

ABSTRACT
Glioma is the most malignant primary brain cancer which frequently occurred in adults. In recent years, long-non coding RNAs (lncRNAs) have been demonstrated to play pivotal roles in human cancers. However, the role of most lncRNAs in gliomagenesis has not been probed. Presently, through TCGA, a novel lncRNA LINC01198 was found to be up-regulated and associated with clinical outcomes in glioblastoma multiforme (GBM). In our study, LINC01198 was proved to be up-regulated in glioma cell lines, and silenced LINC01198 curbed glioma cell proliferation and accelerated cell apoptosis. Importantly, we corroborated that LINC01198 activated the PI3 K/AKT pathway to aggravate glioma progression by targeting PIK3 CA and PTEN. Subsequently, LINC01198 was validated to localize in both cytoplasm and nucleus of glioma cells. Through mechanistic exploration, we illustrated that LINC01198 increased PIK3CA expression by sponging miR-129-5p in the cytoplasm. Furthermore, PTEN was transcriptionally repressed by REST/RCOR1/HDAC2 complex. More importantly, LINC01198 accelerated the assembly of REST/RCOR1/HDAC2 complex and recruited such complex to PTEN promoter so as to impair PTEN expression in glioma. Finally, we further verified that LINC01198 hindered glioma tumour growth in vivo through AKT-dependent manner. Jointly, LINC01198 activates PI3 K/AKT signalling to exert oncogenic function in gliomagenesis by regulating PIK3CA and PTEN, which highlights a new approach for glioma treatment.
KEYWORDS: LINC01198, gliomagenesis, PI3K/AKT pathway, PIK3CA, PTEN
Introduction
Glioma is recognized as a frequently diagnosed malignancy of central nervous system (CNS) and originates from glial cells of the spine or brain [1]. Glioma, with a high rate of both recurrence and mortality, accounts for approximately 80% of all malignant intracranial tumours [2]. The progression of gliomagenesis is biologically complicated, including pathophysiological changes, gene expression changes and signalling pathway disorders [3,4]. To date, despite the advance in multimodal treatments encompassing surgical extirpation, conventional chemotherapy and local irradiation, glioma remains rarely curable due to deficiency of novel effective therapies [5]. Thus, a better understanding of the molecular mechanism involved in gliomagenesis is of great significance for proposing new strategies to lengthen the life span of patients with glioma.
Long non-coding RNAs (lncRNAs) are the evolutionarily conserved RNA transcript longer than 200 nucleotides without protein-coding ability [6–8]. They exert essential functions in multiple aspects of glioma cellular biological behaviours [9,10]. Existing studies indicated that lncRNAs could modulate gene expressions in various manners, such as transcriptional regulation, post-transcriptional modulation and chromatin modification [11,12]. Long intergenic non-protein coding RNA 1198 (LINC01198) is a newly-found lncRNA located on chromosome 13. A recent report indicated that the expression of LINC01198 increases with glioma tumour grade and also has prognostic value for glioma patients [13]. However, the functional and potential mechanistic role of LINC01198 in gliomagenesis remains to be uncovered.
In the current study, we probed the functional effect of LINC01198 on cell proliferation and apoptosis in glioma, and later explored the potential mechanism. It was found that LINC01198 aggravated gliomagenesis through activating PI3 K/AKT pathway.
Materials and methods
Cell culture and reagent
Normal human astrocytes (NHAs), human embryonic kidney cell (HEK-293 T) and human GBM cells (T98 G, LN229, U87, U251) were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) adding Penicillin-Streptomycin Solution (Solabio, Shanghai, China) plus 10% foetal bovine serum (FBS; PAN-Biotech, Aidenbach, Germany) at 37°C in 5% CO2 with an exchange of medium every 3 days. SC79 were purchased from MedChemExpress (Monmouth Junction, NJ, USA).
Cell transfection
Specific shRNAs against LINC01198 (shLINC01198#1 and shLINC01198#2) and their corresponding NC, along with the pcDNA3.1 vector targeting LINC01198 or HDAC2 or REST or RCOR1 and the empty vector, were constructed into the pLKO.1 vector (Addgene #10,878) by Genechem (Shanghai, China). Additionally, miR-129-5p mimics and NC-mimics were similarly acquired from GenePharma (Shanghai, China). Thereafter, above lentiviral constructions were individually transfected into U87 or U251 cells for 48 h via Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA). Finally, transfected cells were processed with 48 h of puromycin treatment to obtain stable subpopulations.
qRT-PCR
Relative expression of genes in indicated cells or xenografts was assessed via qRT-PCR analysis. In brief, total RNA was acquired by using TRIzol Reagent (Invitrogen). Thereafter, RNA was reversely transcribed into first-stranded cDNA utilizing PrimeScript RT Master Mix (Takara, Kusatsu, Japan), followed by carrying out qRT-PCR with an SYBR Premix Ex Taq kit (Takara). U6 or GAPDH was adopted as references and 2−ΔΔCt approach was applied for calculating relative expression.
Cell viability assay
MTT assays were performed to determine cell viability. Cells under indicated conditions were added with MTT solution (Bio-Rad, Hercules, CA, USA) after incubating for 0, 24, 48, 72, 96 h. Upon aspiration of the supernatant, formazan product was dissolved in dimethyl sulphoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA). Thereafter, cell viability was read via a microplate spectrophotometer (BioTek, Winooski, VT, USA) through detecting the absorbance at 490 nm.
EdU assay
Cell proliferation under diverse conditions was examined by EdU assays. Indicated cells were added into 24-well plates and incubated with EdU (RiboBio, Guangzhou, China). Upon that, cells were fixed by 4% paraformaldehyde (PFA; Solarbio) and then dyed by Apollo Dye Solution (RiboBio). Nucleic acid was stained applying Hoechst 33,342 (Sigma-Aldrich). EdU-positive cells with red and blue signals were observed and imaged under the inverted fluorescence microscope (Carl Zeiss, Jena, Germany). The cell proliferative ratio was assessed as the rate of EdU-positive cells to Hoechst-stained cells.
Colony forming assay
Colony formation assay was conducted to detect cell proliferation as well. Transfected U87 or U251 cells treated with or without SC79 were plated in 6-well culture dishes and cultured for 14 days. Upon 15 min fixation with 4% formaldehyde (Sigma-Aldrich), colonies with over 50 cells were dyed in crystal violet (Sigma-Aldrich). Twenty minutes later, the coloured colonies were counted manually.
TUNEL assay
TUNEL assay was carried out in order to measure apoptosis in glioma cells facing different situations. Apoptotic cells were fixed for 25 min by formaldehyde and permeabilized for 5 min in TritonX-100 (Sigma-Aldrich). After that, cells were equilibrated by Equilibration buffer (Sigma-Aldrich). Cells were labelled by TdT reaction mix (Beyotime, Shanghai, China). Subsequently, SSC buffer (Sigma-Aldrich) was applied for stopping the reaction. Cell nuclei were stained by the use of DAPI (Sigma-Aldrich). Images were attained by a fluorescence microscopy (Leica, Mannheim, Germany). Afterwards, per cent of TUNEL-positive cells was evaluated via the number of TUNEL-stained cells divided by total cell number (number of DAPI-coloured cells).
Western blot
The level of indicated proteins in different samples was analysed via Western blot. Cells were lysed using RIPA lysis buffer (KeyGEN, Nanjing, China) and then the protein concentration of each sample was quantified via a BCA protein assay kit (Thermo Fisher Scientific). Thereafter, separation of protein extract was conducted with sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad), after which protein was transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corporation, Billerica, MA, USA). Blocking of membranes was accomplished with 5% non-fat milk, after which the membranes were incubated overnight at 4°C with antibodies against AKT (ab179463, Abcam, Cambridge, USA), p-AKT (ab131443 Abcam), Bcl-2 (ab32124, Abcam), Bax (ab32503, Abcam), Caspase 3 (ab13847, Abcam), Cleaved Caspase 3 (ab2302, Abcam), REST (ab21635, Abcam), RCOR1 (SAB2104148, Sigma-Aldrich), HDAC2 (ab32117, Abcam), PIK3CA (ab40776, Abcam), PTEN (ab32199, Abcam), EGFR (ab52894, Abcam), PI3 K (GW21071, Sigma-Aldrich), PIK3CG (K3641, Sigma-Aldrich), PIK3CB (AV32117, Sigma-Aldrich) and GAPDH (ab245356, Abcam), individually. Membranes were sequentially incubated with secondary antibodies at room temperature for 1 h. After being rinsed by TBST solution for three times, signals were visualized with an ECL kit (Thermo Fisher Scientific).
Fluorescence in situ hybridization (FISH)
FISH assay was carried out to identify the subcellular localization of LINC01198 in glioma cells by the use of the RiboTM Fluorescent in situ Hybridization Kit (Ribo, Guangzhou, China) plus the RiboTM lncRNA FISH Probe Mix as previously described [14].
RIP assay
RIP assay was carried out to determine RNAs bound to indicated proteins via a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore Corporation). In short, cells were lysed via RIP Lysis Buffer. Meanwhile, pre-rinsed magnetic beads were incubated with antibodies against Ago2 (Millipore Corporation), REST, RCOR1, HDAC2 and IgG (Millipore Corporation) for 30 min at room temperature. IgG group was the negative controls, whereas input (10% cell lysates) acted as positive controls. Afterwards, cell lysate was added into tubes with magnetic bead-antibody complexes in 900 μl of RIP Immunoprecipitation Buffer and then incubated at 4°C overnight. Finally, the precipitated RNAs were purified and examined by qRT-PCR.
RNA pull-down assay
The interactions between miRNAs and LINC01198 (or PIK3CA mRNA) were assessed by conducting RNA pull-down assay. The Pierce RNA 3ʹ End Desthiobiotinylation Kit (Thermo Fisher Scientific) was used to label sequences of miR-129-5p-WT/Mut or LINC01198 sense/antisense. RNA pull-down assay was conducted via a Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific). Briefly, cell lysates were incubated with magnetic beads pre-coated with biotinylated RNAs overnight (with 10% of cell lysates served as input). Then, the RNA-binding protein complexes were washed and eluted, and the detection of bound RNAs or proteins was accomplished via qRT-PCR or Western blot.
Luciferase reporter assay
Luciferase reporter assay was employed to analyse the impact of genes on their potential targets. LINC01198-WT/Mut or PIK3CA-WT/Mut was inserted into the pmirGLO dual-luciferase plasmid (Promega, Madison, WI, USA) to construct pmirGLO-LINC01198-WT/Mut or pmirGLO-PIK3CA-WT/Mut. The pmirGLO-LINC01198-WT/Mut was co-transfected into HEK-293 T cells with miR-129-5p mimics or NC-mimics. The pmirGLO-PIK3CA-WT/Mut was co-transfected into HEK-293 T cells with miR-129-5p mimics or miR-129-5p mimics+pcDNA3.1/LINC01198 or NC-mimics. PTEN promoter or PIK3CA promoter was sub-cloned into pGL3 reporter vector (Invitrogen) to form pGL3-PTEN promoter or pGL3-PIK3CA promoter. The pGL3-PTEN promoter was co-transfected with pcDNA3.1/REST or pcDNA3.1/RCOR1 or pcDNA3.1/HDAC2 or pcDNA3.1 into HEK-293 T cells. The pGL3-PTEN promoter or pGL3-PIK3CA promoter was co-transfected with shLINC01198 or shCtrl into U87 or U251 cells. Luciferase activities were explored through the Dual-Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation (ChIP)
The interaction between transcription factors and the promoter of their targets was validated through ChIP assay. The Global Histone H3 Acetylation Assay Kit (Epigentek, Farmingdale, NY, USA) was utilized to conduct ChIP. Briefly, after sonication of cross-linked chromatin DNA into 200–1000-bp fragments, fragmented chromatin was, respectively, immunoprecipitated with antibodies against RNA pol Ⅱ, HDAC2, H3 K27ac, REST, RCOR1 and IgG. Normal mouse IgG was the negative control and input containing 10% cell lysates was the positive control. RNA pol Ⅱ is an important protein for transcription initiation [15] and thus served as a positive control for REST/RCOR1/HDAC2 complex. Besides, the promoters of two genes chromosomally adjacent to PTEN, including MINPP1 and RNLS, were used as the controls for PTEN promoter. qRT-PCR was eventually performed to determine fragments precipitated in each group.
Co-immunoprecipitation (Co-IP)
Co-IP was carried out to assess interactions among REST, RCOR1 and HDAC2 in glioma cells in line with a previous description [16].
Tumour xenograft
The in vivo growth of glioma cells was assessed via in vivo xenograft experiments. Briefly, 12 nude mice (4-week-old) were acquired from SLAC Experimental Animal Center (Shanghai, China). U87 cells transfected with shCtrl, shLINC01198#1/#2 or shLINC01198#1/#2 plus myr/AKT were injected into the right limbs of mice. Tumours volume was evaluated every 4 days calculated by the formula: Volume = 0.5 × length × width [2]. After 4 weeks, mice were killed and tumour weight was measured. Tumour tissues were maintained for follow-up haematoxylin-eosin (HE) or immunohistochemical (IHC) staining.
IHC assay
Tumours obtained from in vivo experiments were fixated in 4% paraformaldehyde, dehydrated via ethanol solutions, inset in paraffin and finally sliced into 4-μm thickness. Afterwards, pieces were cultured with Ki67 antibodies overnight at 4°C, followed by cultivation with HRP-conjugated secondary antibodies for 1 h. Finally, all these sections were visualized and corresponding images were captured under the microscope (Olympus).
Statistical analysis
Data derived from thrice performed experiments were presented as mean ± standard deviation (SD). Statistical analyses were undergone by utilizing SPSS 22.0 (IBM, Armonk, NY, USA). Differences in groups were analysed with Student’s t-test and one-way or two-way ANOVA, as needed. P < 0.05 was deemed to indicate a statistically significant difference between groups.
Results
LINC01198 is a lncRNA that is overexpressed in gliomas
To understand the potential role of LINC01198 in glioma development, we were firstly curious about its expression patterns in glioma and normal conditions. As a result, UCSC (http://genome.ucsc.edu/) data suggested an interesting phenomenon that LINC01198 was always under-expressed in 52 of the 53 human normal tissues (except normal tests) (Fig. 1A). In subsequence, similar results were also represented by data from NCBI (https://www.ncbi.nlm.nih.gov) and NONCODE (http://www.noncode.org/) (Fig. 1B, C). Importantly, it was indicated that LINC01198 had no potential to encode protein itself, as a result of online Coding Potential Assessment Tool (CPAT) (http://lilab.research.bcm.edu/cpat/index.php). Moreover, TCGA database (http://gepia.cancer-pku.cn/) revealed that LINC01198 was significantly overexpressed in GBM (glioblastoma multiforme) and SKCM (skin cutaneous melanoma), whereas it was obviously down-regulated in TGCT (testicular germ cell tumours) (Fig. 1D). Furthermore, data from TCGA unveiled that LINC01198 was distinctly up-regulated in GBM and its up-regulation seemed to be closely associated with a poor clinical outcome of patients with GBM (Fig. 1E, F). Based on these data, we believed that LINC01198 might participate in the tumorigenesis and development of glioma.
Figure 1.
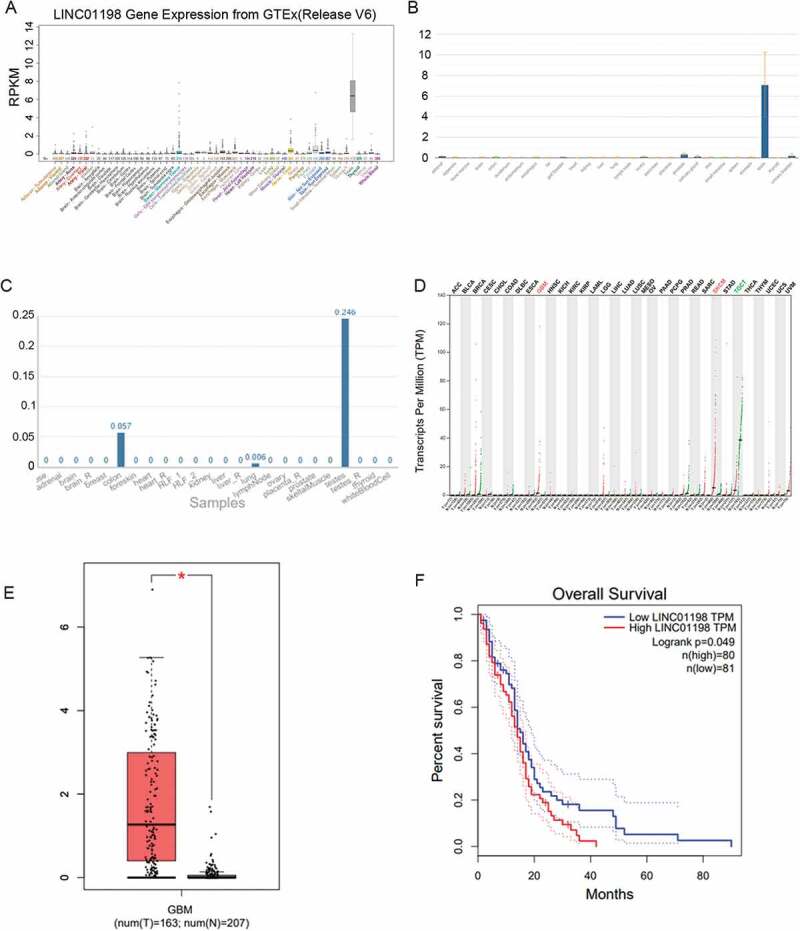
LINC01198 was up-regulated in GBM. (A–C) LINC01198 expression profiles in human normal tissues from UCSC, NCBI and NONCODE. (D) LINC01198 expression pattern in different cancer types. Data were obtained from TCGA. (E, F) LINC01198 expression in GBM tissues and its association with GBM prognosis were all obtained from TCGA database.
Silencing LINC01198 confines cell proliferation and induces cell apoptosis in glioma
For identifying LINC01198 function in glioma, we then evaluated its effect on the malignant phenotypes of glioma cells. Consistent with the results above, LINC01198 was highly expressed in glioma cell lines in comparison with that in the normal NHAs (Fig. 2A). Subsequently, successful silencing of LINC01198 by transfecting shRNAs targeting LINC01198 (shLINC01198#1 and shLINC01198#2) was validated through qRT-PCR (Fig. 2B). Thus, loss-of-function assays were performed in U251 and U87 cells with the highest endogenous LINC01198 expression. Resultantly, the viability of these two glioma cells was lessened by both shLINC01198#1 and shLINC01198#2 (Fig. 2C). Likewise, we also observed that LINC01198 knockdown hampered glioma cell proliferation, as the per cent of EdU-positive cells and the number of colonies were noticeably reduced in both U87 and U251 cells transfected with shLINC01198#1/2 (Fig. 2D, E). Conversely, depletion of LINC01198 by shLINC01198#1/2 markedly increased the proportion of TUNEL-positive cells and the protein levels of pro-apoptotic Bax and cleaved caspase 3 but reduced the protein level of anti-apoptotic Bcl-2 (Fig. 2F, G), demonstrating the stimulation of LINC01198 silence on glioma cell apoptosis. Altogether, LINC01198 aggravated cell proliferation and impaired cell apoptosis in glioma.
Figure 2.
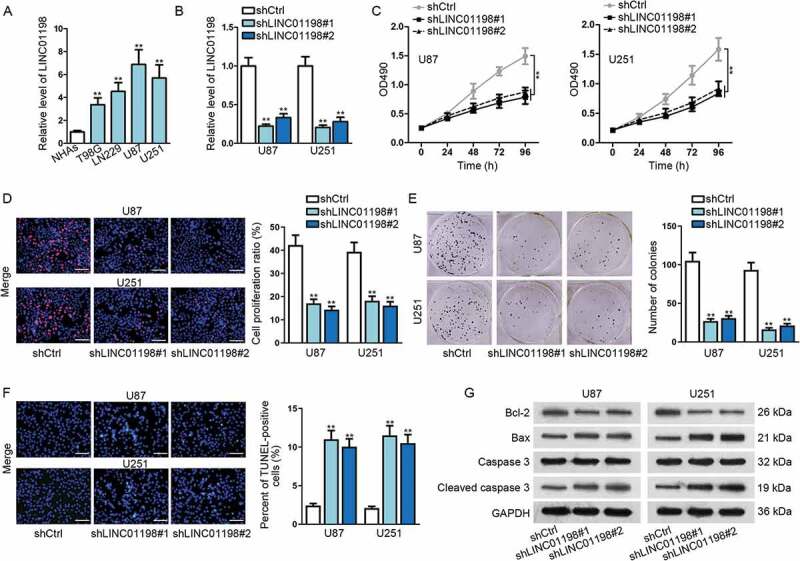
LINC01198 was highly expressed and promoted cell proliferation in glioma. (A) qRT-PCR results showed that LINC01198 was up-regulated in glioma cells. (B) The transfection efficiency of shLINC01198#1 or shLINC01198#2 was assayed in U87 and U251 via qRT-PCR. (C–F) MTT, EdU (scale bar = 100 μm), colony formation and TUNEL (scale bar = 100 μm) assays were performed to detect proliferative and apoptotic cells after silencing LINC01198 expression by shLINC01198#1/2. (G) Apoptosis-related proteins (Bcl-2, Bax and Caspase 3) were examined by Western blot in U87 and U251 cells with or without LINC01198 inhibition. **P < 0.01.
LINC01198 facilitates tumorigenesis in glioma by activating PI3 K/AKT pathway
PI3 K/AKT signalling is a well-known oncogenic pathway which has been reported to aberrantly activate in a variety of human malignancies, including glioma. Here, we wondered whether LINC01198 affected gliomagenesis through this pathway. Therefore, we detected the impact of LINC01198 depletion on AKT activation, the key process in activating PI3 K/AKT pathway. As a consequence, the levels of phosphorylated AKT (p-AKT) at T308 site and its downstream p-mTOR were notably diminished while those of total AKT and mTOR were unaffected in both U87 and U251 cells upon LINC01198 suppression, which was consistent with the result in groups under MK2206 (AKT inhibitor) treatment (Fig. 3A), implying that LINC01198 exerted functions in glioma development by regulating AKT activation. To further validate our assertion, an AKT activator (SC79) was applied to conduct rescue assays in LINC01198-silenced U87 and U251 cells. Undoubtedly, down-regulated LINC01198 repressed the activation of AKT signalling and such phenomenon was rescued in both the two cells under SC79 treatment (Fig. 3B). Intriguingly, both the restrained viability and proliferation in two glioma cells with LINC01198 down-regulation were restored by AKT activation (Fig. 3C, E), whereas ascending apoptosis caused by LINC01198 suppression was attenuated after activating AKT (Fig. 3F, G). According to these findings, we concluded that LINC01198 contributes to gliomagenesis via activating PI3 K/AKT signalling pathway.
Figure 3.

LINC01198 activated PI3 K/AKT signalling pathway in glioma. (A, B) The protein levels of AKT, p-AKT (T308), mTOR and p-mTOR in glioma cells under indicated transfections were tested by Western blotting. (C) The effect of SC79 on shLINC01198#1 or shLINC01198#2-affected glioma cell proliferation and apoptosis was measured by MTT, EdU (scale bar = 100 μm), colony formation, TUNEL (scale bar = 100 μm) and Western blot assays in both U87 and U251 cells. **P < 0.01.
LINC01198 regulates PI3 K/AKT pathway through modulating PIK3CA and PTEN
Then, we launched investigations on the in-depth mechanism by which LINC01198 affected PI3 K/AKT pathway. Firstly, the influence of LINC01198 on the expressions of several genes involving in PI3 K/AKT activation was examined by qRT-PCR and Western blot analyses. The results exhibited that LINC01198 had a positive effect on the expression (at both mRNA and protein levels) of PIK3CA but a negative impact on that of PTEN. However, the expressions of EGFR, PI3 K, PIK3CG, PIK3CB and AKT were not affected (Fig. S1A–C). More interestingly, knockdown of LINC01198 had no impact on the luciferase activity of PIK3CA promoter but significantly reduced that of PTEN promoter (Fig. S1D), implying LINC01198 regulated PIK3CA expression by a post-transcriptional manner but modulated PTEN expression at the transcriptional level. Further, we proved a direct interaction between LINC01198 and PTEN promoter (Fig. S1E), suggesting a high potential that LINC01198 modulated PTEN transcription in glioma. In consequence, we speculated that LINC01198 regulated PI3 K/AKT pathway by targeting PIK3CA and PTEN.
LINC01198 prompts PIK3CA expression in the cytoplasm of glioma cells through miR-129-5p sequestration
Given that lncRNAs exerted various functions dependent on their cellular localizations [17], we assessed the distribution of LINC01198 in glioma cells. According to the prediction of lncLocator (http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/), LINC01198 was indicated to exist in both the cytoplasm and the nucleus, and such prediction was confirmed in both U87 and U251 cells via FISH analysis (Fig. 4A). Besides, we previously indicated a post-transcriptional regulation of LINC01198 on PIK3CA in glioma. Meanwhile, reports have proposed that cytoplasmic lncRNAs regulate gene expression at the post-transcriptional level by sponging miRNAs [18]. Therefore, we guessed that LINC01198 might affect PIK3CA expression via a competing endogenous RNA (ceRNA) network.
Figure 4.
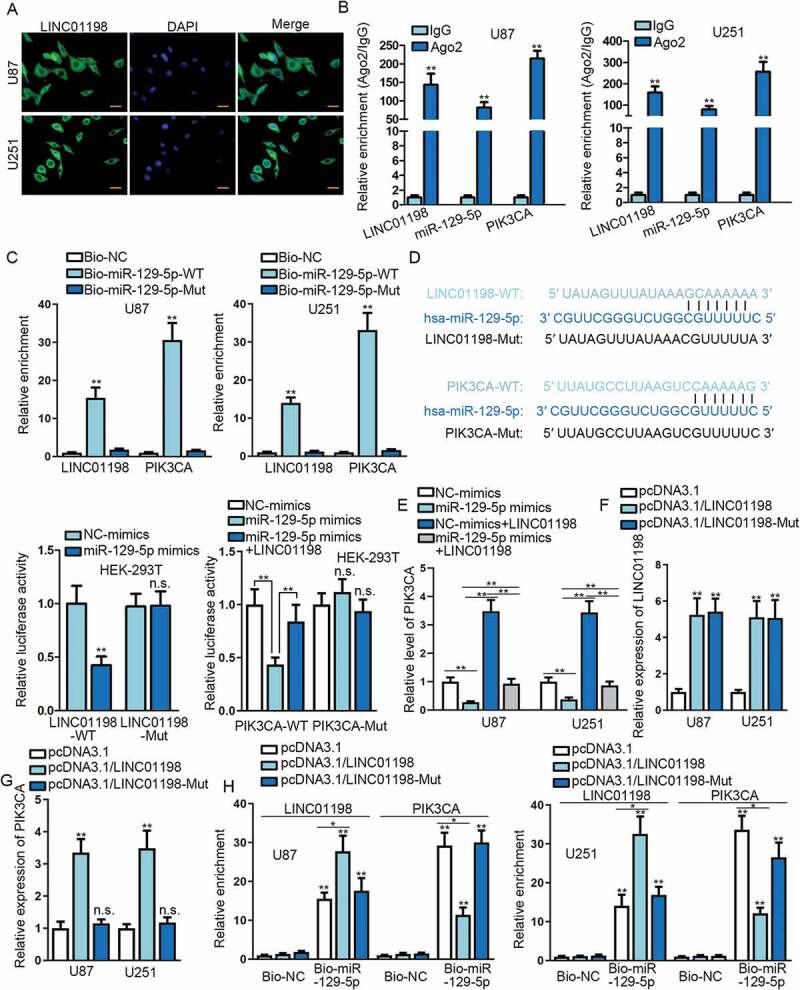
LINC01198 enhanced PIK3CA expression by sponging miR-129-5p in glioma. (A) The distribution of LINC01198 in glioma cells was examined by FISH assay (scale bar = 50 μm). (B–D) Ago2-RIP, RNA pull-down and luciferase reporter assays were conducted for evaluating the interaction between miR-129-5p and LINC01198 or PIK3CA in glioma cells. (E) The expression of PIK3CA in indicated U251 and U87 cells was measured by qRT-PCR. (F–G) The levels of LINC01198 and PIK3CA in U251 and U87 cells with overexpression of full-length or mutant LINC01198 were analysed by qRT-PCR. (H) RNA pull-down assay unveiled a decline of miR-129-5p-targeted PIK3CA mRNA upon up-regulation of full-length LINC01198 rather than mutated LINC01198. *P < 0.05, **P < 0.01, n.s. meant no significance.
Through bioinformatic analysis, 12 miRNAs were predicted to targeting PIK3CA by three online tools including TargetScan (http://www.targetscan.org/vert_72/), starBase (http://starbase.sysu.edu.cn/) and DIANA (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex). Meanwhile, we screened out 170 LINC01198-bound miRNAs by using DIANA tool (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex). Fortunately, after combining two subsets obtained above, two miRNAs (miR-129-5p and miR-9-3p) were suggested to potentially interact with both PIK3CA and LINC01198. Through qRT-PCR, it was turned out that miR-129-5p was generally down-regulated while miR-9-3p was up-regulated in glioma cell lines compared to normal NHAs (Fig. S1 F). Besides, the expression of miR-129-5p but not miR-9-3p was remarkably enhanced in LINC01198-silenced glioma cells (Fig. S1 G). Moreover, overexpression of miR-129-5p led to a decrease of LINC01198 expression in glioma cells (Fig. S1 H). In this regard, we speculated that LINC01198 sponged miR-129-5p to release PIK3CA mRNA from miR-129-5p-comprised RNA-induced silencing complexes (RISCs).
Since Ago2 is one of the core component proteins of RISCs [19], RIP experiment was carried out to precipitate RNAs involved in RISCs by using anti-Ago2 (with anti-IgG as negative control). As expected, LINC01198, miR-129-5p and PIK3CA mRNA were all obtained in anti-Ago2-immunoprecipitated compounds in both U87 and U251 cells (Fig. 4B). Moreover, the direct interaction of miR-129-5p with both LINC01198 and PIK3CA mRNA was verified by RNA pull-down assay (Fig. 4C). Further, the competition between LINC01198 and PIK3CA mRNA in interacting with miR-129-5p was proved by luciferase reporter assay (Fig. 4D). Of note, we also unveiled that PIK3CA expression was silenced by miR-129-5p overexpression whereas overexpression of LINC01198 increased PIK3CA expression in two glioma cells, with an interesting finding that LINC01198 up-regulation mitigated the inhibition of overexpressed miR-129-5p on PIK3CA expression in these cells (Fig. 4E).
Subsequently, we planned to further ensure the competition between LINC01198 and PIK3CA mRNA by interacting with miR-129-5p. Hence, pcDNA3.1 containing LINC01198 with (pcDNA3.1/LINC01198) or without sequences (pcDNA3.1/LINC01198-Mut) bound to miR-129-5p was applied to transfect glioma cells. As indicated in Fig. 4F, the expression of LINC01198 was evidently elevated in both U87 and U251 cells under the transfections of either pcDNA3.1/LINC01198 or pcDNA3.1/LINC01198-Mut (without apparent differences between these two groups), compared with vectors-transfected controls. However, the expression of PIK3CA was only boosted under the overexpression of full-length LINC01198 rather than up-regulation of mutated LINC01198 (Fig. 4G). More interestingly, such phenomenon could be explained that overexpression of wild-type LINC01198 reduced the interaction of PIK3CA mRNA with miR-129-5p whereas mutant LINC01198 lost this function (Fig. 4H). Collectively, LINC01198 boosts PIK3CA expression in glioma via competitively interacting with miR-129-5p in the cytoplasm.
PTEN is transcriptionally repressed by REST/RCOR1/HDAC2 complex
Previously, we uncovered that LINC01198 had a repressive impact on PTEN transcription through direct interaction; therefore, we wondered whether LINC01198 affected PTEN transcription via regulating certain transcription factors of PTEN. Interestingly, UCSC suggested that PTEN transcription might be modulated by REST, RCOR1 (co-repressor of REST, also called CoREST) and HDAC2 (Fig. S2A), and such three factors always assembled as a transcriptional repressor complex. Meanwhile, the RPISeq (http://pridb.gdcb.iastate.edu/RPISeq/) predicted that LINC01198 might interact with all the three proteins. Furthermore, we observed that there was no impact of LINC01198 on the expressions of these three factors (Fig. S2B). Hence, we assumed that LINC01198 might influence PTEN expression via interacting with REST, RCOR1 and HDAC2.
To prove the assumption above, we first investigated whether PTEN was regulated by REST, RCOR1 and HDAC2 in glioma cells. As anticipated, PTEN was negatively regulated by REST, RCOR1 and HDAC2, evidenced by that the expression level of PTEN was dramatically dampened no matter which one of the three genes was overexpressed (Fig. 5A, C). In addition, it was unmasked that all the three proteins interacted with PTEN promoter region, as the sequences that PTEN promoter could be observed in the complexes precipitated by anti-REST, anti-RCOR1 and anti-HDAC2, but not anti-IgG (Fig. 5D). Further, the luciferase activity of PTEN promoter was drastically lessened by up-regulation of REST, RCOR1 or HDAC2 (Fig. 5E), verifying the repression of REST, RCOR1 and HDAC2 on PTEN transcription. Moreover, we certified that HDAC2-binding to PTEN promoter was overtly strengthened by overexpressing one of the three genes, which resulted in a strikingly erased acetylation on histone 3 of PTEN promoter (Fig. 5F, G). In sum, PTEN is negatively regulated by REST/RCOR1/HDAC2 at the transcriptional level.
Figure 5.
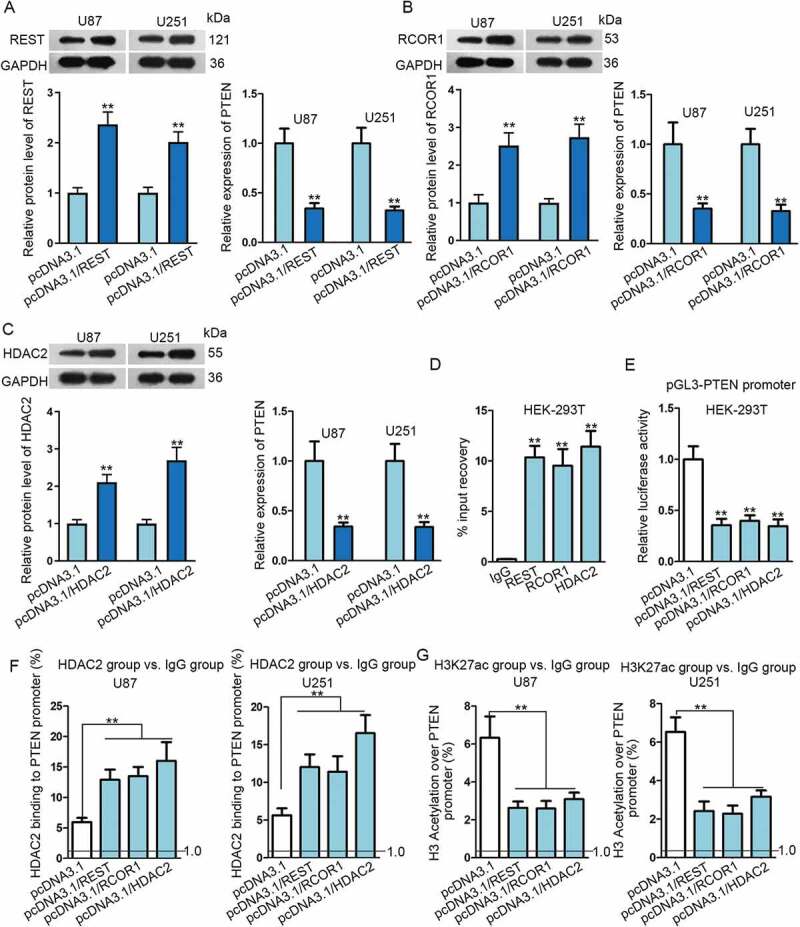
PTEN transcription was inhibited by the REST/RCOR1/HDAC2 complex in glioma. (A–C) Transfection efficiency of pcDNA3.1/REST, pcDNA3.1/RCOR1 or pcDNA3.1/HDAC2 in U87 and U251 cells were assayed via Western blot. Relative expression of PTEN in U87 and U251 cells transfected with above transfection plasmids was determined by qRT-PCR. (D–G) The inhibition of REST/RCOR1/HDAC2 complex on PTEN transcription was verified through ChIP and luciferase reporter assays. **P < 0.01.
LINC01198 reinforces the assembly of REST/RCOR1/HDAC2 complex to hinder PTEN expression
Recently, it has been largely proposed that lncRNAs acted as a molecular scaffold for the interaction between protein and protein (or DNA) [20–22]. Thereafter, we explored whether LINC01198 affected PTEN expression via REST/RCOR1/HDAC2 complex. The RIP assay confirmed that LINC01198 was observably precipitated by REST, RCOR1 and HDAC2 (Fig. 6A). In addition, RNA pull-down assay demonstrated that REST, RCOR1 and HDAC2 proteins were all pulled down by LINC01198 (Fig. 6B). These findings indicated the absolute interactions of LINC01198 with the three proteins. Interestingly, it was uncovered that the interactions among REST, RCOR1 and HDAC2 were blocked in both U87 and U251 cells responding to LINC01198 inhibition (Fig. 6C). Furthermore, the binding of REST, RCOR1 and HDAC2 to PTEN promoter was obviously impaired whereas that of RNA pol Ⅱ to PTEN promoter scarcely affected by LINC01198 depletion (Fig. 6D). More interestingly, such effects enhanced acetylation level on PTEN promoter but did not influence histone acetylation on promoters of either MINPP1 or RNLS, two genes chromosomally adjacent to PTEN (Fig. 6E). All findings illustrated that LINC01198-inhibited PTEN expression in glioma via promoting the assembly of REST/RCOR1/HDAC2 and thereby repressed PTEN transcription.
Figure 6.
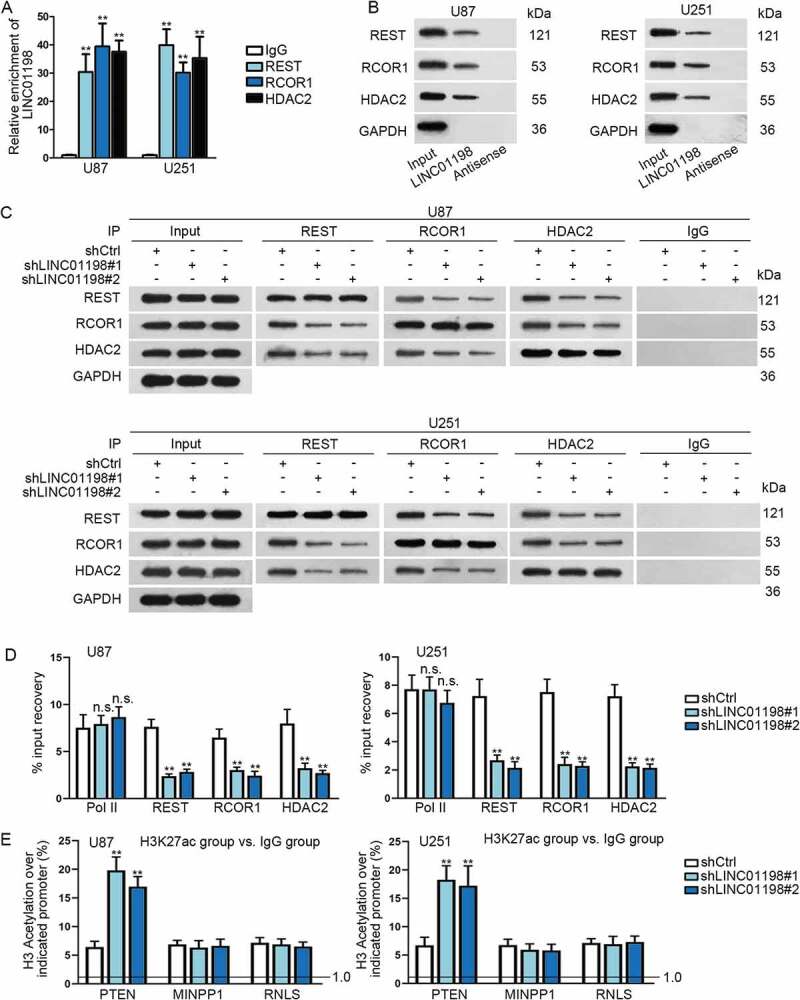
LINC01198 promoted the formation of REST/RCOR1/HDAC2 complex in glioma. (A–C) The interaction of LINC01198 with REST, RCOR1 or HDAC2 was explored via RIP assay, RNA pull-down and Co-IP assay. (D) ChIP assay was carried out to evaluate the impact of LINC01198 silence on the binding of REST, RCOR1 and HDAC2 to PTEN promoter. RNA pol Ⅱ was the negative control. (E) Influence of LINC01198 inhibition on histone acetylation of PTEN promoter was assessed by ChIP assays. Promoters of MINPP1 and RNLS were negative controls for PTEN promoter. **P < 0.01, n.s. meant no significance.
LINC01198 controls glioma cell growth in vivo by targeting AKT signalling
To further confirm whether the contribution of LINC01198 to glioma development was mediated via AKT pathway, we inoculated indicated U87 cells into the left flank of nude mice. As presented in Fig. 7A, the size of tumours from LINC01198-silenced glioma cells was greatly reduced and the tumour growth rate was sharply lowered compared to that of control group, whereas such reduction caused by LINC01198 depletion was countervailed by constitutive active AKT. Likewise, the weight of tumours with LINC01198 inhibition was much lighter than that of those without LINC01198 silence, but such phenomenon further offset under constitutive active AKT (Fig. 7B). In accordance with the observations in vitro assay, the expression of LINC01198 was markedly lessened in tumours under LINC01198 depletion, resulting in reduced expression of PIK3CA and increased level of PTEN, which was finally attenuated via constitutive AKT activation (Fig. 7C). Also, abated AKT activation along with decreased PIK3CA level and increased PTEN level in LINC01198-inhibited xenografts compared with control group were all restored by constitutive AKT activation (Fig. 7D). Moreover, LINC01198 suppression by both shLINC01198#1 and shLINC01198#2 notably reduced the staining of Ki67 (proliferation marker) in vivo, while constitutive AKT activation counteracted the impact of LINC01198 silence on Ki67 staining in above tumours (Fig. 7E). Thus, we concluded that LINC01198 impeded the growth of glioma cells in vivo via acting on AKT pathway.
Figure 7.

LINC01198 promoted glioma tumour growth in vivo through AKT-dependent pathway. (A) Representative images and the corresponding growth curves of tumours from mice injected with indicated U87 cells. (B) Weight of abovementioned tumours. (C–D) The expressions of LINC01198, PIK3CA and PTEN, as well as protein levels of PIK3CA, PTEN, AKT and p-AKT (T308) in these xenografts, were detected by qRT-PCR and Western blot, respectively. (E) Tumour histology was studied by HE staining (scale bar = 200 μm), while the proliferation marker Ki67 was estimated by IHC staining (scale bar = 200 μm). (F) Schedule model of the regulatory mechanism of LINC01198 in gliomagenesis. **P < 0.01.
Discussion
Over the decades, lncRNAs have been increasingly reported to be implicated in the initiation and development of numerous human cancers [23], including glioma [24,25]. In the present study, we discovered a novel lncRNA, LINC01198, which has never been probed in cancers as yet. Intriguingly, we found that LINC01198 was lowly expressed in most of the normal human tissues including normal brain tissues but obviously overexpressed and correlated with prognosis in GBM. Therefore, we focused on LINC01198 and wondered whether it participated in gliomagenesis. Results indicated that LINC01198 was a tumour promoter in glioma, indicating that LINC01198 was a potential target for glioma diagnosis and treatment, just as previously reported lncRNAs [26–28]. Further, we validated that LINC01198 played an accelerative role in glioma through activating PI3 K/AKT pathway, a well-identified oncogenic signalling which has been reported to aberrantly activate in glioma [29–31].
Subsequently, we aimed at digging out the detailed mechanism that how LINC01198 regulated PI3 K/AKT pathway. It was proved that LINC01198 affected the expressions of PIK3CA and PTEN which were upstream genes of this pathway. PIK3CA is an activator of PI3 K/AKT pathway [32,33], and its activation on this pathway has also been recognized in glioma [34]. Besides, the repression of PTEN on AKT activation has been well identified in multiple human diseases [35,36], including glioma [37,38]. Importantly, we disclosed that LINC01198 was a non-protein coding RNA that located in both the cytoplasm and the nucleus of glioma cells, and it affected PIK3CA expression via a post-transcriptional regulation but modulated PTEN at the transcriptional level. LncRNAs usually exert their disparate functions according to different subcellular localization [39,40]. In detail, cytoplasmic lncRNAs have been proposed as sponges of miRNAs to up-regulate protein-coding genes at post-transcriptional level [41,42], whereas nuclear lncRNAs are recognized to modulate gene transcription by coordinating regulatory proteins, such as RNA-binding proteins (RBPs), transcription factors and the co-activators or co-repressors [43–45]. Herein, we uncovered that LINC01198 acted as a ceRNA of PIK3CA in the cytoplasm by absorbing miR-129-5p, a validated tumour suppressor in glioma [46]. Meanwhile, we also revealed that PTEN was transcriptionally repressed by REST and REST-recruited REST/RCOR1/HDAC2 complex, which was reported to hinder gene transcription previously [47–49]. Of importance, we further testified that LINC01198 impeded PTEN expression in glioma by promoting the assembly of REST/RCOR1/HDAC2 complex in the nucleus and recruiting such complex to PTEN promoter. In addition, we certified that LINC01198 functioned as a scaffold for the formation and recruitment of REST/RCOR1/HDAC2 complex to PTEN promoter, and the scaffolding role of lncRNAs has already been reported by several researches [20–22,50,51].
In conclusion, we explained that LINC01198 interacted with miR-129-5p to boost PIK3CA expression in cytoplasm and interacted with REST/RCOR1/HDAC2 complex to repress PTEN expression in nucleus, leading to enhanced AKT activation and therefore accelerated gliomagenesis (Fig. 7F). Our research offered the first evidence that LINC01198 served as an oncogene in cancer and suggested LINC01198 as a novel potential target for glioma treatment as well. However, the significance of LINC01198 in clinical glioma treatment is still poorly evidenced, which needs to be further intensified in future studies.
Supplementary Material
Acknowledgments
Thanks very much for all the participators.
Disclosure statement
No conflicts of interest exist.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ludwig K, Kornblum HI.. Molecular markers in glioma. J Neurooncol. 2017;134:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chinnaiyan P, Kensicki E, Bloom G, et al. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012;72:5878–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dimitrov L, Hong CS, Yang C, et al. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 2015;12:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pan W, Li G, Yang X, et al. Revealing the potential pathogenesis of glioma by utilizing a glioma associated protein-protein interaction network. Pathol Oncol Res. 2015;21:455–462. [DOI] [PubMed] [Google Scholar]
- [5].Van Meir EG, Hadjipanayis CG, Norden AD, et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu Y, Zhou D, Li G, et al. Long non coding RNA-UCA1 contributes to cardiomyocyte apoptosis by suppression of p27 expression. Cell Physiol Biochem. 2015;35:1986–1998. [DOI] [PubMed] [Google Scholar]
- [7].Raveh E, Matouk IJ, Gilon M, et al. The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer. 2015;14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014;9:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839:1097–1109. [DOI] [PubMed] [Google Scholar]
- [10].Kiang KM, Zhang XQ, Leung GK. Long non-coding RNAs: the key players in glioma pathogenesis. Cancers (Basel). 2015;7:1406–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Misawa A, Takayama KI, Inoue S. Long non-coding RNAs and prostate cancer. Cancer Sci. 2017;108:2107–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kondo Y, Shinjo K, Katsushima K. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. 2017;108:1927–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang W, Yang F, Zhang L, et al. LncRNA profile study reveals four-lncRNA signature associated with the prognosis of patients with anaplastic gliomas. Oncotarget. 2016;7:77225–77236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Z, Ji N, Chen Z, et al. Next generation sequencing for long non-coding RNAs profile for CD4(+) T cells in the mouse model of acute asthma. Front Genet. 2019;10:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cinghu S, Yang P, Kosak JP, et al. Intragenic enhancers attenuate host gene expression. Mol Cell. 2017;68(104–17.e6). DOI: 10.1016/j.molcel.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin Y, Kong F, Li Y, et al. The tumor suppressor OVCA1 is a short half-life protein degraded by the ubiquitin-proteasome pathway. Oncol Lett. 2019;17:2328–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol IF11908. 2017;18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Qi X, Zhang D-H, Wu N, et al. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52:710–718. [DOI] [PubMed] [Google Scholar]
- [19].Iwasaki S, Sasaki HM, Sakaguchi Y, et al. Defining fundamental steps in the assembly of the Drosophila RNAi enzyme complex. Nature. 2015;521:533–536. [DOI] [PubMed] [Google Scholar]
- [20].Sun -T-T, He J, Liang Q, et al. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 2016;6:784–801. [DOI] [PubMed] [Google Scholar]
- [21].Zhang E, Han L, Yin D, et al. H3K27 acetylation activated-long non-coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 2017;45:3086–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sun M, Nie F, Wang Y, et al. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016;76:6299–6310. [DOI] [PubMed] [Google Scholar]
- [23].Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253. [DOI] [PubMed] [Google Scholar]
- [24].Deguchi S, Katsushima K, Hatanaka A, et al. Oncogenic effects of evolutionarily conserved noncoding RNA ECONEXIN on gliomagenesis. Oncogene. 2017;36:4629. [DOI] [PubMed] [Google Scholar]
- [25].Zheng J, Liu X, Wang P, et al. CRNDE promotes malignant progression of glioma by attenuating miR-384/PIWIL4/STAT3 axis. Mol Ther. 2016;24:1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [26].Yang C, Zheng J, Xue Y, et al. The effect of MCM3AP-AS1/miR-211/KLF5/AGGF1 axis regulating glioblastoma angiogenesis. Front Mol Neurosci. 2018;10:437. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Katsushima K, Natsume A, Ohka F, et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat Commun. 2016;7:13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang F, Shen Y, Zhang W, et al. An androgen receptor negatively induced long non-coding RNA ARNILA binding to miR-204 promotes the invasion and metastasis of triple-negative breast cancer. Cell Death Differ. 2018;25:2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lv D, Jia F, Hou Y, et al. Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 2017;77:6190–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Catanzaro G, Besharat ZM, Miele E, et al. The miR-139-5p regulates proliferation of supratentorial paediatric low-grade gliomas by targeting the PI3K/AKT/mTORC1 signalling. Neuropathol Appl Neurobiol. 2018;44:687–706. [DOI] [PubMed] [Google Scholar]
- [31].Lv B, Yang X, Lv S, et al. CXCR4 signaling induced epithelial-mesenchymal transition by PI3K/AKT and ERK pathways in glioblastoma. Mol Neurobiol. 2015;52:1263–1268. [DOI] [PubMed] [Google Scholar]
- [32].Tang J, Zhong G, Wu J, et al. SOX2 recruits KLF4 to regulate nasopharyngeal carcinoma proliferation via PI3K/AKT signaling. Oncogenesis. 2018;7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tang J, Zhong G, Zhang H, et al. LncRNA DANCR upregulates PI3K/AKT signaling through activating serine phosphorylation of RXRA. Cell Death Dis. 2018;9:1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schaefer T, Ramadoss A, Leu S, et al. Regulation of glioma cell invasion by 3q26 gene products PIK3CA, SOX2 and OPA1. Brain Pathol. 2019;29:336–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283. [DOI] [PubMed] [Google Scholar]
- [36].Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195. [DOI] [PubMed] [Google Scholar]
- [37].WWP1 . Inhibition rescues PTEN tumor-suppressive activity. Cancer Discov. 2019;9:825. [Google Scholar]
- [38].Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2006;13:84. [DOI] [PubMed] [Google Scholar]
- [39].Chen -L-L. Linking long noncoding RNA localization and function. Trends Biochem Sci. 2016;41:761–772. [DOI] [PubMed] [Google Scholar]
- [40].Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756. [DOI] [PubMed] [Google Scholar]
- [41].Song Y-X, Sun J-X, Zhao J-H, et al. Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat Commun. 2017;8:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang R, Zhang S, Chen X, et al. CircNT5E acts as a sponge of miR-422a to promote glioblastoma tumorigenesis. Cancer Res. 2018;78:4812–4825. [DOI] [PubMed] [Google Scholar]
- [43].Yap K, Mukhina S, Zhang G, et al. A short tandem repeat-enriched RNA assembles a nuclear compartment to control alternative splicing and promote cell survival. Mol Cell. 2018;72:525–40.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu Y-W, Kang C-M, Zhao -J-J, et al. LncRNA PLAC2 down-regulates RPL36 expression and blocks cell cycle progression in glioma through a mechanism involving STAT1. J Cell Mol Med. 2018;22:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen Q, Cai J, Wang Q, et al. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/β-catenin pathway by scaffolding EZH2. Clin Cancer Res. 2018;24:684–695. [DOI] [PubMed] [Google Scholar]
- [46].Diao Y, Jin B, Huang L, et al. MiR-129-5p inhibits glioma cell progression in vitro and in vivo by targeting TGIF2. J Cell Mol Med. 2018;22:2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [47].Paonessa F, Criscuolo S, Sacchetti S, et al. Regulation of neural gene transcription by optogenetic inhibition of the RE1-silencing transcription factor. Proc Natl Acad Sci U S A. 2016;113:E91–E100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gu H, Roizman B. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J Virol. 2009;83:181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ballas N, Battaglioli E, Atouf F, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. [DOI] [PubMed] [Google Scholar]
- [50].Li Y, Wang Z, Shi H, et al. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-myc. Cancer Res. 2016;76:293–304. [DOI] [PubMed] [Google Scholar]
- [51].Ma M, Zhang Y, Weng M, et al. lncRNA GCAWKR promotes gastric cancer development by scaffolding the chromatin modification factors WDR5 and KAT2A. Mol Ther. 2018;26:2658–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.