

ABSTRACT
piRNAs are small non-coding RNAs known to play a main role in defence against transposable elements in germ cells. However, other potential functions, such as biogenesis and differences in somatic and germline expression of these regulatory elements, are not yet fully unravelled. Here, we analysed a variety of piRNA sequences detected in mouse male and female primordial germ cells (PGCs) and gonadal somatic cells at crucial stages during embryonic differentiation of germ cells (11.5–13.5 days post-coitum). NGS of sncRNA and bioinformatic characterization of piRNAs from PGCs and somatic cells, in addition to piRNAs associated with TEs, indicated functional diversification in both cell types. Differences in the proportion of the diverse types of piRNAs are detected between somatic and germline during development. However, the global diversified patterns of piRNA expression are mainly shared between germ and somatic cells, we identified piRNAs related with molecules involved in ribosome components and translation pathway, including piRNAs derived from rRNA (34%), tRNA (10%) and snoRNA (8%). piRNAs from both tRNA and snoRNA are mainly derived from 3ʹ and 5ʹ end regions. These connections between piRNAs and rRNAs, tRNAs or snoRNAs suggest important functions of specialized piRNAs in translation regulation during this window of gonadal development.
KEYWORDS: piRNA, PGC, embryonic gonads, rRNA, tRNA, snoRNA, transposable elements
Introduction
In mouse, sex differentiation of germ cell occurs in primordial germ cells (PGCs) during the embryonic developmental period between 11.5 days post-coitum (dpc) and 13.5dpc, with the participation of gonadal somatic cells (SCs). While female PGCs enter meiosis by the action of retinoic acid signalling, in male, PGCs are mitotically arrested as prospermatogonia [1]. The interstitial/stromal somatic cells lead to Sertoli and Leydig cells in the testis and to granulosa and theca cells in the ovary. In both the ovary and testes, interactions between somatic and germ cells are crucial for the correct gonadal development and the production of functional gametes [2,3]. We recently identified, in mouse, the participation of microRNAs (miRNAs) in the programming and germ cell differentiation in both sexes [4]. However, the patterns of expression of piRNAs in gonadal cells as well as their potential role in the sexual dimorphism in both PGCs and SCs have not been characterized yet.
Piwi associated RNAs (piRNAs) are small non-coding RNAs (sncRNAs), which form a complex with a protein from the PIWI subfamily of Argonaute nucleases (in mice MIWI, MILI, and MIWI2). piRNAs have very broad and diverse functions, however, they are not well understood. The most well-characterized function of the piRNAs is the suppression of transposable elements (TEs) in the germline. This function is regulated by transcriptional and post-transcriptional silencing mechanisms [5]. The relevance of this mechanism is based on the fact that in mouse more than 90% of TEs are retrotransposons [6]. It is known that piRNAs are processed from piRNA-precursors, generally encoded by distinct genomic regions known as piRNA clusters [7]. However, several recent reports showed that the presence and functions of piRNAs in different taxons of metazoan are neither exclusive of the germline or exclusively involved in TE silencing [8], opening a wide landscape of gene regulatory functions not yet unravelled [9–13]. In mammals, the biogenesis and functions of multiple forms of piRNAs are also emerging as genetic and genomic regulators in cell development, homoeostasis and pathological processes, such as cancer [14,15]. In gametogenesis, ablation of elements of the piRNA pathway in testis leads to male infertility by the arrest of spermatogenesis [16]. In mature mouse oocytes, the amount of piRNAs is relatively low [17,18]. However, the potential regulatory roles of piRNAs in the onset of germ cell differentiation during early embryonic development have not been assessed. Previously, we generated a non-redundant piRNA database (IPpiRNA-db) from PIWI immunoprecipitation assays [19] that allowed the identification of different piRNA classes. Using the same nomenclature that we used previously, we called RapiRNAs (repeated associated piRNAs) those piRNAs mapping in the repeated regions of the genome and NRapiRNA those mapping in non-repeated regions. RapiRNAs are mainly related to TEs and other types of repetitive genome elements such as tRNA-derived or snoRNA-derived [19]. In addition, other piRNAs showed sequences complementary with mRNAs. This landscape greatly expands the potential functionality of the piRNAs beyond the action upon TEs.
A comparative analysis of the developmental gonads in the period 11.5–13.5 dpc in the mouse compiles crucial sex-determined differentiation processes in both germ and somatic cells. Consequently, studying the diversity and patterns of piRNAs underlying the process could be critical to understanding the multiple functions of these sncRNAs. In this work, we dissect the piRNAs expression patterns of somatic gonadal cells and PGCs from both sexes during the developmental window from 11.5 to 13.5 dpc. Using next-generation sequencing (NGS), we assessed the roles of the different types of piRNAs and their potential targets during this crucial period of germ cell determination. Both RapiRNAs and NRapiRNAs revealed a close connection between PGCs and nursing somatic cell of the gonads in both sexes, mainly in the most abundant piRNAs (RapiRNAs). We observed high expression of piRNAs rRNA-derived in both types of cells and sex at these early developmental stages. We also detected the expression of specific piRNAs derived from tRNAs and snoRNAs, which displayed differential expression patterns, suggesting potential functional interactions with key elements of the translation machinery such as rRNAs, tRNAs and snoRNAs.
Results
The sncRNAs comprise a group of abundant untranslated RNAs typically with length lower than 100 nt. This class of RNAs includes miRNAs and piRNAs, with important functional roles in germline differentiation. We previously studied the differential expression patterns of miRNA in PGCs and SCs from gonadal embryo in both sexes at 11.5, 12.5, and 13.5 dpc [4].
Characterization of RapiRNAs versus NRapiRNAs during gonadal development
Identified piRNAs were first classified as ‘Repeated associated piRNAs’ (RapiRNAs) and ‘non-repeated associated piRNAs’ (NRapiRNAs) [19]. We first comparatively evaluate the accumulation of these two types of piRNAs in relation to sex, PGCs versus SCs and developmental processes between 11.5 dpc and 13.5 dpc embryos. Globally, as expected, about 90% of all detected piRNA reads were RapiRNAs (Fig. 1A), these sequences showed short length bias, between 17 and 21 nt (Fig. 1B), but are present in the whole piRNA length spectrum. However, NRapiRNA reads representing only about 10% of total reads, showed a narrower profile of length distribution, lacking short lengths but displaying a peak around 31–33 nt.
Figure 1.

Global comparison between RapiRNA vs NRapiRNA reads. (A) Total proportion. (B) Length distribution. (C) In each cell type (PGCs and SC). (D) RapiRNA reads by sample in female (F) and male (M) gonads from 11.5 (11), 12.5 (12) to 13.5 (13) dpc. (E) NRapiRNA reads with same notation.
Comparison of RapiRNA and NRapiRNA reads in each developmental stage, showed a trend to increase its expression from day 12.5 to 13.5 in RapiRNAs. RapiRNA accumulation suggested the participation of piRNAs in the control of TEs as was reported in germ cells in testis including foetal testis [20–23]. Surprisingly, the pattern of RapiRNA distribution along this developmental window is similar in PGCs and in SCs. (Fig. 1D). This suggested that, at least in foetal gonads, these piRNAs are not exclusively or mainly expressed in germ cells in either sexes, allowing us to hypothesize the existence of close communication or interaction between somatic and germ cells for this type of small RNAs. Conversely, this distribution throughout development has not been clearly detected in NRapiRNAs (Fig. 1E). Length distribution and pattern expression of RapiRNA and NrapiRNAs in PGCs and SCs during sexual development suggest different functions between these types of piRNAs.
As expected, the majority of RapiRNAs showed association with LINE transposable elements (71%) and in minor quantity other retrotransposons as LTR (11%) and SINE (6%) (Table 1). However, the expression profiles in PGCs and SCs showed different signatures detected by three different approaches: heatmap, principal component analysis (PCA), and scatter plot showing the differential expression between two groups (Fig. 2). This differential expression could also be considered in the context of the depth changes, for example, DNA methylation controlling specific TE expression that occurs in the different cell types analysed in this developmental period.
Table 1.
Distribution of RapiRNAs mapping different repetitive elements.
| Repetitive elements | PGC | SC | |
|---|---|---|---|
| Transposable elements | LINE | 69.88% | 71.44% |
| SINE | 5.04% | 6.34% | |
| LTR | 11.79% | 11.88% | |
| Other repetitive elements | 13.29% | 10.34% | |
Figure 2.

RapiRNA expression patterns. (A) Unsupervised hierarchical clustered heatmap from log2 of RapiRNA expression values. (B) Principal component analysis of same values, with ellipses added around same group samples. (C) Differential expression analysis. RapiRNAs significantly differentially expressed (with a threshold of log2 folds change >1 and minimum expression >100 counts).
Chromosome distribution of RapiRNAs versus NRapiRNAs
We assessed the chromosome origin of RapiRNAs and NRapiRNAs in each sample. The expression distribution by chromosomes showed different patterns for RapiRNAs and NRapiRNAs, but similar profile concerning the cell types and the development period analysed (Supplementary Figure S1 and Supplementary Table S1). While RapiRNAs data indicated a prevailing trend expression from chromosomes 17, 8 and 9, in NRapiRNAs the most remarkable expression was from chromosomes 7 and 10 in most samples. Interestingly, we did not detect piRNA expression from the chromosome Y. These results increased evidence of these two piRNA types having different putative roles.
Genomic landscape of piRNA origin
To analyse the potential origin and functions of the identified piRNAs, we designed a specific bioinformatic pipeline (see details in Material and methods) (Supplementary Figure S2). The results indicated the relevant presence of piRNAs related with molecules involved in the translation machinery such as rRNA, snoRNAs, and tRNAs.
Despite the widely studied piRNA function in preventing genetic damage caused by TEs, our data showed that about half of piRNA reads originate from functional molecules involved in translation such as rRNA (34%), tRNA (10%), and snoRNA (8%) (Fig.s 3A and B). These results supported the existence of new potential roles for piRNAs. Globally, comparing the origins of these piRNAs in germ versus somatic cells, the tRNA-derived are more represented in SCs, while the rRNA-derived piRNAs are in PGCs, in both cases significantly (p < 0.05).
Figure 3.

piRNA genomic source classification. (A) Average proportion of piRNA reads from all situations. (B) piRNA read origins per sample. (C) Ribosomal RNA read analyses by different rRNA origins of detected rRNA-piRNA expression. An important number of piRNAs originates from different functional molecules such as rRNAs, tRNAs or snoRNAs. Specifically, the high expression of distinct piRNAs rRNA-derived could have a regulatory role at development.
Expression of rRNA-derived piRNAs
piRNAs with sequences matching ribosomal RNA (rRNA) were around 34% of identified piRNA reads (Fig. 3C), being the sequences with the highest rate of reads detected, including some sequences with more than 100.000 reads.
As in all databases of sncRNAs, the presence of any potential false-positive annotations of piRNAs cannot be entirely ruled out, making confidence in the full accuracy of the results dependent on subsequent experimental validations. Nowadays, the gold standard for identification of piRNAs is considered to be the immunoprecipitation (IP) of PIWI-containing ribonucleoprotein complexes from cell lysates, followed by RNAseq. To make our mapping more accurate, we used for analysis only those sequences obtained from 23 datasets where the sequences come from IP-RNAseq experiments (https://www.pirnadb.org/about/informations/dataset). To evaluate the robustness of the IPpiRNA-db, used to analyse our results, and discard any false-positives from abundant cellular RNA, such as rRNAs, we have verified that only 1.5% of sequences included in the IPpiRNA-db mapped to the rRNA regions covering all 45 S pre-ribosomal RNA. We observed that the pattern of distribution of piRNAs along the 45 S pre-ribosomal RNA is not uniform along the entire 45 S sequence, with a predominance in the 5ʹ and 3ʹ plus other additional specific internal regions of 18 S, 5.8 S, and 28 S RNAs.
We observed that, in the 45 S pre-ribosomal RNA locus, the piRNA reads from PGC and SC samples represent about 64% of the whole sncRNA reads associated with this locus. Consequently, the distribution of those sncRNAs detected as the piRNAs along the 45 S pre-ribosomal RNA was similar to the whole sncRNAs (data not shown). However, a low percentage of the 45 S pre-ribosomal RNA DNA was covered by an enriched proportion of rRNA-derived piRNAs in restricted regions of the locus, suggesting that they are not a product of simple degradation of rRNA (Fig. 4).
Figure 4.
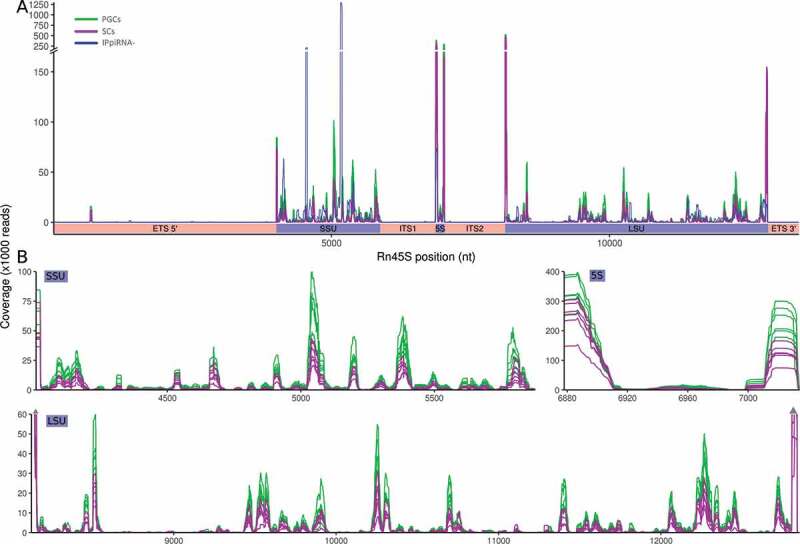
Distribution of the rRNA-derived piRNAs covering the 45 S pre-ribosomal RNA. (A) Distribution of the reads corresponding to the IPpiRNA-db (blue), PGCs (green), and SCs (purple) along the relative distance in nucleotides from the 5ʹ to the 3ʹend. (B) Details of the piRNA distribution in the SSU, 5S, and LSU rRNAs from all 12 different samples of PGCs and SCs.
rRNA transcripts might be originated from each one of the three ribosomal subunits: the small subunit (SSU), the large subunit (LSU), and the 5 S RNA. We observed that the most common origin is from LSU, which covers 50% of the piRNA reads, followed by SSU with 30% of the reads. An assessment of the coverage distribution of the rRNA-derived piRNAs along each type of rRNA sequence showed clusters of piRNA sequences, but different for each one rRNA type, being more abundant in both 5ʹ and 3ʹ ends in LSU but more represented in the 5ʹ end, plus a central region, in the SSU, and almost exclusive in the 5ʹ and 3ʹ for the 5 S type (Fig. 4). These data suggested that rRNA-derived piRNAs could be key regulatory elements in gonadal development in both sexes, putatively related with the global translation process.
Expression of tRF-derived piRNAs
tRNA-derived, termed as tRNA fragments (tRFs), are considered a type of sncRNAs that are processed from tRNAs by non-random fragmentation. Some of these tRFs, called tRF-piRNAs [24], are piRNA sequences derived from both mature and precursor tRNAs. These tRF-piRNAs were characterized previously in different cell types and species by PIWI immunoprecipitation followed by sequencing [19,25–27]. We found that the tRF-piRNAs represent almost 10% of the total piRNAs reads, with a higher representation in SCs (14%) than in PGCs (5%). However, the distribution among the different developmental days is not homogeneous, being slightly more represented at 11.5 dpc in both sexes but more expressed in SCs, which indicated differential dynamic of biogenesis and processing of such tRF-piRNAs for different cell types and developmental periods (Fig. 5A).
Figure 5.

tRFs analyses. (A) tRFs-piRNA expression in each sample in total reads percentage. (B) Schematic view of two tRFs from tRNAs split: 5ʹ and 3ʹ (for 3ʹ fragments the CCA-tail is not considered. (C) Read length distribution for each tRFs (5ʹ and 3ʹ) in PGC and SC. (D) The 15 most expressed tRFs. The tRNA-derived piRNAs are mostly 5ʹ tRFs and have a read length about 28–33 nt. The 95% of these piRNAs derived from most expressed 8 ones.
Four types of tRFs have been established: 5ʹ tRF, 3ʹ CCA-tRF, 3ʹ tRF, and internal tRF (itRF) [28]. The derived from 5ʹ tRF and 3ʹ tRF halves of each tRNA are the most abundant and well known (Fig. 5B). The specific signatures detected as tRF-piRNAs are clearly indicated as functional roles of these tRFs rather than a simple product of tRNA degradations (Table 2). In fact, 98.7% of all detected tRF-piRNAs reads were derived from the 5ʹ arm of tRNAs with a similar proportion in SCs and PGCs.
Table 2.
Top 10 of the most representative piRNAs tRF-derived.
| ORDER | tRNA | READS | % OF tRNAS |
|---|---|---|---|
| #1 | Gly-GCC | 600249 | 32.9% |
| #2 | Glu-CTC | 560191 | 30.7% |
| #3 | Lys-CTT | 248339 | 13.6% |
| #4 | Val-CAC | 131607 | 7.2% |
| #5 | Glu-TTC | 75686 | 4.1% |
| #6 | Lys-TTT | 40141 | 2.2% |
| #7 | His-GTG | 29190 | 1.6% |
| #8 | Val-AAC | 18806 | 1.0% |
| #9 | Leu-CAG | 15033 | 0.8% |
| #10 | Gly-CCC | 14788 | 0.8% |
The length of tRF-piRNAs is in the range of other types of piRNAs, but curiously while the length of 5ʹ tRF-piRNAs was around 28–35 nt, the length of 3ʹ tRF-piRNAs was less than 21 nt (Fig. 5C). Both types of 3ʹ and 5ʹ tRF-piRNAs have been associated with the inhibition of retrotransposition in germ and somatic cells [29,30], which is in agreement with the typical function of piRNAs. However, functional differences between 5ʹ and 3ʹ were also suggested based on the length of the 3ʹ tRF-piRNAs: 18 nt blocking the reverse transcription of retroposons and >22 nt inducing RNAi mechanisms [30]. The results obtained in SCs and PGCs showed these dual length types in 3ʹ tRF-piRNAs, but not in the most abundant 5ʹ tRF-piRNAs.
Potential-specific roles for the different types of tRF-piRNAs include selective recognizing of external retroposons versus endogenous transposons-derived genes, miRNA-like functions, germline versus somatic lines or differentiation and pathological situations. Remarkably, over 95% of tRF-piRNAs derived from only eight tRNAs: Gly-GCC, Glu-CTC, Lys-CTT, Val-CAC, Glu-TTC, LysTTT, His-GTG, Val-AAC, Leu-CAG, and Gly-CCC (Fig. 5D). These tRF-piRNAs showed similar levels of expression in PGCs versus SCs, which initially could suggest close functional cooperation between both types of gonadal cells in both sexes. However, considering different parameters by a heatmap and a principal component analysis among the 12 samples, the results showed differences between PGCs and SCs in both analytical approaches (Fig. 6A and B).
Figure 6.

tRFs expression group comparisons. (A) Unsupervised heatmap showing log2 of tRFs expression values and samples sorted using hierarchical clustering. (B) Principal component analysis of tRFs expression for each sample with an ellipse added around same group samples. The heatmaps reordering and PCA shows a clear differentiation between PGC and SC groups.
SnoRF-piRNAs
RNAs are frequently modified during their translation or maturation. It is known that nucleosides of rRNAs are mainly modified by two well-studied mechanisms: 2ʹ-O-methylation and pseudouridylation [31]. The main types of molecules involved in such modifications are from two families of snoRNAs (small nucleolar RNAs), termed C/D and H/ACA, which contain specific boxes and complementary sequences with rRNAs. snoRNAs are small non-coding RNAs, which after the formation of the RNA duplex and the participation of specific nucleoproteins direct the modifications of both SSU and LSU rRNAs or other types of RNAs such as tRNAs and sncRNAs [32–34]. snoRNAs are also identified as a particular type of genetic mobile elements, which might have evolved and diversified in vertebrates after retroposition and genetic drift [35]. This particular characteristic could also raise the potential link between piRNAs and processed snoRNAs.
Even as rRNAs and tRNAs, the snoRNAs can generate fragments, that we term snoRFs. These fragments can be associated with Piwi proteins (snoRF-piRNAs) as we previously described in mouse gametes and zygotes [19]. To assess whether the particular low size of piRNAs (16–21 nt) detected in all samples could result as products from any particular degradation of rRNAs, tRNAs, or snoRNAs, we evaluated their size distribution (in addition to the threshold established in global analysis), the results showed that these piRNAs: rRNA, tRNA or snoRNA-derived, do not have a biased distribution in short sequences (Supplementary Figure S3). Globally, in the foetal gonads analysed, snoRF-piRNAs represented 6.8% and 8.9% from the whole normalized reads in PGCs and SCs, respectively. Over 2000 different snoRNAs have been identified in humans, being the members of C/D family more abundant than the H/ACA family [36]. We detected in mouse up to 285 different snoRF-piRNAs, of which 173 corresponded to C/D family, 106 from H/ACA and 6 orphan snoRNAs, which represented 88.4%, 11.3%, and 0.3% from total reads, respectively (Fig. 7A). Using a non-supervised hierarchical clustering and PCA analysis, we observed that PGCs and SCs showed different snoRF-piRNAs signatures, indicating the presence of two distinguishable cell populations except for PGC11 F population (Fig. 7B and C). Considering both populations (PGCs and SCs) we assessed differential expression of snoRF-piRNAs displaying over two-fold change. Only 27 snoRF-piRNAs showed this level of difference (22 expressed higher in SCs and 5 in PGCs) (Fig. 7D). Interestingly, the profiles of the most expressed snoRF-piRNAs covered the 5ʹ regions (most of them) or 3ʹ but very rarely the central region of each snoRNA which derive therefrom. This also indicated that the biogenesis of the snoRF-piRNAs was not the consequence of random cleavages of the snoRNAs. Furthermore, such snoRF-piRNAs corresponded to putative functional regions of the snoRNAs associated with rRNAs. Moreover, each specific snoRNA trigger a particular profile of expression of snoRF-piRNAs, very similar for all PGC and SC samples, sex, or developmental stages for the C/D and H/ACA families or orphan snoRNAs (Fig. 8).
Figure 7.

snoRF-piRNAs analyses. (A) Boxplots of each snoRNA class showing global read distribution by snoRNA families. (B) Heatmap generated with snoRNA expressions in all samples sorted using hierarchical clustering. (C) PCA analysis of snoRF-piRNAs expression values, with ellipses grouping PGC or SC samples. (D) Differential expression plot showing log2 of expression values for each group in each axe. In red are the snoRF-piRNAs over two-fold change and expression above 100 copies.
Figure 8.

snoRF-piRNAs reads coverage plots of 12 samples (6 PGC in green and 6 SC in purple), highlighted regions are known snoRNA boxes (in red) and snoRNA-target RNA binding sites (in blue). Graphs are grouped by snoRNA classes; (A) C/D, (B) H/ACA and (C) Orphan. The coverage plots indicate defined cleavage sites.
By analysing the snoRF-piRNA origins we detected that the vast majority of identified snoRF-piRNA sequences were localized in the intronic regions of coding genes, which were included in the networks of ribonucleoproteins and eukaryotic initiation factors (EIFs), acting as stabilizers of the ribosomal formation, along with other genes related with gametogenesis such as Tex14, Cep55, Tcp1 or heat shock proteins (HSPs) (Supplementary Figure S4). The enrichment analysis of the overrepresented GO terms is indicated in Table 3. It is remarkable that most of them are ribosome-related: rRNA binding, structural constituent of ribosome, translation initiation factor activity, and mRNA binding.
Table 3.
GO terms significantly overrepresented in the gene dataset.
| GO molecular function terms | Hits | Expected | Fold-Enrichment | FDR |
|---|---|---|---|---|
| rRNA binding | 16 | 0.44 | 36.40 | 2.23E-16 |
| Structural constituent of ribosome | 27 | 1.05 | 25.75 | 3.37E-25 |
| Translation initiation factor activity | 6 | 0.3 | 19.91 | 3.50E-04 |
| Ran GTPase binding | 4 | 0.22 | 18.20 | 1.36E-02 |
| mRNA binding | 19 | 1.63 | 11.64 | 9.23E-12 |
Discussion
In this work, we characterize piRNA sequences detected in mouse male and female PGCs and gonadal SCs, at crucial stages for sex differentiation during embryonic development (11.5–13.5 dpc). Obtaining large samples of PGCs is greatly limited by the intrinsic characteristics of the sample. The characteristics include a highly complex sample and low cell numbers per gonad. We understand this limitation, and to overcome the lack of duplicates for NGS, we followed these strategies: (1) in order to minimize potential ‘noise’ in the sequencing data, the empirical cut-off value of 100 reads for each sequence was adopted; (2) in addition, PGCs were collected and pooled from embryos obtained from different mothers. These two strategies by themselves should be considered efficient measures to limit, as much as possible, the potential impact of unrelated sequences.
In PGCs, control of TE expression is crucial in the differentiation process. However, to establish the functional architecture of the gonads, close interactions should occur between germ and surrounding somatic cells at very early stages of development in both sexes [37]. It is not surprising, therefore, that some regulatory molecules such as piRNAs can be mostly shared between the two cell lineages. The relative similarity of profiles of some sncRNAs between purified gonadal SCs and PGCs in both sexes could be explained by such interactions. The presence of piRNA in somatic cells from the gonad has been well studied in different organisms. In the Drosophila melanogaster ovary, piRNA production is required in both somatic and germ cells. Moreover, somatic follicular cells have a high level of piRNAs [38,39]. Somatic loss of PIWI or mutations in the somatic piRNA clusters, such as flamenco, also results in germline stem cell differentiation defects, suggesting a key role in the control of PGC maintenance [40]. Indeed, a not yet identified signalling that promotes germline stem cell differentiation may modulate up-regulation of TEs in somatic cells [41]. The communications between germ cells and between germ and somatic cells in embryonic gonadal development can be performed through different junctions [42], including intercellular bridges and gap junctions mediated by complex proteins such as TEX14-CEP55 [43,44] and connexin 43 (COX43) [45], respectively. In males, they are expressed in the precursor of Sertoli and Leydig cells [46]. In the developing ovary, the communication between germ cells and between somatic and germ cells is crucial [43,44]. Additionally, the mechanisms of such communications can also be mediated by micro-vesicles or exosomes [47]. The movements of small regulatory RNAs between cells are very well documented [48]. However, it is possible that this type of shuttle of molecules between germ cells and somatic cells is more selective for other sncRNAs, such as miRNAs, [49] as was reported in the vesicle cargo in different cell types, [50,51] and is also suggested by the relative abundance of specific miRNAs in PGCs or gonadal somatic cells that we detected in our previous analysis from embryonic gonads in the same developing period [4]. Furthermore, the differential global pattern detected in PCGs and somatic cells for piRNAs and miRNAs could be also based on the different biogenesis and function between both types of sncRNAs, since miRNAs can be accumulated in stockpiles as double-stranded RNA binding mRNA targets or as duplex of both strands of the complementary single-stranded miRNAs, as we previously demonstrated in mouse preimplantation embryos [52].
In mouse PGCs, global DNA hypomethylation is already detected at 9.5 dpc with maximum erasure of DNA methylation at 13.5 dpc [53,54]. This massive DNA hypomethylation allows the derepression of TEs. The piRNA pathway is activated to generate the piRNAs needed to defend the genome from TE insertions and is also involved in the re-establishment of DNA methylation. We hypothesize that the close interaction between PGCs and gonadal somatic cells determines that both TEs and piRNAs could be transferred, induced, and initiated by the PGC programming [44], to cooperate in the function of those piRNAs involved in the defence mechanisms against transposons and contributing to de novo DNA methylation, in both sexes and maintaining transgenerational epigenetic inheritance [55,56]. However, other specific piRNAs [19] might participate in other germ or somatic cell differentiation in each respective gonadal sex.
tRF-derived sncRNAs have been identified by immunoprecipitation with AGO and PIWI proteins [19,57–59]. In this sense, the competition between tRF-derived piRNAs and other types of piRNAs for PIWI could be a way of regulating complex functional pathways in diverse mechanisms of differentiation and development involving AGO/PIWI availability in the cell. It has been demonstrated that tRFs are not the result of simple random degradation [29]. The functional role of tRF-derived piRNAs in germ cell differentiation can act as relevant regulatory molecules in the germline fate and function, in addition to protecting against TEs [60].
In all samples that we analysed, the highest expressed tRF-piRNA was from tRNA-Gly-GCC 5ʹ (Fig. 5D). This tRNA fragment has been demonstrated to act as a negative regulator of the expression of transcripts driven by endogenous MERVL retroelements in both mouse stem cells and embryos [29]. Experiments interfering specifically with tRNA-Gly-GCC 5ʹ resulted in the upregulation of genes expressed in totipotent cells but not expressed in pluripotent cells, [61] nor specifically in PGCs [62], which indicates the potential participation of these tRF-derived piRNAs in early germ cell differentiation. Curiously, tRF-Gly-GCC is the highest developmental differentially expressed tRF in females of the mosquito Aedes aegypti [63].
Other tRF-derived piRNAs showing very high expression in all samples is derived from tRNA-Glu-CTC 5ʹ. It has been reported that tRF5-Glu, downregulates the expression of the Breast Cancer Anti-Oestrogen Resistance 3 (BCAR3) mRNA and consequently ovarian cancer cell proliferation [64]. BCAR3 regulates BCAR1, and the complex BCAR1-BCAR3 can promote the migratory/invasive phenotype by activation of cell signalling effectors [65–68]. In addition, in mammalian males, oestrogen has relevant roles in spermatogenesis and testis development by different pathways [69]. BCAR3 is also expressed in Sertoli and germ cells during mouse testis development [70]. It is tempting to think that the control of BCAR3, which is involved in cell differentiation versus proliferation in both gonads (and potentially gonadal tumorigenesis) could be mediated by tRF-Glu as a key piRNA during gonadal cell development, coincident with the high level of expression that we detected in both germ and somatic cells in both sexes in early gonadal development.
Globally, tRFs can inhibit translation [71,72]. In human cells, experiments of transfection of 5ʹtRFs but not 3ʹtRFs induce inhibition of global translation [73]. The vast majority of tRF-piRNAs that we detected are from 5ʹtRFs. It has been very recently reported that upregulation of tRNAs-derived in mouse embryonic stem cells is associated with retinoic acid (RA)-induced differentiation. Furthermore, six specific 5ʹtRFs are associated with RA-induced differentiation [74]. Remarkably, our study detected the same five specific 5ʹ tRF-piRNAs from those six 5ʹtRFs identified in the RA-induction in stem cells: tRNA-Gly-GCC, tRNA-Lys-TTT, tRNA-Glu-TTC, tRNA-Val-CAC, and tRNA-Val-AAC, which are in our study among the eight tRF-piRNAs most expressed in all samples (Fig. 5D).
Processed RNA forms, such as rRNA, tRNA, and snoRNA, have been identified in different species [75]. Besides their participation in the defence against TEs, it is tempting to hypothesize that piRNAs derived from rRNAs, tRNAs, and snoRNAs, may act cooperatively to reprogram translation regulation in gonadal cells in both sexes during near pluripotency developmental stages before to generate differentiated germ cells, as other metabolic reprograming occurs in PGC differentiation [76]. It should also be considered that many TEs are evolutionarily integrated into genomic regions of rRNAs [77], which could also explain the high abundance of piRNAs associated with rRNA sequences in our analysis.
tRNAs and snoRNAs are integrated into the translation machinery by interacting with rRNAs. By sequence-specific recognition, snoRF-piRNAs may sequester rRNAs to dynamically modify the translation. Interestingly, some snoRF-piRNAs that we detected have their loci in intron regions of ribonucleoproteins or proteins regulating translation. The ribosome heterogeneity, mainly due to ribosomal protein composition, entails preferential translation of mRNAs and consequently generates a critical gene regulatory function in development and cell differentiation [78]. The results obtained showed a remarkable accumulation of piRNAs derived from elements related to the ribosome and translation machinery (Table 3). Hence, these snoRF-piRNAs could be involved in the generation of ribosomal heterogeneity in PGCs and gonadal SCs, interacting in the process of ribosomal biogenesis and translation regulation. Further studies on the mechanisms of such interaction and functions of all these types of non-canonical piRNAs will be of significant interest in key processes of differentiation such as those that affect the germline.
Materials and methods
Ethics statement
Mus musculus
CD1 mice were used as the experimental model. Animals were maintained under standard controlled conditions (22 ± 1°C, 50-55% of humidity and 12 h light/dark cycle) with water and food available ad libitum. Mice were managed in strict accordance with the recommendations ruled by the European Commission (Directive 2010/63/UE and Directive 86/609/ECC) and the Spanish Royal Legislative Decree RD53/2013 for the Care and Use of Laboratory Animals. The Animal Experiment Bioethics Committee of the Centro de Investigaciones Biológicas (CSIC) approved the protocols (Permit: CAM/PROEX 054/15).
Biological samples, RNA extraction, and NGS (Next-generation sequencing)
Pregnant female mice were euthanized at day 11.5, 12.5, and 13.5 dpc. The embryos were dissected in order to extract the gonads. We obtained and analysed samples from 12 different conditions: male or female, cell type (PGCs or SCs), and developmental stage (11.5, 12.5, or 13.5 dpc.). To minimize potential biological variation between individuals, pools of gonads from at least 50 embryos from 6 pregnant females were used in each sample. For simplicity F and M followed by 11, 12, or 13 were used in Figures and Tables corresponding to female and male gonads of 11.5, 12.5, and 13.5 dpc respectively. PGCs and SCs were sorted using paramagnetic technologies using a PGC-specific antibody (anti-CD15, SSEA-1), obtaining cell-type enrichment over 90%, discerned by alkaline phosphatase reaction [79]. RNA purification and NGS were carried out following the experimental procedures performed in our previous study [4]. Sequencing the fraction of small RNAs (≤100 nt) was performed using MiSeq Sequencing System (Illumina) in single-end mode, with a read length of 75 bp and an average depth of 10 million reads.
Data analysis
Quality of the raw sequencing data was checked using FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed with Trim Galore script (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The quality Phred threshold considered was 28 and the minimum sequence length was 16 nt. In order to minimize processing time and memory usage, identical sequences were collapsed in one item (specific sequence) using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html), maintaining the value of expression of each sequence (number of reads).
Bowtie v1.1.2 short read aligner (http://bowtie-bio.sourceforge.net/index.shtml) was used for sequence alignment. Quantification of piRNA expression was performed using HTSeq-count tool v0.11.1 [80]. It is not specified, -v1 -k1 – best -y parameters were used in Bowtie or -m intersection-strict -o in HTSeq-count software.
Normalization of a total number of reads per sample was performed using size factors calculated with estimateSizeFactors function from DESeq package. We set a strict threshold for further analysis to avoid noise in our data. We only used sequences with at least 100 detected reads.
All heatmaps were generated with heatmap.2 R function using default reordering.
The schematic view of the complete bioinformatic workflow is illustrated in Supplementary Figure S2.
piRNA identification
piRNA sequences were identified and characterized following bioinformatic workflows that we developed in previous studies [4,19,27]. Briefly, after discarding the possible miRNAs by miRBase data, reads were aligned to a custom-made IPpiRNA-db (available at https://github.com/edugenetico/Immunoprecipitation-piRNA-database/) [19], using Bowtie parameters -v 0 -k 1 – best -y. This database is a curated version of piRNA data from piRBase [81] considering as piRNAs those sequences that were identified after immunoprecipitation with PIWI proteins in 23 different assays. The use of the IPpIRNA-db and the consideration of at least 100 detected reads for each sequence trying to minimize potential false-positive sequences. To adjust the expression of multimapping piRNA sequences, reads of each piRNA were divided by the times they mapped in different regions of the genome. Only fully matched sequences of piRNA were included in the database, which means that smaller piRNAs are considered, but a threshold of >100 reads for each identified piRNA was included in the study to minimize potential products of degraded molecules.
Classification by repeated region
The repetitive regions were considered according to repeatmasker results (http://www.repeatmasker.org/) against mm10 assembly of Mus musculus (http://genome.ucsc.edu/cgi-bin/hgTables). This procedure allows us to classify piRNA reads between reads ‘piRNAs associated to repetitive genome elements’ (RapiRNAs) and ‘piRNAs not associated to repetitive elements’ (NRapiRNAs) [19].
piRNA’s genomic origin classification
To identify the genomic origin of the piRNA sequences we used consecutive alignment of non-aligned reads in the previous database (Supplementary Figure S2). Six different databases were used to classify the reads in six feature types: tRNA (from Genomic tRNA Database-GtRNAdb) [82]; snoRNA, lncRNA, and CDS (from Ensemble database version GRCm38, filtering by desired features, available at www.ensembl.org); rRNA and ‘other repeat elements’ (http://genome.ucsc.edu/cgi-bin/hgTables).
To identify the profile of piRNA expression corresponding to the 45 S pre-ribosomal RNA units, as a precursor for the processed 18 S (SSU), 5.8 S, and 28 S (LSU) rRNAs, mouse whole sequence of the 45 S (Genbank accession number BK000964) was used as a reference to identify the detected rRNA-derived piRNAs. To analyse the distribution of piRNAs into this locus, the corresponding reads of the IPpiRNA-db were obtained from the piRBase database. Coverage of piRNAs from the PGC and SC samples was analysed along with the piRNAs from the IPpiRNA-db. (Fig. 4). In order to analyse the coverage of different rRNAs (LSU, SSU, and 5 S) the ‘coverage’ function from BEDtools was used for each rRNA locus from RepeatMasker data. The coverage was represented as a percentage of total sequence in each rRNA type.
tRNA-derived identification
To identify piRNAs tRNAs-derived we aligned piRNAs to tRNA database from Genomic tRNA Database (GtRNAdb) release 17 [82], using Bowtie (-v3 – best -y). The tRNA sequences were previously split into two arms respect to the anticodon region: the 5ʹand the 3ʹ arms (Fig. 5B). HTSeq-count software was used to assess the expression for each tRNA fragment (tRF). The distribution of the length of the tRF-derived arms was also assessed.
snoRNA-derived identification
snoRNA-derived piRNAs (snoRFs-piRNA) were identified using specific snoRNA dataset at Ensemble database (https://www.ensembl.org/Mus_musculus/Info/Index). In order to classify the snoRNA family (C/D, H/ACA, or Orphan) the snoRFs-piRNA sequences were analysed by two tools: MGI webtool (http://www.informatics.jax.org/) and Rfam blast service (http://rfam.xfam.org/search). Additionally, the snoRNAs boxes and RNA binding sites were obtained from snOPY database (http://snoopy.med.miyazaki-u.ac.jp/). The snoRNA-piRNA expression was carried out using HTSeq-count.
To assess in which potential pathway the snoRFs-piRNA could be involved we identified the genes where the snoRFs-piRNA mapped. From the identified genes functional networks were generated by STRING software v11.0 [83], using the parameters: 0.9 confidence level, excluding textmining interactions and hiding non-connected nodes. PANTHER [84] overrepresentation test was used to investigate the potential biological processes (Release 20190711) by means of Gene Ontology (GO) with default settings (False Discovery Rate calculation for multiple testing, p < 0.05 and Fold enrichment >10), and selecting the most representative term.
Supplementary Material
Acknowledgments
We thank the technical assistance of the CIB-Animal Facility staff. We want to thank Bruce Campbell for proofreading the manuscript.
Funding Statement
This study was supported by the Agencia Estatal de Investigación; Ministerio de Ciencia, Innovación y Universidades (BFU2017-87095-R), Spain. ; ; M.A.B-E was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development [4R00HD090289-03] and the Magee Auxiliary Research Scholar (MARS).
Additional information
The datasets supporting the conclusions of this article are available in the Gene Expression Omnibus (GEO) repository, under the accession number GSE98713.
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Nakatsuji N, Chuma S.. Differentiation of mouse primordial germ cells into female or male germ cells. Int J Dev Biol. 2001;45:541–548. [PubMed] [Google Scholar]
- [2].McLaren A. Primordial germ cells in the mouse. Dev Biol. 2003;262:1–15. [DOI] [PubMed] [Google Scholar]
- [3].Murray SM, Yang SY, Van Doren M. Germ cell sex determination: a collaboration between soma and germline. Curr Opin Cell Biol. 2010;22:722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fernandez-Perez D, Brieno-Enriquez MA, Isoler-Alcaraz J, et al. MicroRNA dynamics at the onset of primordial germ and somatic cell sex differentiation during mouse embryonic gonad development. RNA (New York, NY). 2018;24:287–303. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Aravin AA, Lagos-Quintana M, Yalcin A, et al. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5(2):337–350. . [DOI] [PubMed] [Google Scholar]
- [6].Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41(1):331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Toth KF, Pezic D, Stuwe E, et al. The piRNA pathway guards the germline genome against transposable elements. Adv Exp Med Biol. 2016;886:51–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ishizu H, Siomi H, Siomi MC. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012;26(21):2361–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ross RJ, Weiner MM, Lin H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature. 2014;505(7483):353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garcia-Lopez J, Alonso L, Cardenas DB, et al. Diversity and functional convergence of small noncoding RNAs in male germ cell differentiation and fertilization. RNA (New York, NY). 2015;21:946–962. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rajasethupathy P, Antonov I, Sheridan R, et al. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jones BC, Wood JG, Chang C, et al. A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat Commun. 2016;7:13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rouget C, Papin C, Boureux A, et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weng W, Li H, Goel A. Piwi-interacting RNAs (piRNAs) and cancer: emerging biological concepts and potential clinical implications. Biochim Biophys Acta Rev Cancer. 2019;1871:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu Y, Dou M, Song X, et al. The emerging role of the piRNA/piwi complex in cancer. Mol Cancer. 2019;18:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pillai RS, Chuma S. piRNAs and their involvement in male germline development in mice. Dev Growth Differ. 2012;54:78–92. [DOI] [PubMed] [Google Scholar]
- [17].Watanabe T, Totoki Y, Toyoda A, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. [DOI] [PubMed] [Google Scholar]
- [18].Tam OH, Aravin AA, Stein P, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Larriba E, del Mazo J. An integrative piRNA analysis of mouse gametes and zygotes reveals new potential origins and gene regulatory roles. Sci Rep. 2018;8:12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aravin A, Gaidatzis D, Pfeffer S, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. [DOI] [PubMed] [Google Scholar]
- [21].Houwing S, Kamminga LM, Berezikov E, et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell. 2007;129:69–82. [DOI] [PubMed] [Google Scholar]
- [22].Lau NC, Seto AG, Kim J, et al. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–367. [DOI] [PubMed] [Google Scholar]
- [23].Girard A, Sachidanandam R, Hannon GJ, et al. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. [DOI] [PubMed] [Google Scholar]
- [24].Sobala A, Hutvagner G. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip Rev RNA. 2011;2:853–862. [DOI] [PubMed] [Google Scholar]
- [25].Couvillion MT, Sachidanandam R, Collins K. A growth-essential Tetrahymena Piwi protein carries tRNA fragment cargo. Genes Dev. 2010;24:2742–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Keam SP, Young PE, McCorkindale AL, et al. The human Piwi protein Hiwi2 associates with tRNA-derived piRNAs in somatic cells. Nucleic Acids Res. 2014;42:8984–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Larriba E, Rial E, del Mazo J. The landscape of mitochondrial small non-coding RNAs in the PGCs of male mice, spermatogonia, gametes and in zygotes. BMC Genomics. 2018;19:634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Keam SP, Hutvagner G. tRNA-derived fragments (tRFs): emerging new roles for an ancient RNA in the regulation of gene expression. Life (Basel). 2015;5:1638–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schorn AJ, Martienssen R. Tie-Break: host and retrotransposons play tRNA. Trends Cell Biol. 2018;28:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bachellerie JP, Cavaille J, Huttenhofer A. The expanding snoRNA world. Biochimie. 2002;84:775–790. [DOI] [PubMed] [Google Scholar]
- [33].Bratkovic T, Rogelj B. The many faces of small nucleolar RNAs. Biochim Biophys Acta. 2014;1839:438–443. [DOI] [PubMed] [Google Scholar]
- [34].Dupuis-Sandoval F, Poirier M, Scott MS. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA. 2015;6:381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Weber MJ. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet. 2006;2:e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lestrade L, Weber MJ. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006;34:D158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McLaren A. Germ and somatic cell lineages in the developing gonad. Mol Cell Endocrinol. 2000;163:3–9. [DOI] [PubMed] [Google Scholar]
- [38].Malone CD, Brennecke J, Dus M, et al. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li C, Vagin VV, Lee S, et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell. 2009;137:509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma X, Wang S, Do T, et al. Piwi is required in multiple cell types to control germline stem cell lineage development in the Drosophila ovary. PLoS ONE. 2014;9(3):e90267. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Upadhyay M, Martino Cortez Y, Wong-Deyrup S, et al. Transposon dysregulation modulates dWnt4 signaling to control germline stem cell differentiation in Drosophila. PLoS Genet. 2016;12:e1005918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tazuke SI, Schulz C, Gilboa L, et al. A germline-specific gap junction protein required for survival of differentiating early germ cells. Development. 2002;129:2529–2539. [DOI] [PubMed] [Google Scholar]
- [43].Lei L, Spradling AC. Mouse oocytes differentiate through organelle enrichment from sister cyst germ cells. Science. 2016;352:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Eppig JJ. Reproduction: oocytes call, granulosa cells connect. Curr Biol. 2018;28:R354–R6. [DOI] [PubMed] [Google Scholar]
- [45].Juneja SC, Barr KJ, Enders GC, et al. Defects in the germ line and gonads of mice lacking connexin43. Biol Reprod. 1999;60:1263–1270. [DOI] [PubMed] [Google Scholar]
- [46].Perez-Armendariz EM, Lamoyi E, Mason JI, et al. Developmental regulation of connexin 43 expression in fetal mouse testicular cells. Anat Rec. 2001;264(3):237–246. [DOI] [PubMed] [Google Scholar]
- [47].Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015;25(6):364–372. [DOI] [PubMed] [Google Scholar]
- [48].Jose AM. Movement of regulatory RNA between animal cells. Genesis. 2015;53(7):395–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].da Silveira JC, de Avila A, Garrett HL, et al. Cell-secreted vesicles containing microRNAs as regulators of gamete maturation. J Endocrinol. 2018;236(1):R15–R27. [DOI] [PubMed] [Google Scholar]
- [50].Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4(1):2980. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koppers-Lalic D, Hackenberg M, Bijnsdorp IV, et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014;8(6):1649–1658. . [DOI] [PubMed] [Google Scholar]
- [52].Garcia-Lopez J, del Mazo J. Expression dynamics of microRNA biogenesis during preimplantation mouse development. Biochim Biophys Acta Gene Regulatory Mechanisms. 2012;1819(8):847–854. [DOI] [PubMed] [Google Scholar]
- [53].Matsui Y, Mochizuki K. A current view of the epigenome in mouse primordial germ cells. Mol Reprod Dev. 2014;81(2):160–170. [DOI] [PubMed] [Google Scholar]
- [54].Guibert S, Forne T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012;22(4):633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ashe A, Sapetschnig A, Weick E-M, et al. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell. 2012;150(1):88–99. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shirayama M, Seth M, Lee H-C, et al. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans Germline. Cell. 2012;150(1):65–77. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Goff LA, Davila J, Swerdel MR, et al. Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PLoS ONE. 2009;4(9):e7192. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stark MS, Tyagi S, Nancarrow DJ, et al. Characterization of the melanoma miRNAome by deep sequencing. PLoS ONE. 2010;5(3):e9685. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Venkatesh T, Suresh PS, Tsutsumi R. tRFs: miRNAs in disguise. Gene. 2016;579(2):133–138. [DOI] [PubMed] [Google Scholar]
- [60].Martinez G. tRNA-derived small RNAs: new players in genome protection against retrotransposons. RNA Biol. 2018;15(2):170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Macfarlan TS, Gifford WD, Driscoll S, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487(7405):57–63. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pashai N, Hao H, All A, et al. Genome-wide profiling of pluripotent cells reveals a unique molecular signature of human embryonic germ cells. PLoS ONE. 2012;7(6):e39088. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Eng MW, Clemons A, Hill C, et al. Multifaceted functional implications of an endogenously expressed tRNA fragment in the vector mosquito Aedes aegypti. PLoS Negl Trop Dis. 2018;12(1):e0006186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhou K, Diebel KW, Holy J, et al. A tRNA fragment, tRF5-Glu, regulates BCAR3 expression and proliferation in ovarian cancer cells. Oncotarget. 2017;8(56):95377–95391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cai D, Iyer A, Felekkis KN, et al. AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates Rac, PAK1, and the cyclin D1 promoter. Cancer Res. 2003;63(20):6802–6808. [PubMed] [Google Scholar]
- [66].Near RI, Zhang Y, Makkinje A, et al. AND-34/BCAR3 differs from other NSP homologs in induction of anti-estrogen resistance, cyclin D1 promoter activation and altered breast cancer cell morphology. J Cell Physiol. 2007;212(3):655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wallez Y, Riedl SJ, Pasquale EB. Association of the breast cancer antiestrogen resistance protein 1 (BCAR1) and BCAR3 scaffolding proteins in cell signaling and antiestrogen resistance. J Biol Chem. 2014;289(15):10431–10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wilson AL, Schrecengost RS, Guerrero MS, et al. Breast cancer antiestrogen resistance 3 (BCAR3) promotes cell motility by regulating actin cytoskeletal and adhesion remodeling in invasive breast cancer cells. PLoS ONE. 2013;8(6):e65678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Akingbemi BT. Estrogen regulation of testicular function. Reprod Biol Endocrinol. 2005;3(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pennekamp P, Feldner S, Seesing FJ, et al. Bcar3 is expressed in sertoli cells and germ cells of the developing testis in mice. Sex Dev. 2011;5(4):197–204. . [DOI] [PubMed] [Google Scholar]
- [71].Ivanov P, Emara MM, Villen J, et al. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43(4):613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sobala A, Hutvagner G. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10(4):553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yamasaki S, Ivanov P, Hu G-F, et al. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Krishna S, Yim DG, Lakshmanan V, et al. Dynamic expression of tRNA-derived small RNAs define cellular states. EMBO Rep. 2019;20(7):e47789. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zywicki M, Bakowska-Zywicka K, Polacek N. Revealing stable processing products from ribosome-associated small RNAs by deep-sequencing data analysis. Nucleic Acids Res. 2012;40(9):4013–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sainz de la Maza D, Moratilla A, Aparicio V, et al. Metabolic reprogramming, autophagy, and reactive oxygen species are necessary for primordial germ cell reprogramming into pluripotency. Oxid Med Cell Longev. 2017;2017:4745252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Eickbush TH, Eickbush DG. Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics. 2007;175(2):477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Shi Z, Fujii K, Kovary KM, et al. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol Cell. 2017;67(1):71–83 e7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].De Felici M. Isolation and culture of germ cells from the mouse embryo. In: Celis, JE (Ed.), Cell biology: a laboratory handbook. New York: Academic Press; 1998. p. 73–85. [Google Scholar]
- [80].Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang J, Zhang P, Lu Y, et al. piRBase: a comprehensive database of piRNA sequences. Nucleic Acids Res. 2019;47(D1):D175–D80. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44(D1):D184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D13. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–2141. . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.