

ABSTRACT
RAS proteins function as molecular switches that regulate cellular growth by cycling between active GTP- and inactive GDP bound states. While RAS activity is modulated by factors (guanine nucleotide exchange and GTPase activating proteins) that control levels of active Ras-GTP, RAS proteins also undergo a number of post-translational modifications that regulate their function. One such modification is ubiquitylation. Monoubiquitylation of KRAS at lysine 147 (mUbRAS) enhances Ras activation and promotes signaling through the RAF and Phosphoinositide 3-Kinase (PI3K) signaling pathways. We have previously shown that mUbRAS leads to activation of RAS through a defect in GTPase activating protein (GAP) mediated downregulation, similar to the action of most oncogenic mutations. Consistent with these findings, we now show that mUbRASimpairsRAS binding to the p120 GAP catalytic domain. Mutations in activated G12V RAS that prevent ubiquitylaton at 147 show a decrease in tumorigenesis, suggesting that in addition to activating KRAS, monoubiquitylation at this site may promote downstream signaling and transformation. To investigate whether mUbRAS alters RAS effector interactions, we chemically ubiquitylated KRAS at residue 147 and characterized binding of mUbRAS to RAS binding domains (RBDs) from three distinct downstream effectors that play key roles in RAS-mediated transformation. Results from these studies show a decrease in binding of mUbRAS (7-10-fold) relative to the CRAF RAS Binding Domain (RBD), the catalytic subunit of Phosphoinositide 3-Kinase catalytic gamma (PI3Kcγ) and RALGDS RBD. Intriguingly, we find that mUbRAS shows greatly enhanced (> 40-fold) binding to the CRAF RBD when bound to GDP. These findings, taken together, suggest that mUbRASmay promoteactivation of RAS through a GAP defect, and facilitate RAF association and MAPK signaling in a nucleotide independent manner.
Keywords: RAS GTPase, ubiquitylation, allostery, effector, signal transduction
Introduction
RAS is one of the most frequently mutated proteins in human cancer, with activating mutants identified in ∼30% of tumors.1 Mutations in RAS identified in cancer and RASopathies, promote deregulated RAS signaling by populating the active GTP-bound state. Given the key role of RAS in cellular growth control, the nucleotide bound state of RAS is highly regulated. Once bound to GTP, RAS proteins can engage a number of downstream effectors and promote signaling through pathways that regulate cellular growth, differentiation and apoptosis.2 Binding of GTP, causes a conformational change in two regions of the protein, termed switch I (amino acids 30–37) and switch II (amino acids 60–75). The change in switch conformation facilitates higher affinity binding to GTPase activating proteins (GAPs) and downstream effectors. RAS is capable of hydrolyzing GTP at a slow rate to return to the inactive, GDP bound state. However, nucleotide hydrolysis is greatly accelerated through the action of GAPs.3 Consequently, GAPs are important regulatory proteins that help to limit the amount of activated (GTP–bound) RAS.3,4 Additionally, RAS proteins bind GDP and GTP with high affinity and their intrinsic rate of nucleotide dissociation is quite slow.5 Guanine nucleotide exchange (GEF) factors constitute a distinct class of RAS regulatory proteins, as they greatly stimulate the rate of GDP exchange for GTP to promote RAS activation.6 RAS proteins can also undergo a number of post-translational modifications (PTM) that regulate its function. While posttranslational modifications in the hypervariable domain promote membrane association critical for proper spatial/temporal control of RAS signaling, PTMs within the core guanine nucleotide-binding domain have also been shown to regulate RAS activity, but are less well understood.
Some of the PTMs reported for RAS occur at lysine residues, and include ubiquitylation and acetylation.7 While acetylation at lysine 104 is reported to downregulate RAS activity, monoubiquitylation of KRAS at lysine 147 (mUbRAS) upregulates RAS activity, signaling and tumorigenesis.8-10 Ubiquitylation is a highly regulated post-translational modification that can be reversed by deubiquitinating enzymes.11 The major site of KRAS monoubiquitylation (mUb) occurs at lysine 147.11 Sasaki et al. reported that mUb of KRAS increases GTP loading and activation of the RAF and Phosphoinositide 3-Kinase (PI3K) signaling.11 We followed up on these observations and showed that recombinant mUbRAS,generated using a disulfide ligation strategy, is GAP impaired.12 These findings indicate that mUbRAS can upregulate RAS activity through a GAP defect, similar to the action of most RAS oncogenic mutations. Interestingly, ligation of a PDZ domain to KRAS at position 147, in lieu of ubiquitin, retained the GAP defect12,13 , whereas ligation of a small peptide or change in linker length (> 2 amino acids) between RAS and ubiquitin retained GAP sensitivity.13 These results indicate that the GAP defect associated with mUbRAS has precise requirements on the linker length and protein size, and is not due to specific interactions between RAS and ubiquitin. We also observed differences in switch dynamics upon Ras ubiquitylation, leading to the intriguing possibility that ligation of ubiquitin allosterically regulates switch dynamics and consequently GAP recognition.12 Given that a GAP-defective hyper-activated KRAS G12V mutation containing a K147L mutation that lacks the ubiquitylation site, showed decreased tumor formation in a xenograft model, RAS ubiquitylation may contribute to RAS-mediated tumorigenesis.11 Although mUbRAS shows higher association with specific RAS effectors, RAF and PI3K in HEK293T cells, and upregulates signaling through these kinase cascades, it is currently unclear whether KRAS ubiquitylation directly modulates effector binding.
To evaluate whether mUb of KRAS at position 147 alters binding interactions with regulatory and effector proteins, we conducted a series of fluorescence-based binding assays. We find that mUbRAS impairs GTP-dependent binding interactions with the p120 GAP catalytic domain (GAPcat), PI3Kcγ and both CRAF- and RALGDS RBDs, but displays significantly enhanced binding to the CRAF RBD in its GDP bound state. These observations lead to the intriguing possibility that mUbRAS upregulates RAF signaling in a nucleotide independent manner.
Methods
Protein expression, purification and ubiquitin ligation
Human KRAS C118S (1–169), KRAS C118S/K147C (residues 1–169), Ubiquitin 76C (1–76), CRAF RBD (residues 54–131), and the catalytic domain of p120-RASGAP (GAPcat, residues 715–1047) were subcloned into a pQlinkH vector (Addgene) containing a histidine tag and expressed in Escherichia coli BL21 (DE3) RIPL cells (Novagen).14 Human RALGDS (residues 741–833) construct was generously provided by Mitsuhiko Ikura. RALGDS protein was expressed using a pQlinkH vector (Addgene) with an N-terminal glutathione S-transferase (GST) tag. Cells were grown at 37°C in a shaking culture of lysogeny broth (LB) supplemented with ampicillin and chloramphenicol. Protein expression was induced using 0.1mmol/L Isopropyl β-D-1-thiogalactopyranoside (IPTG). At an OD600 of 0.6, the temperature was reduced to 18°C, the cells incubated for additional 12–15 hours, harvested by centrifugation with pellets stored at −20°C.
Cells were lysed by sonication in 50 mL volumes of 50 mM Hepes, 150 mM NaCl, 20 mM imidazole, 5 mM MgCl2, 2 mM β-mercaptoethanol, and 50 μM GDP pH 7.75 (Buffer A). All RAS C118S/K147C and Ub76C purification buffers contained 2 mM tris(2-carboxyethyl) phosphine (TCEP). KRAS C118S/K147C, KRAS C118S, CRAF RBD, GAPcat proteins were purified using nickel (Ni) Sepharose columns at room temperature. The centrifuged cell lysate was mixed with the Ni sepharose beads and the Ni column washed in Buffer A. The column was subsequently washed with Buffer B containing higher salt (500 mM NaCl and 40 mM imidazole). Finally, the column was washed using Buffer A containing increasing amounts of imidazole (35 mM, 40 mM and 45 mM) before eluting the protein using Buffer C containing 20 mM Hepes, 150 mM NaCl and 250 mM Imidazole, pH 7.75. The histidine tag was cleaved overnight using the His-tagged tobacco etch virus (TEV) protease while dialyzing in Buffer A. The eluted protein was then passed over a nickel-agarose column to remove uncleaved RAS and His-tagged TEV. Further purification of protein from contaminants was achieved using size-exclusion chromatography (Superdex 75 10/300 GL; GE Life Sciences) at 4°C. Purity exceeding 95% was confirmed by SDS-PAGE analysis. RALGDS was purified by mixing the centrifuged lysate with glutathione beads for 1 hour (Amersham Pharmacia Biotech). The column was washed using 300 mL of 30 mM Tris, 10 mM NaCl, 10% glycerol and 1 mM dithiothreitol at pH 7.5. The protein was eluted by the addition of 10 mM reduced glutathione to the wash buffer. The GST-tag was cleaved overnight using thrombin protease while dialyzing in wash buffer. The cleaved RALGDS was then purified using size exclusion chromatography (Superdex 75 10/300 GL; GE Life Sciences). The purified human Phosphoinositide 3-Kinase catalytic subunit, gamma (PI3Kcγ, residues 144–1102) was generously provided by Genentech.
Chemical ubiquitylation of RAS
Monoubiquitylation of KRAS was accomplished using previously published methods.12,15 In brief, a ten-fold excess of purified ubiquitin 76C was mixed with KRAS C118S/K147C and dialyzed against 4L of 30 mM Tris, 100 mM NaCl, 5 mM MgCl2, 50 μM GDP, pH 8.5 at 4°C overnight. The efficiency of chemical ubiquitylation was determined using a non-reducing SDS-PAGE. The reaction was considered complete by the absence of non-modified RAS.
RAS nucleotide loading
The non-hydrolysable fluorescent GTP analog, mant-GppCp (mGppCp), was used in lieu of GTP, to prevent GTP hydrolysis during the binding assays. RAS protein was buffer exchanged into 20 mM Hepes, 50 mM NaCl, 1mM MgCl2 at pH 7.4. The concentration of RAS was adjusted to 100 μM and 500 μM mGppCp was added. The sample was incubated with alkaline phosphatase conjugated to sepharose beads at 10% (vol/vol) in the presence of 1 mM EDTA. The sample was incubated at 4°C for 16 hours and then was dialyzed into BINDING Buffer (20 mM Hepes, 50 mM NaCl, 5mM MgCl2 at pH 7.4). The nucleotide loading efficiency was determined using HPLC.16
Nucleotide loading of mant-GDP (mGDP) required RAS buffer exchange in Buffer B containing 20 mM ammonium sulfate at pH 7.4. Mant-GDP was added at 5:1 (mol mGDP:mol RAS), EDTA (1mol EDTA: 1 mol RAS), incubated at 37°C for 30 minutes and then exchanged into binding Buffer C.
Mant-fluorescence binding assay
The mant-fluorescence binding assay was adapted from a previous protocol.17,18 In brief, KRAS loaded with mGppCp (1.5 μM) was incubated with increasing concentrations of CRAF RBD, RalGDS and PI3Kcγ in assay Buffer D (20 mM Hepes, 50 mM NaCl and 5 mM MgCl2, pH 7.4). Nucleotide dissociation was initiated by addition of 1000-fold excess of unlabeled nucleotide at 25°C. The rate-of-dissociation was monitored by the change in fluorescence using an excitation wavelength of 355 nm and emission at 448 nm. The fluorescence measurements were made using a Spectramax M5 plate reader using a 384 Greiner plate. The nucleotide dissociation curve was fit using a one-phase exponential decay to determine Kobs. The dissociation rate was fit against the ligand concentration using previously published methods.17 A minimum of three independent binding experiments were used to determine the binding affinity and standard error of RAS to each effector.
Results
GAPcat shows a significant loss of binding to mUbRAS
We previously found that mUbRAS is defective in GAP-mediated GTP hydrolysis.12,13 This defect could result from either impairment in binding, catalytic activity or both. To ascertain whether mUbRAS alters GAP binding, we employed a fluorescence-based binding assay. This assay measures the affinity of RAS with the catalytic domain of p120 GAP (GAPcat, residues 714–1047) by monitoring the rate of fluorescent nucleotide dissociation (mGppCp) with increasing concentration of GAPcat.19 RAS was monoubiquitylated at 147 by chemical ligation of Ub76C with KRAS K147C. Using the dissociation assay, we then determined the affinity of mUbRAS (mGppCp) to GAPcat in comparison to non-modified KRAS. Consistent with previous observations, we find that KRAS (wt) loaded with mGppCp binds to GAPcat with a Kd of 1.9 ± 0.2 μM.20,21 However, mUbRAS shows decreased (10-fold) affinity for GAPcat. The loss-of-binding (Table 1) is consistent with the GAP insensitivity reported by Baker et al.12
Table 1.
Comparative binding affinity of wt KRAS and mUbRas with GAP and effector proteins.
| CRAF | RalGDS | PI3Kcγ | GAPcat | CRAF (GDP) | |
|---|---|---|---|---|---|
| wt KRAS | 61.0 ± 0.53 nM | 1.01 ± 0.08 μM | 1.86 ± 0.29 μM | 1.9 ± 0.20 μM | 55.2 ± 6.1 μM |
| mUbRAS | 391 ± 21.20 nM | 7.14 ± 0.51 μM | 18.9 ± 2.02 μM | 19.4 ± 1.4 μM | 1.2 ± 0.2 μM |
Monoubiquitylation of KRAS impairs GTP-dependent RAS-effector interactions
Given that mUbRAS shows decreased binding to the catalytic domain of p120 GAP (GAPcat) coupled with our previous findings that mUbRAS alters RAS switch dynamics, we postulated that monoubiquitylation of KRAS at position 147 alters nucleotide dependent switch conformations resulting in altered GAP and effector interactions. To determine whether mUbRAS alters interactions with a subset of RAS effectors, we loaded mUbRAS with mGppCp and evaluated binding to a subset of RAS effectors that demonstrate important roles in RAS mediated tumorigenesis. It has previously been the CRAF RBD domain binds with high binding affinity to KRAS complexed to GTP.22,23 Consistent with previous observations, we obtained a dissociation constant (Kd) of 61.0 ± 0.53 nM. In contrast, mUbRAS (mGppCp) has a 7-fold reduced affinity for CRAF RBD (Kd = 391 ± 21.2 nM).
We extended these analyses to determine the binding affinity of mUbRAS with effector proteins RALGDS RBD and PI3Kcγ. Each of these effector proteins exhibit weaker binding to KRAS complexed to GTP relative to the CRAF RBD.22,24,25 While effectors RALGDS and PI3Kcγ bind to KRAS(mGppCp) with low micromolar dissociation constants of 1.01 ± 0.08 μM and 1.86 ± 0.29 μM, respectively (Fig. 1c), consistent with previous results,25 we find that both RALGDS and PI3Kcγ show reduced binding to mUbRAS. In particular, RALGDS binds with significantly reduced (∼7-fold) affinity (Kd = 7.14 ± 0.51 μM) to mUbRAS (mGppCp). The binding interaction of RAS with PI3Kcγ is unique among effectors as the binding pocket of RAS includes contacts with both switch I and switch II.25 While the affinity of KRAS (wt) with PI3Kcγ was measured to be moderately weak (Kd = 1.86 ± 0.29 μM), the binding affinity of mUbRAS (mGppCp) with PI3Kcγ reflected previous trends observed for GAPcat, CRAF and RalGDS, resulting in a significant decrease (∼7-fold) in binding affinity (Fig. 1c) (Kd = 18.9 ± 2.02 μM) of mUbRAS (mGppCp) with PI3Kcγ relative to KRAS (wt).
Figure 1.
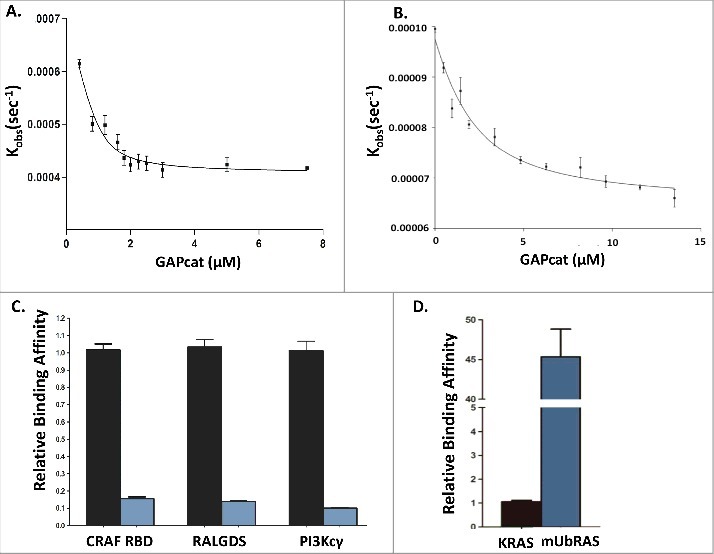
Panel A: Fluorescence binding curve of KRAS (mGppCp) with GAPcat (Kd = 1.9 ± 0.2 μM) compared with Panel B: Fluorescence binding assay of mUbRAS (mGppCp) with GAPcat (Kd = 19.4± 1.4 μM). Panel C: Relative binding affinity of KRAS (mGppCp, black bars) and mUbRAS (mGppCp, blue bars) with effectors CRAF RBD, RALGDS, and PI3Kcγ. Monoubiquitylated KRAS (mGppCp) shows a 7 to 9-decrease in affinity to the CRAF RBD, RalGDS RBD, and PI3Kcγ. Statistical error was determined from 3–5 independent experiments. Panel D: In contrast, mUbRAS (GDP shows 45-fold higher affinity relative to wt KRAS (GDP). Statistical error was determined from 3–5 independent experiments.
Monoubiquitylation of KRAS(GDP) shows greatly enhanced binding to the CRAF RBD
RAS effectors associate with higher affinity to the GTP-bound form of wt RAS relative to the GDP-bound state.26 To determine whether mUbRAS alters binding to RAS effectors in a nucleotide dependent manner, we conducted binding assays on wt RAS and mUbRAS complexed to GDP (mGDP). We were unable to quantitatively measure binding of PI3Kcγ and RALGDS with RAS(mGDP), as these lower affinity interactions (Kd ∼100 μM) were outside of the sensitivity range of the fluorescence-based assay. However, we were able to measure the affinity of KRAS (wt, mGDP-bound) to CRAF RBD, with a Kd = 55.2 ± 6.1 μM, in agreement with previously published data.26-28 The affinity of the CRAF RBD for KRAS(GDP) is ∼ 900-fold weaker than for KRAS (GTP). Intriguingly, the interaction of mUbRAS(GDP) with CRAF RBD is 45-fold tighter (Kd = 1.2 ± 0.2 μM) compared to wt KRAS (GDP). Given the greatly enhanced affinity (45-fold) for mUbRAS(GDP) and the lower binding to mUbRAS (GTP), the difference in GTP versus GDP specificity of mUbRAS for CRAF differs by only 3-fold relative to 900-fold for wt KRAS, rendering the interaction less nucleotide dependent.
Discussion
Monoubiquitylation has been shown to alter RAS activity, downstream signaling and tumorigenesis.11,12 To better understand how ubiquitylation upregulates RAS activity by conferring a defect in GAP mediated downregulation, we measured the binding affinity between mUbRAS and the p120 RASGAP catalytic domain, and found greatly impaired (10-fold) binding to GAPcat. The significantly decreased binding is consistent with our previous findings that mUbRAS shows a 10-fold impairment in GAP-mediated GTP hydrolysis relative to wt KRAS.12 As mUbRAS also promotes downstream signaling and tumorigenesis, we investigated whether mUb of KRAS at 147 alters effector binding. We find that mUbRAS results in a decrease in binding affinity (7 to 9-fold) for CRAF RBD, RALGDS RBD and PI3Kcγ. While this may be seemingly contradictory to the enhanced RAF and PI3K signaling observed for mUbRAS, the decrease in RAS(GTP) binding to effectors may be compensated by an increase in RAS(GDP) binding. In fact, we observe a large (45-fold) increase in binding affinity of mUbRAS (GDP) to CRAF RBD. These findings indicate that mUbRAS may bind and activate RAF in a nucleotide independent manner. We were unable to quantify binding of mUbRAS (GDP with effectors RALGDS or PI3Kcγ) given the lower affinity of these interactions relative to CRAF RBD.
How does mUbRAS alter regulatory and effector interactions? Two potential mechanisms may explain these findings. First, ubiquitylation of lysine 147 may alter RAS switch dynamics, allowing for GTP-like binding in the GDP-bound state. In support of this postulate, NMR data obtained on mUbRAS(GDP) indicate that the switch I, p-loop and part of switch II regions show altered backbone dynamics, more similar to slower dynamic motions observed for KRAS(GTP).12 We also find that substitution of a PDZ domain in lieu of ubiquitin retains the GAP defect, and that the length of the linker connecting to KRAS is important for the GAP-insensitivity of mUbRAS.13 The close proximity of the ligated protein to the dynamic switch regions may allow ubiquitin to modify the switch dynamics through an occlusion affect. As the RAS switch regions are in fast exchange on the NMR time scale when RAS is bound to GDP, ubiquitylation at 147 may slow exchange and favor ‘closed’ GTP-like conformations. We have also observed that mUbRAS bound to GTP shows altered switch dynamics by NMR (unpublished). Given the (7–10 fold) decrease in mUbRAS(GTP) binding with regulatory proteins and effectors, we postulate that ubiquitin ligation at 147, alters switch dynamics to lower the population of the ‘active’ conformation(s). As a second potential mechanism, monoubiquitylation of RAS at position 147 may show enhanced binding to RAS (GDP) through additional contacts between RAS, ubiquitin and effector proteins. Ubiquitin contains multiple binding sites that contribute to protein binding interactions,29,30 with dissociation constants in the low-micromolar range.29 Additional NMR structural and dynamic studies in addition to ligation of non-ubiquitin proteins to lysine 147, should provide insight into the mechanism by which mUbRAS alter effector binding and consequently downstream signaling. Moreover, cellular studies that probe nucleotide independent mUbRAS engagement and activation of RAF will aid in determining whether mUbRAS bound to GDP may facilitate MAPK signaling.
Acknowledgements
The research efforts described herein have been supported by NIH GM106227 and NIH CA203657 to SLC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J. Clin. 2009;59(4):225-249. https://doi.org/ 10.3322/caac.20006 [DOI] [PubMed] [Google Scholar]
- 2.Hobbs GA, Der CJ, Rossman KL. RAS Isoforms and Mutations in Cancer at a Glance. J. Cell Sci. 2016;129:1287-1292. https://doi.org/ 10.1242/jcs.182873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bos JL, Rehmann H, Wittinghofer A. Review GEFs and GAPs : Critical Elements in the Control of Small G Proteins. Cell. 2007;129(5):865-877. https://doi.org/ 10.1016/j.cell.2007.05.018 [DOI] [PubMed] [Google Scholar]
- 4.Du X, Sprang SR. Transition State Structures and the Roles of Catalytic Residues in GAP-Facilitated GTPase of Ras as Elucidated by (18)O Kinetic Isotope Effects. Biochemistry 2009;48(21):4538-4547. https://doi.org/ 10.1021/bi802359b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheffzek K, Ahmadian MR, Wittinghofer A. GTPase-Activating Proteins: Helping Hands to Complement an Active Site. Trends Biochem. Sci. 1998;23(7):257-262. https://doi.org/ 10.1016/S0968-0004(98)01224-9 [DOI] [PubMed] [Google Scholar]
- 6.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras Superfamily GEFs and GAPs: Validated and Tractable Targets for Cancer Therapy?. Nat. Rev. Cancer. 2010;10(12):842-857. https://doi.org/ 10.1038/nrc2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahearn IM, Haigis K, Bar-sagi D, Philips MR. Regulating the Regulator : Post-Translational Modification of RAS. Nat. Publ. Gr. 2012;13(1):39-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A et al., Consistent Blind Protein Structure Generation from NMR Chemical Shift Data. Proc. Natl. Acad. Sci. U. S. A. 2008;105(12):4685-4690. https://doi.org/ 10.1073/pnas.0800256105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang MH, Laurent G, Bause AS, Spang R, German N, Haigis MC, Haigis KM. HDAC6 and SIRT2 Regulate the Acetylation State and Oncogenic Activity of Mutant K-RAS. Mol. Cancer Res. 2013;11(September):1072-1078. https://doi.org/ 10.1158/1541-7786.MCR-13-0040-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the Regulator: Post-Translational Modification of RAS. Nat. Rev. Mol. Cell Biol. 2012;13(1):39-51. https://doi.org/ 10.1038/nrm3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki AT, Carracedo A, Locasale JW, Anastasiou D, Takeuchi K, Kahoud ER, Haviv S, Asara JM, Paolo P, March LCC et al., Ubiquitination of K-Ras Enhances Activation and Facilitates Binding to Select Downstream Effectors. Sci. Signal. 2011;4(163):ra13. https://doi.org/ 10.1126/scisignal.2001518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker R, Lewis SSM, Sasaki AT, Wilkerson EM, Locasale JW, Cantley LC, Kuhlman B, Dohlman HG, Campbell SL. Site-Specific Monoubiquitination Activates Ras by Impeding GTPase-Activating Protein Function. Nat. Struct. Mol. Biol. 2013;20(1):46-52. https://doi.org/ 10.1038/nsmb.2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hobbs GA, Gunawardena HP, Baker R, Campbell SL. Site-Specific Monoubiquitination Activates Ras by Impeding GTPase-Activating Protein Function. Small GTPases. 2013;4(3):186-192. https://doi.org/ 10.4161/sgtp.26270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmu L, Lautwein A, Schmitz F, Wittinghofer A. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science. 1997;277(5324):333-338. https://doi.org/ 10.1126/science.277.5324.333 [DOI] [PubMed] [Google Scholar]
- 15.Merkley N, Barber KR, Shaw GS. Ubiquitin Manipulation by an E2 Conjugating Enzyme Using a Novel Covalent Intermediate. J. Biol. Chem. 2005;280(36):31732-31738. https://doi.org/ 10.1074/jbc.M505205200 [DOI] [PubMed] [Google Scholar]
- 16.Zakaria M, Brown PR. High-Performance Liquid Column Chromatography of Nucleotides, Nucleosides and Bases. J. Chromatogr. 1981;226:267-290. https://doi.org/ 10.1016/S0378-4347(00)86062-4 [DOI] [PubMed] [Google Scholar]
- 17.Herrmann C, Horn G, Spaargaren M, Wittinghofer A. Differential Interaction of the Ras Family GTP-Binding Proteins H-Ras, Rap1A, and R-Ras with the Putative Effector Molecules Raf Kinase and Ral-Guanine Nucleotide Exchange Factor. J. Biol. Chem. 1996;271(12):6794-6800. https://doi.org/ 10.1074/jbc.271.12.6794 [DOI] [PubMed] [Google Scholar]
- 18.Block C, Janknecht R, Herrmann C, Nassar N, Wittinghofer A. Quantitative Structure-Activity Analysis Correlating Ras/Raf Interaction in Vitro to Raf Activation in Vivo. Nat. Struct. Biol. 1996;3(3):244-251. https://doi.org/ 10.1038/nsb0396-244 [DOI] [PubMed] [Google Scholar]
- 19.Lenzen C, Cool RH, Prinz H, Wittinghofer A. Kinetic Analysis by Fluorescence of the Interaction between Ras and the Catalytic Domain of the Guanine Nucleotide Exchange Factor Cdc25 Mm †. Biochemistry 1998;2960(97):7420-7430. [DOI] [PubMed] [Google Scholar]
- 20.Eccleston JF, Moore KJ, Morgan L, Skinner RH, Lowe P N. Kinetics of Interaction between Normal and Proline 12 Ras and the GTPase-Activating Proteins, p120-GAP and Neurofibromin. The Significance of the Intrinsic GTPase Rate in Determining the Transforming Ability of Ras. J. Biol. Chem. 1993;268(36):27012-27019 [PubMed] [Google Scholar]
- 21.Gideon P, John J, Frech M, Lautwein A, Clark R, Scheffler JE, Wittinghofer A. Mutational and Kinetic Analyses of the GTPase-Activating Protein (GAP)-p2l Interaction : The C-Terminal Domain of GAP Is Not Sufficient for Full Activity. Mol. Cell. Biol. 1992;12(5):2050-2056. https://doi.org/ 10.1128/MCB.12.5.2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiel C, Serrano L, Herrmann C. A Detailed Thermodynamic Analysis of Ras/effector Complex Interfaces. J. Mol. Biol. 2004;340(5):1039-1058. https://doi.org/ 10.1016/j.jmb.2004.05.050 [DOI] [PubMed] [Google Scholar]
- 23.Wohlgemuth S, Kiel C, Krämer A, Serrano L, Wittinghofer F, Herrmann C. Recognizing and Defining True Ras Binding Domains I: Biochemical Analysis. J. Mol. Biol. 2005;348(3):741-758. https://doi.org/ 10.1016/j.jmb.2005.02.048 [DOI] [PubMed] [Google Scholar]
- 24.Kiel C, Serrano L. Structure-Energy-Based Predictions and Network Modelling of RASopathy and Cancer Missense Mutations. Mol. Syst. Biol. 2014;10:727. https://doi.org/ 10.1002/msb.20145092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Crystal Structure and Functional Analysis of Ras Binding to Its Effector Phosphoinositide 3-Kinase Gamma. Cell. 2000;103:931-943. https://doi.org/ 10.1016/S0092-8674(00)00196-3 [DOI] [PubMed] [Google Scholar]
- 26.Kiel C, Filchtinski D, Spoerner M, Schreiber G, Kalbitzer H R, Herrmann C. Improved Binding of Raf to Ras.GDP Is Correlated with Biological Activity. J. Biol. Chem. 2009;284(46):31893-31902. https://doi.org/ 10.1074/jbc.M109.031153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ford B, Boykevisch S, Zhao C, Kunzelmann S, Bar-Sagi D, Herrmann C, Nassar N. Characterization of a Ras Mutant with Identical GDP- and GTP-Bound Structures. Biochemistry. 2009;48(48):11449-11457. https://doi.org/ 10.1021/bi901479b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakhaeizadeh H, Amin E, Nakhaei-rad S, Dvorsky R, Ahmadian MR. The RAS-Effector Interface : Isoform-Specific Differences in the Effector Binding Regions. Public Libr. Sci. One. 2016;11(12):1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hurley JH, Lee S, Prag G. Ubiquitin-Binding Domains. Biochem. J. 2006;399(3):361-372. https://doi.org/ 10.1042/BJ20061138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher RD, Wang B, Alam SL, Higginson DS, Robinson H, Sundquist WI, Hill CP. Structure and Ubiquitin Binding of the Ubiquitin-Interacting Motif. J. Biol. Chem. 2003;278(31):28976-28984 [DOI] [PubMed] [Google Scholar]