sequences containing putative interacting guide and target RNAs. This dataset has been used for finding tRFs in an earlier paper [10], reporting thousands of tRF-containing chimeras. However, tRF binding patterns inferred from CLASH were not the main focus of that work, which has provided examples of just five targets as an illustration. In our study, we performed a comprehensive bioinformatic analysis of the CLASH chimeras and identified 1321 tRF isoforms (including hundreds of isoforms not detected earlier [10]) using them to infer tRF targeting patterns on a tRFome-wide scale.
We found that Ago1-loaded tRFs target a wide range of transcripts including coding and ncRNAs. We report a novel phenomenon – a large number of putative interactions between tRFs and intronic sequences, consistent with the evidence of Ago function in the nucleus [23,24]. We also found that tRFs may be operating as guide molecules enabling Ago interactions with a specific group of short introns, recently identified as agotrons [25].
We analysed sequences of chimeras formed in vivo between tRFs and their targets to identify clusters of RNA-RNA interaction signatures. We catalogued possible binding patterns between different types of tRF guides and targeted sequences and identified motifs that may be responsible for these interactions. Finally, we compared our computational predictions of target interaction sites with those found in a recent experimental screen. Strikingly, for three common tRFs the predictions matched the seed location determined in luciferase assays [26], demonstrating the predictive power of our approach. Our results support the emerging view of the Ago-enabled tRF action and demonstrate the possibility of inferring the binding regions and mechanisms of tRF/target interactions computationally.
Results
1. Ago-1 loaded tRNA fragments
To investigate tRF/target RNA interactions, we analysed a series of CLASH (Cross Linking and Sequencing of Hybrids) libraries, originally used to study miRNAs and their interactome in HEK293 cells [22]. Similar to miRNAs and their targets, CLASH captures exact tRF/target RNA interactions in vivo, and this allowed us to identify high confidence interactions. This dataset has been previously used [10] to identify a subset of tRFs and their targets. However, we found that a large portion of them has been missed in that earlier study (we detected >3-fold higher numbers of tRF isoforms, including 258 tRF-5, 406 tRF-3, 160 tRF-1 and 497 tRF-i). Further, our analysis provides additional insights into the tRF mechanism of action by using the sequences of tRF/target pairs to systematically find their binding patterns and potential interaction sites.
We identified hybrid reads starting with a tRF sequence and used BLAST [27] to find the best match for the remainder of each read. Potential guide tRFs were mapped to the full tRNA sequences, including mature tRNAs. Hybrid reads that passed all the quality control filters (see Materials and Methods) were considered tRF/target RNA chimeras and used for downstream analyses. We observed a total of 41,219 CLASH chimeras (supported by at least two different reads) containing guide tRFs and a variety of target RNAs. After collapsing all combinations of one tRF with the same sequences of the same target, we obtained 6,117 unique chimeras, with most of the tRFs being of nuclear origin. Unlike the prominent presence of Ago-loaded mitochondrial tRFs (mt-tRFs) in Drosophila [12] and in human tRF databases [9], only 44 unique CLASH chimeras contained mt-tRFs.
We noted an overwhelming excess of tRF-3 that derived from mature tRNAs (with added CCA), followed by tRF-5 and tRF-i (Fig. 1A). The least frequent chimeras were formed between tRF-1 and target RNAs (Fig. 1A), in agreement with earlier results [10]. We then asked if CLASH chimeras contained non-random pairs of tRFs with their targets and considered the specificity of their pairing based on binding energy, using RNAhybrid [28] to calculate the minimum free energy (MFE) of hybridization for each tRF/target chimera. Binding within CLASH chimeras was significantly stronger than that for simulated chimeras of observed tRFs with random RNA (difference of 10.7 kcal/mol. p-value < 10−16, Fig. 1B). Further, we compared the MFE distributions of CLASH chimeras for each tRF type with the MFE of simulated shuffled chimeras, where targets were randomly picked from chimeras of three other tRF types. The tRF-3, tRF-1 and tRF-5 MFE showed the largest and most significant differences (mean energy gains of 8.1, 4.7 and 2.7 kcal/mol, respectively) in such chimera reshuffling, while tRF-i MFE gain was less significant (Fig. 3, energy differences and p-values in the rightmost column).
Figure 1.
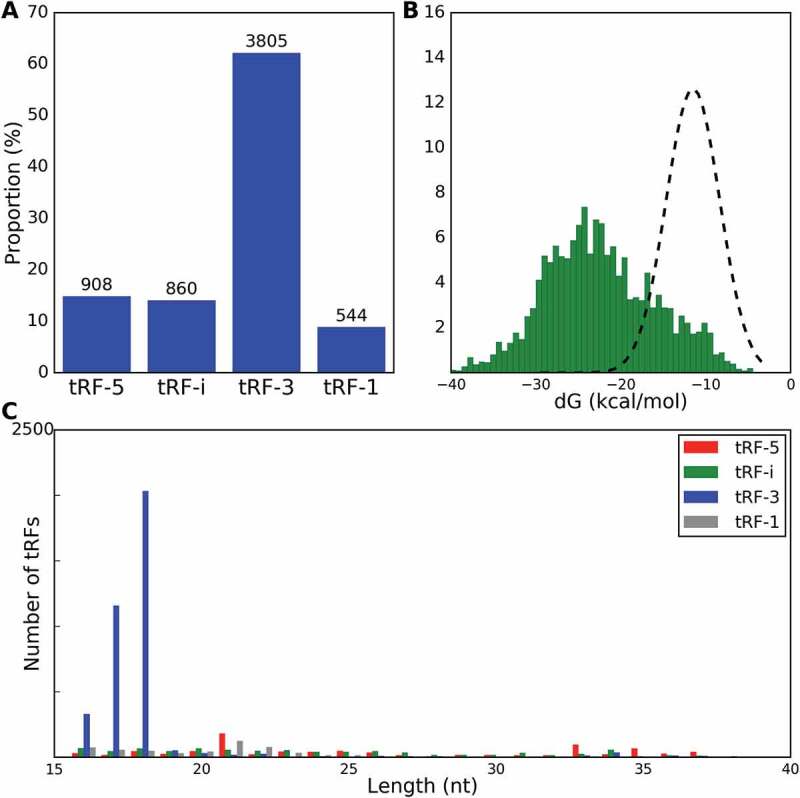
Ago-1 loaded tRFs. A) Distribution of guide tRF types identified from CLASH RNA chimeras (%% on the y-axis and actual counts of unique chimeras containing specific tRF types given above the histogram bars). B) Minimum Free Energy (MFE) histogram for tRF/target RNA chimeras (green histogram) and for randomly generated control interactions (black line). C) tRF length distribution in chimeras identified from CLASH data.
Figure 3.
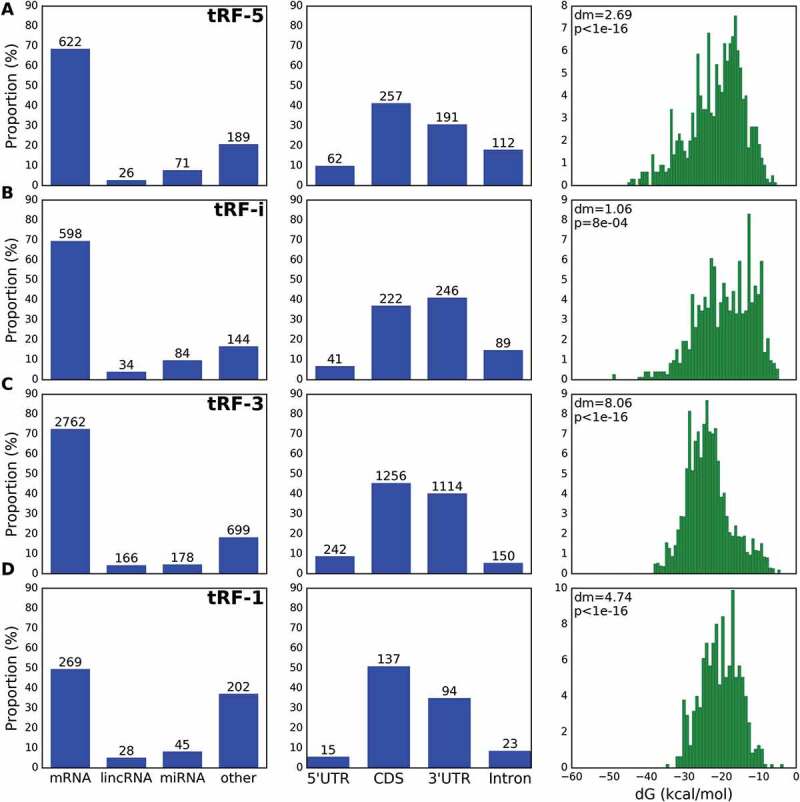
tRFs guide Ago1 to a variety of RNA targets. tRF target distribution plots (left, %% on the y-axis and actual unique chimera counts given above the histogram bars), targeting frequency of mRNA regions (middle) and MFE histograms of tRF/target RNA unique chimeras (right). Mean MFE of matching tRF and target pairs were lower than the MFE of randomly matched pairs, as indicated by dm, difference of means, and t-test p-value. Rows depict tRF-5 (A), tRF-i (B), tRF-3 (C) and tRF-1 (D).
Next, we examined whether tRFs are generated by cleavage at specific sites and noted clear differences in length distribution and abundance of different tRFs, resembling our earlier results in rat brains [13]. We observed a narrow peak of tRF-3, with the most prominent peak for fragments of length 18 nts (Fig. 1C) and much wider distributions for other tRF types, with the tallest peaks in tRF-5 and tRF-1 close to miRNA sizes (Fig. S1).
As another test for non-randomness of tRF generation processes, we considered potential correlation between the levels of mature tRNAs and their corresponding fragments in HEK293 cells. We compared the abundance of Ago1-loaded tRFs with tRNA abundance determined by hydro-tRNAseq [29] and also with tRF levels from small RNA [30] sequencing (not Ago-loaded) in the same cell line. We found no correlation between Ago1-loaded tRFs and total cytoplasmic tRF abundance or tRNA levels (Table S1). This was consistent with the unequal loading of tRFs from the same tRNA gene to Ago1 that we observed (Fig. 2).
Figure 2.

Abundance heatmap for tRFs generated from mature nuclear tRNAs. The scale on the right represents the count of unique chimeric reads found in CLASH data that contained each specific type of tRF as a guide sequence.
Taken together, these results suggest that distinct types of tRFs are likely to be generated by multiple mechanisms of cleavage, in agreement with our previous findings for divergent changes in abundance with age for different tRFs in Drosophila [12] and rat brains [13]. This further supports the notion that tRFs have specific cleavage sites and are not byproducts of random degradation.
2. General features of tRF/target interactions
We analysed all interactions between tRFs and their respective RNA targets, identified as distinct transcript fragments within the same chimera. Following the logic of CLASH experiments, we considered frequent occurrences of the same tRF/target RNA pair as evidence of interaction with a target. We did not restrict ourselves to protein coding genes (for an illustrative set of 100 most frequent mRNA targets see Table S2) and took into account every possible hybrid read formed between a tRF and its target RNA. We found that tRFs interact with a wide variety of RNAs including mRNAs, lincRNAs and miRNAs (Fig. 3). Similarly, mRNA and lincRNAs have been earlier identified as targets of miRNAs [22]. Less frequently targeted transcripts were categorized under ‘Other’ and most of them were annotated in Ensembl transcriptome as miscellaneous RNAs (‘misc_RNAs’, 30% of targets), processed transcripts (23%) and pseudogenes (12%).
Next, we calculated the targeting frequency of individual regions of mRNAs by tRFs. We found that almost all types of tRFs primarily targeted coding sequences (CDS) and 3ʹ untranslated regions (UTRs) of mRNAs followed by intronic regions and 5ʹ UTRs (Fig. 3), expanding beyond the canonical 3ʹUTR targeting mode of miRNAs [31,32]. We have previously predicted intron targeting based on k-mer conservation [12], and this is the first report of introns as tRF targets based on CLASH experiments. Our observation is compatible with the evidence that Ago proteins also localize in the nucleus [23,24].
In our analysis we considered possibilities of finding functional elements in introns, such as miRNAs or other ncRNAs and it is conceivable that such yet unannotated elements are being targeted by tRFs. None of the intron targeting cases reported here contained currently annotated RNAs. However, there was one striking exception. We found that tRFs can potentially guide Ago to a specific type of short introns called agotrons. Agotrons are relatively rare (82 are known), they are defined based on their length (<150 nts) and association with Ago proteins: Ago-2 has been reported to bind ~30 nts on the 5ʹ end of the agotrons [25]. We found five known agotrons in chimeras with tRFs in Ago1, with targets starting precisely at agotron 5ʹ ends (Fig. S2). Such coincidence suggests a mechanism whereby tRFs may guide Ago proteins to the agotron borders and drive their excision (or interact with agotrons after the excision).
We reasoned that if such mechanism indeed existed, there could be other cases of such tRF/agotrons chimeras at the borders of unknown agotrons, and we searched for such events. We detected 28 total unique chimeras (Table S3) joining tRFs or miRNAs with agotrons borders (or a 1 nt offset in six border cases, including those in Fig. S2). Notably, two of these miRNAs had almost the same sequence as tRFs (1 nt missing or added to the 3ʹ end) and may be misannotated miRbase entries (Table S3). In four other cases, however, genuine miRNAs (one of which, hsa-mir-6747 was transcribed from the intron of EEF1G gene) appeared to be interacting with agotron borders. It remains to be determined if these introns are real agotrons, although they show typical characteristics of this class [25]. Nevertheless, to the best of our knowledge, none of the cases of tRF or miRNA interaction with agotrons (or such agotrons candidates) has been reported. The agotron-targeting tRFs shared similar sequences and a 12-nt motif with strong triple C at the 5ʹ end was revealed for the tRFs (Fig. S2). Interestingly we observed uncommon G richness at the 5ʹ end and C richness at the 3ʹ end of these specific introns targeted by the tRFs (Table S4).
In addition to sense transcripts, we found that tRFs potentially also target Natural Antisense Transcripts (NATs) [33–35], with 1,373 unique interactions between tRFs and NATs. We observed that the tRF/NAT chimeras (i) were less favoured energetically compared to chimeras with sense transcripts, (ii) included longer isoforms of tRF-5 and tRF-i and (iii) showed much lower tRF-3 abundance (Fig. S3).
3. tRF/target hybridization modes
We inferred binding modes of tRFs in CLASH chimeras as follows. We predicted the hybridization patterns between tRFs and their CLASH targets with RNAhybrid [28] and encoded each tRF base as target-binding (1) or not binding (0). We selected tRF-3 (as forming the most of unique interactions) of the length 18 nts (the highest peak in the respective length distribution in Fig. 1C) and applied k-means clustering to reveal distinct binary signatures of interactions. We considered chimeras with targets differing by <5 nts in length at either end as identical (i.e. a target overhang of 5 or more bases was considered a different chimera). This gave us 1,687 unique chimeras with 18-nt tRF-3.
Five clusters of consistent and similarly shaped binding patterns between the nucleotides of guide tRFs and their target RNAs (Fig. 4) were different in size and in their average MFE of hybridization. These cluster shapes revealed several main recurrent themes in tRF/target interactions.
Figure 4.
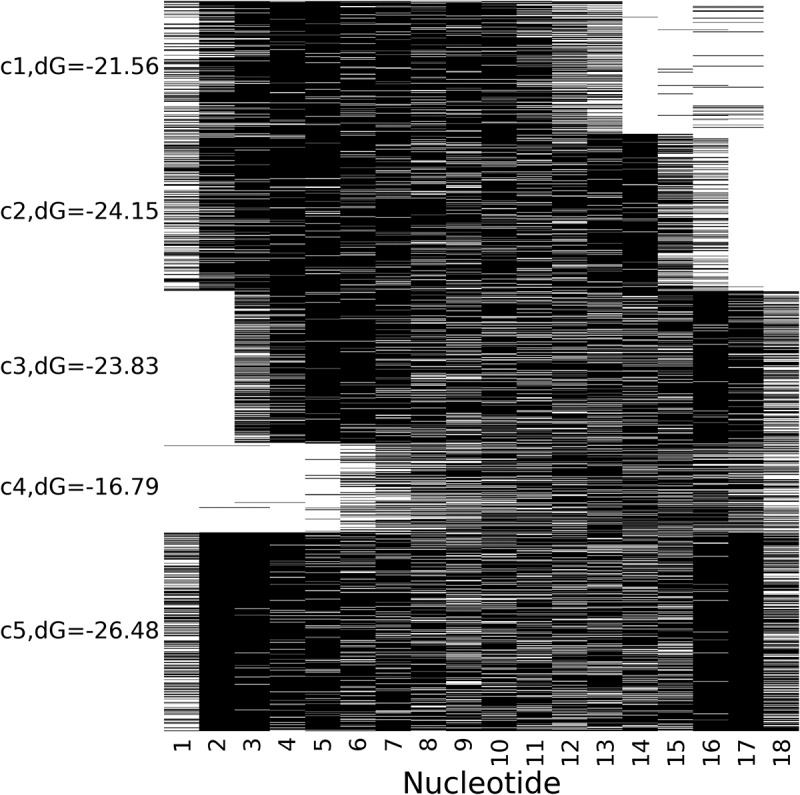
Base-pairing patterns for unique tRF-3/target chimeras. Each line represents a guide tRF from a unique CLASH chimera. Paired nucleotides are depicted in black and unpaired nucleotides are shown in white. The labels C1 through C5 mark the vertical centre points of the five identified clusters and the average MFE for the interactions in each cluster is shown.
Many chimeras showed binding primarily on the 5ʹ end of the respective tRFs, often involving their 3ʹ end as well (especially nts 16–17) to enable the strongest interactions. Cluster 4 contained chimeras with no 5ʹ binding nucleotides yielding the weakest interactions. Overall, these binding patterns include nucleotides located across the whole length of a tRF, consistent with what has been reported for miRNAs [21].
tRFs targeting the same gene were typically found in the same cluster and more than 90% of the targets in Fig. 4 specifically interacted with tRFs in one cluster. The Gene Ontology analysis of targets revealed that they many of them were involved in regulation and some may have a certain cluster-specific functionality (with enrichment p-values below at least 10−4). E.g. genes in clusters 1 and 5 were enriched for protein transport and localization and in cluster 3 – for protein methylation. Targets regulating various aspects of the cell cycle were in clusters 1, 2 and 5. The targets of tRFs in cluster 5 showed enrichment for embryo development, including the brain development, similar to the ageing-associated tRFs in flies and rat brain [12,13]. Additionally, genes in that cluster regulated and participated in mRNA processing (and 3ʹ-end processing), its splicing and export from the nucleus.
4. Analysis of tRF/target pairs reveals interaction sites for tRFs
To determine whether tRF/target interactions can be driven by seed regions or motifs, we used MEME [36] to find statistically overrepresented sequences among the target RNAs for each major tRF isoform that had a minimum of five distinct target genes (longest target RNA was selected for each gene). For each such overrepresented sequence we checked whether it matched a reverse complementary sub-sequence of the respective guide tRF using FIMO [37] to identify likely interacting regions.
We found that targets of 26 tRFs of all types (20 tRF-3, four tRF-5, one tRF-i and one tRF-1) contained an enriched motif and a reverse complement match the respective guide tRF; we considered these as seeds/interacting sites for tRFs (Fig. S4). We used the following notation for each tRFs
geneID-origin-type-position,
which reflects its host tRNA gene ID, Nuclear or Mitochondrial origin of the tRNA (N or M), followed by the tRF type represented as 5p/3p/Mi/3t (for tRF-5/tRF-3/tRF-i/tRF-1, respectively) and specific start-end nucleotide positions in the tRNA gene). For AlaAGC, CysGCA and GluCTC, we found multiple isoforms from tRNA genes with slight differences in sequence (and in seed region) but many different targets. Seed regions were found mostly at the 5ʹ end of a tRF, with instances of central interaction sites (LysTTT, SerCGA, ThrATG, ThrTGT and AlaAGC), as also reported for miRNAs [21,38]. Two tRFs, AspGTC and LysCTT, contained a likely interaction site at the 3ʹ end, while one motif matched both 5ʹ and 3ʹ end of the LeuAAG.
The location of these motifs generally matched the hybridization patterns of the respective tRFs (Fig. S4). We also plotted for each 7-mer along the tRF length the frequency of its occurrence in gene regions (5ʹUTR, CDS, 3ʹUTR) conserved across 100 animal genomes (using the approach we applied to fly and mammalian genomes earlier [12,13]). We observed that mostly major but sometimes minor peaks of 7-mer conservation appeared right next to the starts of motifs/interaction sites or within them (Fig. S4). Such agreement between the three independent lines of evidence (motifs found in sets of targets, hybridization patterns of tRFs/targets and 7-mer conservation) strongly suggests that our predictions represent functional interaction sites compatible with the regulatory mode of action of Ago-guide RNA complexes.
Recently, 5ʹ seed regions (of at least 6 nts in length) have been experimentally validated for three different tRF-3 (Leu-AAG, Leu-TAA, Cys-GCA) in HEK293 cells [26]. These three tRFs were the most abundant among Ago1-loaded ones (Fig. 2). Notably, we observed significant motifs for all three of them (Fig. 5 and Fig. S4). Furthermore, for all three tRFs the interaction sites we predicted were found on the 5ʹ end of the tRF in the experiment. This is a striking agreement and a very strong combined evidence that all three tRFs recognize targets with a 5ʹ seed. However, despite this match in the location of the seed sequence, its length, as predicted by our pipeline, can vary. We report motifs of 5–13 nts and note that some nucleotides on the 3ʹ end of the guide tRF might be required for effective binding to target RNAs, in agreement with the clustering results (Fig. 4). Interestingly, for Leu-AAG we found the same motif on both ends of the tRF (underlined in Fig. 5).
Figure 5.

Computationally and experimentally identified interaction sites for tRF-3 type tRFs. Computationally predicted interaction site locations are shown in red boxes. The 5ʹ tRF end has been proposed as the location of the seed regions for these three tRFs on the basis of luciferase assays [26], and nucleotides at positions 2–8 are shown in bold letters.
Given that guide binding may be imperfect and involve bulges, we also searched for motifs with GLAM2 [39], allowing for gapped motifs, and mapped them back to tRFs using GLAM2SCAN [39]. Most of the MEME motifs were also found using GLAM2 and their potential gap/bulge locations are shown with black arrows in Fig. S4.
Discussion
In this report we characterized tRFs and their respective targets loaded to Ago1 proteins and identified putative targeting modes of tRFs using CLASH dataset from a human HEK293 cell line. We showed that tRFs target a wide variety of transcripts through distinct modes of hybridization, as summarized by several clusters of typical patterns (Fig. 4). Some of the interaction sites, as shown before [10,12,13], are similar to 5ʹ-localized seed sequences previously identified for miRNAs. We also observed other sites, located in 3ʹ-end and central parts of tRFs or extending across the whole tRF molecule (Fig. S4). We found all types of tRFs in CLASH chimeras (including tRF-i, not detected previously [10]), with tRF-3 originating from mature tRNAs being vastly abundant. The most frequent tRFs were 17 to 21 nts long, but longer isoforms (>30 nt) were also frequent among tRF-5 and tRF-i.
In the past, tRFs have been often neglected as products of random degradation of parental tRNA molecules. As an argument against randomness, earlier studies have pointed to the lack of correlation between the levels of different tRFs from the same gene [10] or between the tRNA gene copy numbers and tRF levels [40–42], although not in Ago1. Here, we extended this argument further using CLASH data to compare in HEK293 cells (Table S1) levels of tRFs loaded to Ago1, the total cellular tRFs fraction and the expression of the parental tRNA gene measured using hydro-tRNAseq [29]. Consistent lack of correlation between tRNA and tRF levels in different experimental systems supports the view that tRFs are not products of random degradation and different types of tRFs may be produced through different mechanisms. Our results also highlight the difference between the spectra of tRF isoforms in the total cellular tRFs fraction and in complex with Ago1.
The main targets of tRFs were mRNAs, followed by various ncRNAs (comprising some 1/3 of the targets). Further, multiple ribosomal proteins (RPL35A, RPLP0, RPS14, RPS19 and RPL7L1) and such genes as a translation initiation factor EIF3C were among the most frequent targets of Ago-loaded tRFs (Table S2). This observation is consistent with a hypothesis put forth in a recent Drosophila study [43], suggesting that tRFs may be involved in global translational control in addition to posttranscriptional regulation of specific RNAs. Interference with translation via ribosome targeting has also been reported for tRFs [44]. On the other hand, we noted plentiful targets not directly involved in such global translational control. Among mRNA regions, we observed that CDS and 3ʹ UTRs were most frequently targeted by tRFs followed by introns and 5ʹ UTRs.
We report that tRFs also target miRNAs when loaded to Ago1. The original CLASH study has shown that Ago-loaded miRNAs can target tRFs [22]. Taken together, these findings suggest that tRFs and miRNAs may regulate each other in addition to their respective coding or non-coding targets, creating additional layers of regulation.
For an unbiased view of tRF targeting modes, we selected unique tRF/target RNA chimeras and performed clustering analysis for the most abundant type of tRFs. We found a variety of binding patterns in tRFs, not always limited to specific 5ʹ nucleotides positions of 2–8 (P2-8) in miRNA-like seeds (Fig. S4). Clusters of binding patterns involving nucleotides located on the 5ʹ end of the guide tRF generally showed stronger binding (lower MFE) compared to clusters with hybridizing nucleotides in the middle of the tRF. Hybridization of 3ʹ located nucleotides also seems to play an important role since it was evident for the largest proportion of interactions (Fig.4, c3-5). Thus, in addition to the 5ʹ P2-8 miRNA-like seed sequence [12,13,19,20], tRFs likely recognize their targets through multiple binding regions.
To further detail the trends highlighted by the clusters, we used an ab initio approach to search for motifs/interaction sites across tRFs without restrictions to the canonical P2-8 seed binding. Overall, there were some 2/3 of the motifs found close to the 5ʹ end of tRFs with some extensions to the 3ʹ end, two cases of 3ʹ motifs, with the rest of motifs centrally located (Fig. S4). Several cases of 5ʹ tRF seeds at P2-8 or close coordinates have been reported, both in computational and experimental studies [10,12,13,19,20,26]. Also, 3ʹ tRF seeds have been found computationally [12,13] in other organisms and experimentally in human [20] but we did not find this exact tRF isoform in CLASH chimeras. We know of no reports of central binding sites, sites at multiple locations or those extending across 2/3 of the length of a tRF. We note that the sites we detected reflect the existing sampling of tRFs and targets in the CLASH dataset and might change somewhat as the full set of targets becomes known.
Our predicted interaction sites were in agreement with the experiment for all three tRFs, for which the minimal 5ʹ seeds have been recently validated [26]. However, we observed variations in the length of the seeds we identified computationally, compared to the uniform size proposed in that work. For distinct Cys tRF isoforms we saw further variability in the interaction site, and the longest motif extended further towards the 3ʹ end of a tRF (Fig. S4). Additionally, for Leu-AAG we observed that the enriched motif was present both at the 5ʹ and 3ʹ end of the tRF. It is worth noting that the experimental data (see Fig. 4A in Kuscu et al. [26]) also show support for binding over a part of the 3ʹ instance of this motif.
Our analysis was based on finding motifs in multiple targets of individual tRFs, thus we focused on the functionality related to target binding. In terms of chimera yield, MFE and detecting seed regions, our results are overwhelmingly in favour of a target-binding functional role of tRF-3 isoforms, with lower support of tRF-5 (as was the case in rat, with 4-fold more seeds in tRF-3 vs tRF-5 [13]) and tRF-1 plus the weakest and often non-significant evidence for tRF-i. However, this does not completely negate a possible cellular role of the latter tRF class. It is conceivable that functionality of some tRFs requires not conserved but species-specific seed matches (hence no conserved motifs), or simply a displacement of other guide RNA from Ago (hence randomly paired chimeras and weaker MFE). For example, a very different mechanism, involving a formation of tetramolecular RNA G-quadruplexes, has been reported for certain tRFs [45].
There is evidence on the nuclear localization of Ago proteins [24,46] and tRF-5 [10] as well as potential roles of miRNAs in the nucleus [47]. Remarkably, in CLASH analysis we observed a high relative frequency of interaction of tRF-5 with intronic regions, compatible with such earlier evidence. However, we note that other tRF types also have intronic targets, thus they may also be present in the nucleus. Ago proteins in complexes with miRNAs and siRNAs have been shown to be actively involved in transcriptional regulation and pre-mRNA splicing [24,46–48], and tRFs may also be utilized by Ago in these processes. Changes in gene expression in the progeny of low-protein or high-fat diet fathers conferred by the tRF fragments in mouse sperm [49,50] may be related to such mechanisms.
Interestingly, our results suggest a possible role for tRFs in guiding Ago to the 5ʹ end of short introns, recently classified as agotrons [25]. Agotrons are identified based on their length (< 150 nts) and interaction with Ago2 in the first 30 nts of their 5ʹ end. We found that tRF-3 in chimeras next to the borders of agotrons in Ago1 and therefore are likely to be involved in agotron biogenesis or to interact with them.
Our finding of agotron-like targets of tRFs may shed further light on this intron family and its biogenesis and function. The 14 potential novel agotrons we identified have no overlap with reported mirtrons [51] although we cannot be certain whether they are in fact Dicer-independent agotrons or undiscovered Dicer-dependent mirtrons. An exception for miRNA to bypass the Dicer has been reported, whereby the pre-miR-451 could be loaded into Ago2 in mice and zebrafish and cleaved by its endonucleolytic slicer activity [52,53]. Although murine Ago1 may load but not cleave the pre-miR-451 [52], it is still possible that the pre-miRNA can function in Ago1 as an agotron-like species using its 5ʹ part, such example has been reported for pre-miR-151a [25]. Together, our observation of agotron-like targets of tRFs in Ago1 raises an intriguing possibility of a new mechanism for the Ago1 facilitated by tRFs to participate in excision of agotrons. Alternatively, tRFs may interact with already excised agotrons, targeting their 5ʹ end in Ago1 complex. Notably, we did not detect agotrons as guide sequences in Ago1 chimeras.
We also found other characteristics in addition to those reported (Ago associated, highly structured and GC enriched) for agotrons [25]. These specific GU-AG introns have G-rich 5ʹ-end following the consensus donor site sequence GURAGU (R = purine) which is responsible for interacting with the 5ʹ terminus of the U1 snRNA in the early splicing. Another observation for the agotrons is the C-rich 3ʹ-end followed by the consensus AG acceptor site. The upstream of the acceptor site, named as polypyrimidine (PY) tract, is usually U-rich in human and acts as a canonical binding site for the U2 snRNP auxillary factor U2AF65 [54]. The weak C-rich binding sites may require additional cis-elements or motifs such as the G-rich or G-rich motifs upstream of the weak PY tract [54]. The G-rich 5ʹ end and C-rich 3ʹ end have also been observed for specific groups of mirtrons (those hairpin boundaries are intron boundaries) reported in [51]. Hence, our finding of tRFs targeting the 5ʹ end of the introns in CLASH may indicate that these unexpected molecules as well as Ago1 facilitate the splicing of specific introns without strong U2AF65 binding sites. Given the mirtron hairpin example [51], it is tempting to speculate that the C-rich motif of tRFs (Fig. S2) may be interacting with potential hairpin elements in agotrons in this process.
Unlike miRNAs, tRFs are produced from tRNA genes, which have a very different function. Could this function or its evolutionary consequences (e.g. codon usage) affect tRF generation? In addition to showing no correlation between the tRNA and tRF levels, we checked if there was a link between the codon utilization in transcripts and the transcript being targeted by the respective tRFs. We could not detect any clear connection (e.g. increased or decreased codon usage in the targets) between these two measures. That is further illustrated by a comparison of the codon usage plots for the targets of the three tRFs with identical anticodon, CysGCA (Fig. S5). The largest differences between these transcripts occur in unrelated codons, while the usage of TGC codon itself shows clearly distinct trends between the targets of all three tRFs (below, equal or higher than background, respectively). Thus, at least in the CLASH screen, we found no evidence of a consistent over- or underutilization of a tRF codon the corresponding targets.
In conclusion, we note that our results strongly support the emerging complex regulatory functionality of tRFs and demonstrate the possibility of inferring the seed regions and mechanisms of tRF/target interactions computationally. We are aware of only a few human tRF interaction sites described and validated to some extent since the report of tRFs by Lee at al [6]. Including the three of our bioinformatic predictions matching validated seed locations, we provide here integrated evidence for 26 binding regions. The motifs/seed sequences we report can be directly assayed for tRF binding or utilized for predicting and validating additional potential tRF targets.
Materials and methods
CLASH data analysis
CLASH data for HEK293 cells were downloaded from the GEO database (GSE50452) [22]. We used fastx_toolkit 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/) to remove barcode and adapter sequences and collapse identical reads. We used an in house developed aligner script to identify tRFs from hybrid reads, allowing no mismatches, indels or end dissimilarities and giving preference to longer tRF isoforms. In detail, the aligner determines if a hybrid read starts with a known tRNA sequence and checks if the next nucleotide can still be part of the tRF sequence, stopping at the first mismatch. This way, the longest tRF isoform (≥16 nts) is identified as the guide sequence and the remainder of the hybrid read is considered the targeted sequence.
For tRF-5 we selected matches that started within the first 5 nucleotides of known tRNA sequences. For tRF-3, we selected matches that ended within the 5 nucleotides at the end of mature tRNA sequences (including the additional CCA). For tRF-1, we selected matches that ended within 3 nts to 40 nts of the tRNA trailer sequences (to distinguish them from tRF-3). All the identified tRF containing hybrids were confirmed not to be full tRNAs or pre-tRNAs by running blastn, word size 7, default scoring matrix against the union of tRNA sequences from two independent databases [2,55]. The portion of the hybrid read following the tRF sequence was considered the targeted sequence and it was searched against the human transcriptome [56] and the human genome (hg38) using blastn, word size 7, default scoring matrix and 10 maximum hits. Reads were considered chimeras if a hit had an e-value less than or equal to 0.1 and the combined length of the targeted sequence and the tRF sequence was greater than or equal to 75% of the total length of the chimeric read. Chimeras supported by at least two different reads were included in the analysis. Chimeras containing rDNA targets (105 reads) were excluded to minimize bias in the results.
Small RNA sequencing and hydro-tRNAseq data analysis
Hydro-tRNAseq data for HEK293 cells and small RNA sequencing data from whole cytoplasmic fraction of HEK293 cells were downloaded from the GEO database (GSE95683 and GSE75136) [29,30]. fastx_toolkit 0.0.13 was used to remove adapter sequences and collapse identical reads. Sequencing read alignments were performed using bowtie 1.1.1 aligner (http://bowtie-bio.sourceforge.net/manual.shtml). We aligned the sequenced reads against the human genome (version hg38) and also to the union of human tRNA sequences from two independent databases [2,55]. For each replicate, the raw read counts were normalized by the total number of reads that mapped to the human genome. For hydro-tRNAseq we allowed up to two mismatches as in the original publication [29].
Hybridization pattern analysis
We used RNAhybrid 2.1.2 [28] with default parameters to calculate minimum free energy for observed tRF/target RNA interactions and for random controls. To examine the binding mode of tRFs, we utilized the secondary structures for unique tRF/target chimeras obtained using RNAhybrid. We encoded each nucleotide across the tRF/target RNA chimera as 0 (if it was predicted not to bind) or 1 (if it was predicted to bind with a nucleotide from the target RNA) and we performed clustering analysis for the most abundant isoforms (with regards to fragment length) for each type of tRFs. We used scikit-learn (http://scikit-learn.org/) to perform unsupervised clustering using k-means algorithm. To explore the functional difference of tRFs with specific interaction pattern, we selected the genes targeted by tRFs in each cluster and performed the Gene Ontology (GO) analysis [57] to find enriched terms in the target genes list.
Seed and target motif enrichment analysis
In order to identify enriched motifs within tRF targets (Fig. S4), we selected for every tRNA gene a representative major isoform of each tRF type if they were identified on the tRNA gene. tRF isoforms of the same type and same tRNA gene but located > 5 nt away (based on the start and end coordinates) were classified into different group using Wards’ minimum variance clustering method. We took into account tRFs with at least 5 unique target genes according to CLASH data. We used MEME [36] with default parameters (e-value < 0.01) and searched for enriched motifs longer than 5 nucleotides across the longest targeted sequences for each target gene for a given tRF isoform. Next, we used FIMO [37] with default parameters (p-value < 0.001) to match such over-represented motifs back to tRF sequences to find potential seeds/interaction sites. To search for gapped motifs in the same tRFs and targeted sequences, we also used GLAM2 [39] with default parameters and found the reverse complementary match of the motif with the highest score on the tRF sequence using GLAM2SCAN [39] with default parameters.
To investigate if the seed of tRFs have enriched matches in the conserved genomic regions, we generated 7-mer sub-sequences of tRFs by applying a 7-nt sliding window and shifting by one nucleotide from the 5ʹ to the 3ʹ end. We calculated the number of exact matches for each of these sub-sequences in the 5ʹUTR, CDS and 3ʹUTR of all human protein coding genes that were conserved across 100 vertebrates. The frequency of matches in human genome (hg38, masked repeats excluded) was used as a background frequency.
Supplementary Material
Acknowledgments
Work in A.G.’s lab is supported by the grants from the National Science Foundation (DBI-1458202), National Institutes of Health (R15CA220059) and Rutgers GAIA.
Funding Statement
This work was supported by the Rutgers, The State University of New Jersey [GAIA].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Abe T, Inokuchi H, Yamada Y, et al. tRNADB-CE: tRNA gene database well-timed in the era of big sequence data. Front Genet. 2014;5:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chan PP, Lowe TM.. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37(Database issue):D93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fu H, Feng J, Liu Q, et al. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583(2):437–442. [DOI] [PubMed] [Google Scholar]
- [4].Thompson DM, Parker R. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J Cell Biol. 2009;185(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yamasaki S, Ivanov P, Hu G-F, et al. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee YS, Shibata Y, Malhotra A, et al. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009;23(22):2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saikia M, Krokowski D, Guan B-J, et al. Genome-wide identification and quantitative analysis of cleaved tRNA fragments induced by cellular stress. J Biol Chem. 2012;287(51):42708–42725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Telonis AG, Loher P, Honda S, et al. Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget. 2015;6(28):24797–24822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pliatsika V, Loher P, Telonis AG, et al. MINTbase: a framework for the interactive exploration of mitochondrial and nuclear tRNA fragments. Bioinformatics. 2016;32(16):2481–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kumar P, Anaya J, Mudunuri SB, et al. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014;12:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shigematsu M, Honda S, Kirino Y. Transfer RNA as a source of small functional RNA. J Mol Biol Mol Imaging. 2014;1(2). [PMC free article] [PubMed] [Google Scholar]
- [12].Karaiskos S,Naqvi AS, Swanson KE, et al. Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol Direct. 2015;10(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Karaiskos S, Grigoriev A. Dynamics of tRNA fragments and their targets in aging mammalian brain. F1000Res. 2016;5:2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Anderson P. Ivanov, tRNA fragments in human health and disease. FEBS Lett. 2014;588(23):4297–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cole C, Sobala A, Lu C, et al. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA. 2009;15(12):2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Z, Ender C, Meister G, et al. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012;40(14):6787–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miyoshi K, Miyoshi T, Siomi H. Many ways to generate microRNA-like small RNAs: non-canonical pathways for microRNA production. Mol Genet Genomics. 2010;284(2):95–103. [DOI] [PubMed] [Google Scholar]
- [18].Pekarsky Y, Balatti V, Palamarchuk A, et al. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc Natl Acad Sci U S A. 2016;113(18):5071–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maute RL, Schneider C, Sumazin P, et al. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Q, Lee I, Ren J, et al. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther. 2013;21(2):368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shin C, Nam J-W, Farh KK-H, et al. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010;38(6):789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Helwak A, Kudla G, Dudnakova T, et al. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153(3):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schraivogel D, Meister G. Import routes and nuclear functions of Argonaute and other small RNA-silencing proteins. Trends Biochem Sci. 2014;39(9):420–431. [DOI] [PubMed] [Google Scholar]
- [24].Taliaferro JM, Aspden JL, Bradley T, et al. Two new and distinct roles for Drosophila Argonaute-2 in the nucleus: alternative pre-mRNA splicing and transcriptional repression. Genes Dev. 2013;27(4):378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hansen TB, Venø MT, Jensen TI, et al. Argonaute-associated short introns are a novel class of gene regulators. Nat Commun. 2016;7:11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kuscu C, Kumar P, Kiran M, et al. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA. 2018;24(8):1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(suppl_2):W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gogakos T, Brown M, Garzia A, et al. Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017;20(6):1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wissink EM, Fogarty EA, Grimson A. High-throughput discovery of post-transcriptional cis-regulatory elements. BMC Genomics. 2016;17:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hausser J, Syed AP, Bilen B, et al. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013;23(4):604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee I, Ajay SS, Yook JI, et al. New class of microRNA targets containing simultaneous 5ʹ-UTR and 3ʹ-UTR interaction sites. Genome Res. 2009;19(7):1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khorkova O, Myers AJ, Hsiao J, et al. Natural antisense transcripts. Hum Mol Genet. 2014;23(R1):R54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Werner A, Swan D. What are natural antisense transcripts good for? Biochem Soc Trans. 2010;38(4):1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wight M, Werner A. The functions of natural antisense transcripts. Essays Biochem. 2013;54:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bailey TL, Boden M, Buske FA, et al. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(suppl_2):W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. [DOI] [PubMed] [Google Scholar]
- [39].Frith MC, Saunders NFW, Kobe B, et al. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol. 2008;4(5):e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kawaji H, Nakamura M, Takahashi Y, et al. Hidden layers of human small RNAs. BMC Genomics. 2008;9:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mahlab S, Tuller T, Linial M. Conservation of the relative tRNA composition in healthy and cancerous tissues. RNA. 2012;18(4):640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martens-Uzunova ES, Olvedy M, Jenster G. Beyond microRNA – novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013;340(2):201–211. [DOI] [PubMed] [Google Scholar]
- [43].Luo S, He F, Luo J, et al. Drosophila tsRNAs preferentially suppress general translation machinery via antisense pairing and participate in cellular starvation response. Nucleic Acids Res. 2018;46(10):5250–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gebetsberger J, Zywicki M, Künzi A, et al. tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea. 2012;2012:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lyons SM, Gudanis D, Coyne SM, et al. Identification of functional tetramolecular RNA G-quadruplexes derived from transfer RNAs. Nat Commun. 2017;8:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ameyar-Zazoua M, Rachez C, Souidi M, et al. Argonaute proteins couple chromatin silencing to alternative splicing. Nat Struct Mol Biol. 2012;19:998. [DOI] [PubMed] [Google Scholar]
- [47].Catalanotto C, Cogoni C, Zardo G. MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci. 2016;17(10):1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Allo M, Buggiano V, Fededa JP, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol. 2009;16(7):717–U43. [DOI] [PubMed] [Google Scholar]
- [49].Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen Q, Yan M, Cao Z, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351:397–400. [DOI] [PubMed] [Google Scholar]
- [51].Wen J, Ladewig E, Shenker S, et al. Analysis of nearly one thousand mammalian mirtrons reveals novel features of dicer substrates. PLoS Comput Biol. 2015;11(9):e1004441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cheloufi S, Dos Santos CO, Chong MMW, et al. A Dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465(7298):584–U76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cifuentes D, Xue H, Taylor DW, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328(5986):1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Murray JI, Voelker RB, Henscheid KL, et al. Identification of motifs that function in the splicing of non-canonical introns. Genome Biol. 2008;9(6):R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jühling F, Mörl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37(Database issue):D159–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Aken BL, Achuthan P, Akanni W, et al. Ensembl 2017. Nucleic Acids Res. 2017;45(D1):D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.