

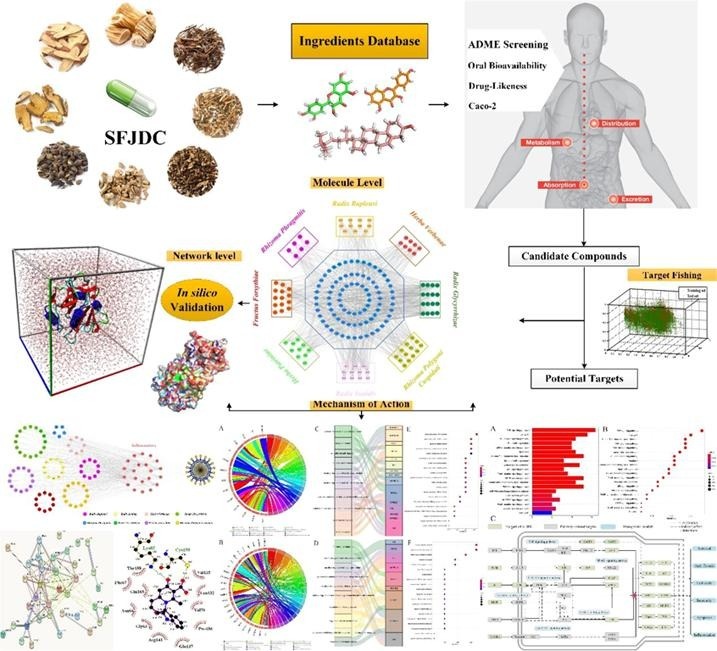
Graphical abstract
Keywords: Shufeng Jiedu Capsule, SARS-CoV-2, Coronavirus Pneumonia, Immunomodulation, Anti-inflammatory, Mechanism of action
Highlights
-
•
A systematic study integrated is proposed to illustrate the potential immune and anti-inflammatory mechanisms of SFJDC against SARS-CoV-2.
-
•
SFJDC regulates related targets, showing the potential anti-novel coronavirus effect.
-
•
The work can provide a better understanding of the therapeutic mechanism of SFJDC for treating SARS-CoV-2.
Abstract
The outbreak of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)-caused pneumonia (Coronavirus disease −19, COVID-19), has resulted in a global health emergency. However, there is no vaccine or effective antiviral treatment against the newly emerged coronavirus and identifying the available therapeutics as soon as possible is critical for the response to the spread of SARS-CoV-2. Shufeng Jiedu Capsule (SFJDC), a well-known prescription of Traditional Chinese Medicine (TCM) in China, has been widely used in treating upper respiratory tract infections and acute lung injury, owing to its immunomodulatory and anti-inflammatory effects. Despite the definite evidence of effective use of SFJDC in the diagnosis and treatment of pneumonia caused by SARS-CoV-2, the underlying action mechanism remains unknown. Currently, a systematic study integrated with absorption, distribution, metabolism and excretion (ADME) evaluation, target prediction, network construction and functional bioinformatics analyses is proposed to illustrate the potential immune and anti-inflammatory mechanisms of SFJDC against SARS-CoV-2. Additionally, to further validate the reliability of the interactions and binding affinities between drugs and targets, docking, Molecular dynamics Simulations (MD) simulations and Molecular Mechanics/Poisson-Boltzmann Surface Area approach (MM-PBSA) calculations were carried out. The results demonstrate that SFJDC regulates the immunomodulatory and anti-inflammatory related targets on multiple pathways through its active ingredients, showing the potential anti-novel coronavirus effect. Overall, the work can provide a better understanding of the therapeutic mechanism of SFJDC for treating SARS-CoV-2 pneumonia from multi-scale perspectives, and may also offer a valuable clue for developing novel pharmaceutical strategies to control the current coronavirus.
1. Introduction
At the end of 2019, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)-caused pneumonia (Coronavirus disease −19, COVID-19) is a highly infectious disease. The World Health Organization announces the COVID-19 pandemic on March 11, 2020 (CDC COVID-19 Response Team et al., 2020). Cluster evidence of infected family members and medical staff confirms the existence of person-to-person transmission through droplets, contact and poisonous gas, causing an infection of>8000,000 people worldwide, with > 435,000 deaths and the WHO has declared the epidemic to be a global public health emergency (Li et al., 2020, Cui et al., 2018). Although researchers are racing against time to develop vaccines and test anti-viral drugs in clinical trials, there is no appropriate drug or effective vaccine against the current coronavirus until now (Wang et al., 2020). In the absence of an effective treatment for this novel virus, finding novel drugs or new therapeutics to control the replication and spread of SARS-CoV-2 is urgently required.
However, the development of these novel drugs or therapeutics is a long process and may take months or even years. For this specific indication, Traditional Chinese Medicine (TCM) can be considered as an alternative measure, due to its proven clinical performance in curing many diseases, even the severe acute respiratory syndrome (SARS) (Wu et al., 2020). Interestingly, genetic and in silico analysis for SARS-CoV-2 indicates a close relationship with the SARS coronavirus, further showing the potential use of TCM in the current outbreak (Ling, 2020). In fact, the Chinese government has encouraged doctors to consider combining Western antiviral drugs with TCM remedies to fight the new type of coronavirus pneumonia and now, more and more TCM herbs have been recommended and extensively utilized in China for preventing and treating COVID-19.
Shufeng Jiedu Capsule (SFJDC) is a prescription of TCM in China, which has been extensively used in treating upper respiratory tract infections and acute lung injury over 30 years. Eight medicinal herbs make up this capsule, which contains Rhizoma Polygoni Cuspidati, Fructus Forsythiae, Radix Isatidis, Radix Bupleuri, Herba Patriniae, Herba Verbenae, Rhizoma Phragmitis and Radix Glycyrrhizae (Li et al., 2017). Evidence from clinical and basic research indicates that SFJDC alone or in combination with other chemotherapeutics shows anti-inflammatory and immunomodulatory functions (Ji et al., 2020). Notably, it has been well documented that the general treatments for COVID-19 patients are very important to enhance host immune response against RNA viral infection (Zhang & Liu, 2020). Thus, SFJDC could also be considered as a choice to enhance host immunity and anti-inflammatory effect against the infection of novel coronavirus. Actually, in the latest version of diagnosis and Treatment of Pneumonia Caused by SARS-CoV-2 (version 7) (Wang et al., 2020), SFJDC has been recommended for combating the present disease. It has been reported that four patients with mild and severe cases of SARS-CoV-2 infection have been cured or have significant improvement in their respiratory symptoms after combing with SFJDC on the base of supportive care (Lu, 2020). Besides, according to a recent news report, the symptoms of 108 patients with COVID-19 pneumonia are milder because they were treated with SFJDC in the early stage (Qu et al., 2020), Despite the definite evidence of effective use of SFJDC, its mechanism of action against SARS-CoV-2 have not been elucidated.
In the present study, we proposed a systematic study focusing on exploring the potential immune and anti-inflammatory mechanisms of SFJDC on treating COVID-19. Firstly, a series of computational absorption, distribution, metabolism, and excretion (ADME) screening and target fishing models employed to obtain the active ingredients of herbs in SFJDC as well as their corresponding protein targets. Then, functional bioinformatics analyses and network construction carried out to clarify the pharmacological mechanisms of actions involved in the herb functions. Besides, to further verify the reliability of the interactions between the obtained drugs and targets at molecular level, the docking and molecular dynamics (MD) were carried out. This work gives a valuable clue for developing novel pharmaceutical strategies to control the pandemic of SARS-CoV-2.
2. Materials and methods
2.1. Workflow of the systems investigation approach
Our protocol includes the following main steps: (1) The bioactive constituents of SFJDC was firstly identified via an ADME-screening model which including the Oral Bioavailability (OB), Drug-Likeness (DL) and Carcinoma Cell Line (Caco-2) modules together. (2) The active ingredients from the ADME-screening model were then to predict the potential targets by using a systematic prediction of multiple drug-target (SysDT) model (Yu et al., 2012). (3) The corresponding compound-target (C-T) and target-pathway networks were constructed for further analysis. (4) The functional enrichment analysis was carried out to identify the important biological processes operating in Shufeng Jiedu Capsule. (5) The docking and MD simulations were performed to validate the C-T networks and further probe the binding mode and binding affinity from the C-T interactions. An overview of the whole experimental procedure is presented in Fig. 1 .
Fig. 1.

The work scheme of this study.
2.2. Database construction and molecular modeling
The ingredients of all herbs in SFJDC herbal medicines were mainly obtained from Encyclopaedia of traditional Chinese medicine (ETCM), Integrative Pharmacology-based Research Platform of Traditional Chinese Medicine (TCMIP) and TCM Systems Pharmacology Database (TCMSP). The chemical structures were obtained from the NCBI PubChem database, and all structures of these compounds were optimized for each backbone structure along with energy minimization by using Autodock 4.1 and GROMACS 2019 software (Berendsen, van der Spoel, & van Drunen, 1995) for subsequent analysis.
2.3. Pharmacokinetic screening models
In the present study, to evaluate the pharmacokinetic properties of the active ingredients of the SFJDC, three pharmacokinetic screening models were performed. The OB, DL and Caco-2 permeability were applied to prescreen for the active compounds.
2.3.1. Oral Bioavailability
In this work, a robust in-house tool OBioavail1.1 model was carried out to compute the OB value for candidate compounds (Xu et al., 2012) (Wang et al., 2017). The multiple linear regression (MLR), partial least square (PLS) and support vector machine (SVM) modes were conducted to determine correlations between variables in an optimal model. PreOB is actually an effective in-house previously developed model for predicting the OB of herbal ingredients and integrates information about cytochrome P450 3A4 and P-glycoprotein (Xu et al., 2012). In the present work, the OB threshold is defined as 30%, and the ingredients with OB ≥ 30% were adopted as the candidate compounds.
2.3.2. Drug-Likeness
To screen out the drug-like chemicals, a Predict Drug-Likeness (PreDL) (Willett, Barnard, & Downs, 1998) model developed was carried out to dis-criminate between drug-like and nondrug-like of each compound by using the Tanimoto coefficient. PreDL is used to calculate the DL index, which can help us estimate ADME characteristics of the chemical substance under study (Yang et al., 2017). The drug-likeness evaluation approach is as follows:
| (1) |
where A denotes the molecular properties of ingredient, and B represents the average drug-likeness index of all compounds in DrugBank database. Presently, the compounds with the value of DL ≥ 0.15 are selected as active ingredients from the SFJDC.
2.3.3. Caco-2
Many computerized drug absorption models using in vitro Caco-2 permeability have been widely established and used in drug discovery and development processes (Pham The et al., 2011). Here, we use the robust computer simulation Caco-2 permeability prediction model preCaco-2 (Wessel, Jurs, Tolan, & Muskal, 1998), which is composed of 100 drug molecules and has satisfactory statistical results (R2 > 0.8) to predict drug absorption. In this section, a Caco-2 permeability model was carried out to predict the fraction of oral dose absorbed and drug’s intestinal absorption (Pham The et al., 2011). Considering that the compounds with Caco-2 < -0.4 are considered to be not permeable, the permeability of Caco-2 ≥ -0.4 are selected as active compounds.
The compounds which simultaneously satisfied the conditions of OB ≥ 30%, DL ≥ 0.15 and Caco-2 ≥ -0.4 and were sorted out into the candidate compound pool.
2.4. Target prediction
In order to get the potential targets of active compounds, an in-house developed model SysDT based on RF and SVM algorithm was performed to predict the potential targets with the threshold of RF score ≥ 0.8 and SVM ≥ 0.7 (Yu et al., 2012). The SysDT model integrates a large scale information including chemistry, genomics and pharmacology, which shows impressive reliability of prediction for drug-target interactions (Yu et al., 2012).
In this model, two indicators were obtained, indicating the probability of each candidate object interacting with the target under the RF and SVM methods. The model shows appropriate predictive performance for drug-target interactions, in which the consistency, sensitivity and specificity are 82.23%, 81.33% and 93.62%, respectively. All targets information including their gene names, functions, organisms, tissues, etc., are obtained from the above two steps and were mapped to UniProt (http://www.uniprot.org) database for normalization of their name and organisms. Only the targets of Homo sapiens were reserved for further analysis.
2.5. Functional enrichment analysis
To explore meaningful functional annotation and vital biological process of the identified targets, Gene Ontology (GO) analysis was performed by Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources 6.8 software. The screening of significantly enriched GO terms with p < 0.05 and FDR < 0.05 were selected for further research. In addition, to further investigate the achieved targets, the GOplot package was performed for investigating their mutual relations. The results were visualized by the Goplot v1.0.2 and R 3.6.2 packages.
2.6. Network construction
In this work, to gain further insight into the involvement of multi-component therapeutic features from SFJDC and further to explore the biological effects of drugs on the pathway level, active compounds of SFJDC and their corresponding targets were carried to construct the C-T and Compound-Target-Function (C-T-F) networks for systematically pharmacology analysis. In addition, all ingredients were extracted from ADME screening and their corresponding targets were obtained from the SysDT model. Then, by mapping to KEGG, the obtained target profile is organized into several pathways. All maps of the network are generated and visualized by the open source software of the bioinformatics package Cytoscape 3.6.0 (Shannon, 2003).
2.7. Docking for target validation
To validate the C-T interactions, the active ingredients were docked to its predicted targets by the Lamarckian genetic algorithm program AutoDock version 4.1 package with default settings (Morris et al., 2009). Ligand docking is a key method to deal with the C-T interactions, which generate posture and dimensionless fitness scores in structural molecular biology and computer-aided drug design (Yang et al., 2018). The binding pocket was defined as the volume of the co-crystallized ligand in the template protein. The radius around the ligand was set to 10 Å in the crystal structure as a binding site, and the top 10 docking positions were selected for further analysis. All conformations of the compounds were evaluated with Score fitness function, which are calculated considering contributions of hydrogen-bonding energy, and van der Waals interactions and ligand torsion strain between targets and the compounds, which indicate the veracity of the conformations (Arooj, Sakkiah, Kim, Arulalapperumal, & Lee, 2013).
2.8. Molecular dynamics simulations
To further explore the mechanism of binding information and provide more insights into the interactions between active ingredients and targets, six C-T interactions from the hub compounds and targets were selected for MD validations by using the GROMACS 2019 software (Berendsen et al., 1995) with the ambersb99 force field (Hornak et al., 2006). The X-ray crystal structures of six targets ESR2 (PDB: 5TOA), GSK3β (PDB: 6GN1), NOS2 (PDB: 3E7G), NR3C1 (PDB: 6DXK), PPARG (PDB: 6ONI) and TOP2A (PDB: 1ZXM), were extracted from RCSB Protein Data Bank.
The C-T complex was placed in a cubic box and solvated with SPC water, and the minimum distance between the protein surface and the box wall was set to 10. With the Berendsen thermostat method, the temperature was set to a constant value. The value of the isothermal compression ratio was set to 4.5 × 10-5 bar−1, and the Parrinello-Rahman scheme was implemented to maintain the pressure at 1 bar (Lin, Perryman, Schames, & McCammon, 2002). By using the steepest descent and deepest descent (SD) algorithm under periodic boundary conditions for 10,000 steps, the entire system was minimized in energy, and then the system was equilibrated by 200 ps MD simulation at 300 K. Finally, 100 ns long simulations were run with 2 fs time step.
2.9. Calculations of binding free energy
Binding free energy is the thermodynamic property that can accurately reflect the binding affinity when the drug-target interaction reaches equilibrium. And the process of drug molecule recognition target is precisely controlled by the change of free energy, which also determines that free energy has become one of the most physical quantities in drug discovery (Kollman et al., 2000). In the present study, the binding free energy for eight compound-protein complexes from the MD simulations was calculated by the g_mmpbsa (Kumari, Kumar, & Lynn, 2014) tool of GROMACS 2019 based on the Molecular Mechanics/Poisson-Boltzmann Surface Area approach (MM-PBSA). For each molecular species including complexes, ligands and proteins, the difference in binding free energy from protein-ligand binding affinities were calculated. Binding free energy is computed using the following equations:
| ΔGbind = ΔEMM + ΔGsol - TΔS | (2) |
| ΔEMM = ΔEinternal + ΔEelectrostatic + ΔEvdw | (3) |
| ΔGsol = ΔGPB + ΔGSA | (4) |
where ΔEMM is the interaction energy between the compound and the target. ΔGsol is the solvation energy, which is the sum of the electrostatic solvation free energy (ΔGPB) from the polar contribution and the non-electrostatic solvation component (ΔGSA) from the non-polar contribution
2.10. Bioactivity prediction
The binding free energy obtained from molecular dynamics simulations was used to calculate the inhibitory constant (Ki), which is based on the following equation (Jedrzejas et al., 1995, Jangam et al., 2019):
(5)
Where ΔGbind is the binding free energy and R represents the gas constant (1.987 × 10–3 kcal/K-mol), and T (298.15 K) represents the absolute temperature of the target-compound complex.
2.11. Target-Pathway mapping
To further elucidate the synergistic biological mechanisms how cellular targets affect disease by regulating multiple metabolic pathways of SFJDC for SARS-CoV-2, an incorporated pathway was established. This presently a pathways selection were obtained from Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Through Fisher's exact test in the DAVID database, the rich KEGG pathway of targets with a higher false discovery rate of FDR < 0.05 was examined. As a result, a better understanding of the relatively complete on immunomodulation and anti-inflammatory related pathways were manually assembled to further characterize the synergistic mechanisms of the compounds.
3. Results and discussion
3.1. Statistical analysis of herbal medicines in SFJDC
SFJDC is a traditional Chinese medicine containing eight herbs, which have been summarized in Table 1 . The number of ingredients and active compounds and the corresponding abbreviations of each herb are also depicted. Using the approach described in 2.2 section, totally 1124 compounds in these herbs are obtained after eliminating the duplicate chemicals. As can be observed, Radix Bupleuri (CH) has the largest number of compounds (number = 349), followed by Radix Glycyrrhizae (GC, number = 199) and Radix Isatidis (BL, number = 169). Among them, Rhizoma Phragmitis (LG) has the littlest number of ingredients compounds (number = 31).
Table 1.
The herbal medicines contained in SFJDC and the number of corresponding compounds in each herb.
| Name | Number | Abbreviation | ||
|---|---|---|---|---|
| Latin | Pinyin | Compounds | Active compounds | |
| Rhizoma Polygoni Cuspidati | Huzhang | 62 | 13 | HZ |
| Fructus Forsythiae | Lianqiao | 165 | 14 | LQ |
| Radix Isatidis | Banlanggen | 169 | 12 | BL |
| Radix Bupleuri | Chaihu | 349 | 13 | CH |
| Herba Patriniae | Baijiangcao | 52 | 10 | BJ |
| Herba Verbenae | Mabiancao | 58 | 9 | MB |
| Rhizoma Phragmitis | Lugen | 31 | 7 | LG |
| Radix Glycyrrhizae | Gancao | 199 | 16 | GC |
3.2. Candidate compounds screening
Employing the screening criteria of OB ≥ 30%, we obtained totally 85 active ingredients from the herbal medicines of SFJDC. Among them, 13 are from Rhizoma Polygoni Cuspidati (HZ), 12 in Fructus Forsythiae (LQ), 12 in BL, 13 in CH, 8 in BJ, 6 in Herba Verbenae (MB), 5 in LG, and 16 in GC. Actually, these active compounds with satisfactory pharmacokinetic parameter are in good agreement with the pharmacological data reported, confirming the reliability of the OB screening model. For example, quercetin, one of the important bioflavonoids present in HZ and more than other twenty herbs, can enhance mental/physical performance and decrease the risk of infection, showing the unique biological properties (Li et al., 2016). With the antiviral and antibacterial activity of quercetin, it particularly affects gastrointestinal, respiratory, urinary, and dermal system (Anand David, Arulmoli, & Parasuraman, 2016).
Besides, the DL of drugs also plays an important part in screening the active ingredients, and presently, we obtain a total of 78 potential compounds with DL ≥ 0.15 from the SFJDC. For instance, the main chemical constituent phillyrin from Forsythia suspense, has shown a great inhibitory effect in treating gonorrhea, erysipelas and inflammation (Zhong et al., 2013). Previous study has demonstrated that phillyrin can significantly reduce over inflammatory responses and then turn into decreasing LPS-caused histopathological changes through targeting the MAPK and NF-κB signaling (Zhong et al., 2013). Additionally, phillyrin can also reduce inflammation and inhibiting viral replication, revealing the potential protective effects on influenza A virus-induced infection.
What’s more, 87 potential active molecules possess satisfactory screening criterion of Caco-2 (≥-0.4) value, showing their excellent pharmacokinetic properties. Taking Wogonin in LQ for an example, its various potent pharmacological properties containing antioxidant, anti-inflammatory, cytotoxic, neuroprotective and antidiabetic have been reported (Hassanin, Tolba, Tadros, Elmazar, & Singab, 2019). In addition, there is growing evidence that Wogonin can inhibit the replication of multiple viruses, such as vesicular stomatitis virus (Błach-Olszewska et al., 2008) and varicella-zoster virus (Choi, Lee, Kim, & Shin, 2015) as well as human papillomavirus. Also, through regulating the AMPK pathway, wogonin can suppress both influenza A and B virus replication in human lung epithelial cells (Hassanin et al., 2019).
Although some ingredients have relatively low OB, DL or Caco-2 values, these chemicals also exhibit significant confirmed biological activities and thus were also collected as the active compounds. For example, polydatin (OB = 21.44%, Caco-2 = -0.9), isolated from the root and rhizome of HZ, is a monocrystalline compound and has been demonstrated to show multiple biological activities and pharmacological effects (Ye et al., 2017). Through reducing the expression of proinflammatory mediators, polydatin can ameliorate cerebral ischemia induced oxidative stress, presenting the anti-inflammatory activities. Besides, Saikosaponin D also has the low screening criterions (DL = 0.01, Caco-2 = -1.94). Actually, the anti-inflammatory and anti-infectious activities of saikosaponin D have been reported previously. Saikosaponin D can relieve ventilator-induced lung injury by decreasing oxidative stress and apoptosis and inhibiting the expression of proinflammatory cytokines in rats (Li et al., 2020), indicating that saikosaponin D could be served as a potential candidate for the treatment of inflammatory-related and lung diseases.
Finally, a total of 1124 compounds from the SFJDC were collected. As a result, based on the three filtering parameters (OB ≥ 30%, DL ≥ 0.15 and Caco-2 ≥ -0.4) and considering the compounds with unsatisfactory pharmacokinetic characteristics but showing confirmed biological activities, we obtained totally 94 active ingredients from SFJDC in the present work, which is depicted in Table 2 .
Table 2.
Bioactive compounds with their OB, DL and Caco-2 values predicted.
| ID | Compound Name | OB | DL | Caco-2 | Herbs |
|---|---|---|---|---|---|
| HZ01 | Physovenine | 106.21 | 0.19 | 0.51 | Rhizoma Polygoni Cuspidati |
| HZ02 | Picralinal | 58.01 | 0.75 | 0.23 | Rhizoma Polygoni Cuspidati |
| HZ03 | Anthraquinoe | 56.10 | 0.14 | 0.86 | Rhizoma Polygoni Cuspidati |
| HZ04 | Cianidanol | 54.83 | 0.24 | −0.03 | Rhizoma Polygoni Cuspidati |
| HZ05 | Rhein | 47.07 | 0.28 | −0.2 | Rhizoma Polygoni Cuspidati |
| HZ06 | Quercetin | 46.43 | 0.28 | 0.05 | Rhizoma Polygoni Cuspidati |
| HZ07 | cis-Resveratrol | 41.13 | 0.11 | 0.75 | Rhizoma Polygoni Cuspidati |
| HZ08 | Ambrettolide | 40.59 | 0.14 | 1.36 | Rhizoma Polygoni Cuspidati |
| HZ09 | β-sitosterol | 36.91 | 0.75 | 1.32 | Rhizoma Polygoni Cuspidati |
| HZ10 | Luteolin | 36.16 | 0.25 | 0.19 | Rhizoma Polygoni Cuspidati |
| HZ11 | Coumarin | 29.17 | 0.04 | 1.20 | Rhizoma Polygoni Cuspidati |
| HZ12 | Oleanolic acid | 29.02 | 0.76 | 0.59 | Rhizoma Polygoni Cuspidati |
| LQ01 | Bicuculline | 69.67 | 0.88 | 0.72 | Fructus Forsythiae |
| LQ02 | Phillyrin | 36.40 | 0.86 | −0.79 | Fructus Forsythiae |
| LQ03 | Lactucasterol | 40.99 | 0.85 | 0.88 | Fructus Forsythiae |
| LQ04 | Arctiin | 34.45 | 0.84 | −0.73 | Fructus Forsythiae |
| LQ05 | Betulinic acid | 55.38 | 0.78 | 0.73 | Fructus Forsythiae |
| LQ06 | β-Amyrin acetate | 42.06 | 0.74 | 1.36 | Fructus Forsythiae |
| LQ07 | Sitogluside | 20.63 | 0.62 | −0.14 | Fructus Forsythiae |
| LQ08 | Adhyperforin | 44.03 | 0.61 | 0.93 | Fructus Forsythiae |
| LQ09 | Hyperforin | 44.03 | 0.60 | 0.87 | Fructus Forsythiae |
| LQ10 | Forsythinol | 81.25 | 0.57 | 0.59 | Fructus Forsythiae |
| LQ11 | Myricetin | 13.75 | 0.31 | −0.15 | Fructus Forsythiae |
| LQ12 | Kaempferol | 41.88 | 0.24 | 0.26 | Fructus Forsythiae |
| LQ13 | Wogonin | 30.68 | 0.23 | 0.79 | Fructus Forsythiae |
| LQ14 | Euxanthone | 92.98 | 0.16 | 0.68 | Fructus Forsythiae |
| BL01 | Rosasterol | 35.87 | 0.75 | 1.28 | Radix Isatidis |
| BL02 | Sinoacutine | 49.11 | 0.46 | 0.70 | Radix Isatidis |
| BL03 | Sinensetin | 50.56 | 0.45 | 1.12 | Radix Isatidis |
| BL04 | Isaindigodione | 60.12 | 0.41 | −0.09 | Radix Isatidis |
| BL05 | Hydroxyindirubin | 63.37 | 0.30 | 0.51 | Radix Isatidis |
| BL06 | Ineketone | 37.14 | 0.30 | 0.39 | Radix Isatidis |
| BL07 | Taxifolin | 60.51 | 0.27 | −0.24 | Radix Isatidis |
| BL08 | Hispidulin | 30.97 | 0.27 | 0.48 | Radix Isatidis |
| BL09 | Quindoline | 33.17 | 0.22 | 1.50 | Radix Isatidis |
| BL10 | Methyllinolenate | 46.15 | 0.17 | 1.48 | Radix Isatidis |
| BL11 | Methyl vaccenate | 31.90 | 0.17 | 1.43 | Radix Isatidis |
| BL12 | Elaidolinolenic | 45.01 | 0.15 | 1.21 | Radix Isatidis |
| CH01 | Cedrenol | 108.56 | 0.76 | 1.30 | Radix Bupleuri |
| CH02 | Isoborneol | 86.98 | 0.76 | 1.27 | Radix Bupleuri |
| CH03 | Ledol | 82.78 | 0.66 | 1.32 | Radix Bupleuri |
| CH04 | Borneol | 81.8 | 0.64 | 1.22 | Radix Bupleuri |
| ID | Compound Name | OB | DL | Caco-2 | Herbs |
| CH05 | Spathulenol | 80.01 | 0.53 | 1.26 | Radix Bupleuri |
| CH06 | Sainfuran | 79.91 | 0.41 | 0.90 | Radix Bupleuri |
| CH07 | Scoparone | 74.75 | 0.24 | 0.85 | Radix Bupleuri |
| CH08 | Methyleugenol | 73.36 | 0.23 | 1.47 | Radix Bupleuri |
| CH09 | Hexanoic acid | 73.08 | 0.20 | 0.80 | Radix Bupleuri |
| CH10 | Dibutyl phthalate | 64.54 | 0.13 | 0.80 | Radix Bupleuri |
| CH11 | Isorhamnetin | 49.6 | 0.07 | 0.31 | Radix Bupleuri |
| CH12 | Longikaurin A | 47.72 | 0.05 | 0.08 | Radix Bupleuri |
| CH13 | Saikosaponin D | 34.39 | 0.01 | −1.94 | Radix Bupleuri |
| BJ01 | Oleanolic acid | 29.02 | 0.76 | 0.59 | Herba Patriniae |
| BJ02 | Sitosterol | 36.91 | 0.75 | 1.32 | Herba Patriniae |
| BJ03 | Sinoacutine | 63.39 | 0.53 | 0.72 | Herba Patriniae |
| BJ04 | Asperglaucide | 58.02 | 0.52 | 0.28 | Herba Patriniae |
| BJ05 | Bolusanthol B | 39.94 | 0.41 | 0.29 | Herba Patriniae |
| BJ06 | Villosolside | 14.44 | 0.35 | −1.51 | Herba Patriniae |
| BJ07 | Acacetin | 34.97 | 0.24 | 0.67 | Herba Patriniae |
| BJ08 | Vilmorrianine C | 33.96 | 0.22 | 0.59 | Herba Patriniae |
| BJ09 | Apigenin | 23.06 | 0.21 | 0.43 | Herba Patriniae |
| BJ10 | Villosol | 99.3 | 0.08 | −0.17 | Herba Patriniae |
| MB01 | Dihydroverticillatine | 42.69 | 0.84 | 0.56 | Herba Verbenae |
| MB02 | Ursolic acid | 16.77 | 0.75 | 0.67 | Herba Verbenae |
| MB03 | β-Carotene | 37.18 | 0.58 | 2.25 | Herba Verbenae |
| MB04 | Artemetin | 49.55 | 0.48 | 0.81 | Herba Verbenae |
| MB05 | Cornudentanone | 39.66 | 0.33 | 0.47 | Herba Verbenae |
| MB06 | Aucubin | 4.17 | 0.33 | −1.83 | Herba Verbenae |
| MB07 | Diosmetin | 31.14 | 0.27 | 0.46 | Herba Verbenae |
| MB08 | Hastatoside | 142.41 | 0.12 | −0.54 | Herba Verbenae |
| MB09 | (+)-α-Curcumene | 26.56 | 0.06 | 1.93 | Herba Verbenae |
| LG01 | Stigmasterol | 43.83 | 0.76 | 1.44 | Rhizoma Phragmitis |
| LG02 | Tricin | 27.86 | 0.34 | 0.51 | Rhizoma Phragmitis |
| LG03 | (+)-Thujopsene | 53.81 | 0.12 | 1.85 | Rhizoma Phragmitis |
| LG04 | Arctinal | 58.01 | 0.07 | 1.33 | Rhizoma Phragmitis |
| LG05 | Caffeic acid | 25.76 | 0.05 | 0.21 | Rhizoma Phragmitis |
| LG06 | Eugenol | 56.24 | 0.04 | 1.35 | Rhizoma Phragmitis |
| LG07 | p-Coumaric acid | 43.29 | 0.04 | 0.46 | Rhizoma Phragmitis |
| GC01 | Glycyrol | 90.78 | 0.67 | 0.71 | Radix Glycyrrhizae |
| GC02 | Licoricone | 63.58 | 0.47 | 0.53 | Radix Glycyrrhizae |
| GC03 | Glabridin | 53.25 | 0.47 | 0.97 | Radix Glycyrrhizae |
| GC04 | Isotrifoliol | 31.94 | 0.42 | 0.53 | Radix Glycyrrhizae |
| GC05 | Lupiwighteone | 51.64 | 0.37 | 0.68 | Radix Glycyrrhizae |
| GC06 | Dehydroglyasperin C | 53.82 | 0.37 | 0.68 | Radix Glycyrrhizae |
| ID | Compound Name | OB | DL | Caco-2 | Herbs |
| GC07 | Medicarpin | 49.22 | 0.34 | 1.00 | Radix Glycyrrhizae |
| GC08 | Licochalcone A | 40.79 | 0.29 | 0.82 | Radix Glycyrrhizae |
| GC09 | Kumatakenin | 50.83 | 0.29 | 0.61 | Radix Glycyrrhizae |
| GC10 | Calycosin | 47.75 | 0.24 | 0.52 | Radix Glycyrrhizae |
| GC11 | Formononetin | 69.67 | 0.21 | 0.78 | Radix Glycyrrhizae |
| GC12 | Licochalcone B | 76.76 | 0.19 | 0.47 | Radix Glycyrrhizae |
| GC13 | Pinocembrin | 64.72 | 0.18 | 0.61 | Radix Glycyrrhizae |
| GC14 | Liquiritigenin | 32.76 | 0.18 | 0.51 | Radix Glycyrrhizae |
| GC15 | Echinatin | 66.58 | 0.17 | 0.38 | Radix Glycyrrhizae |
| GC16 | Isoliquiritigenin | 85.32 | 0.15 | 0.44 | Radix Glycyrrhizae |
3.3. The pharmacology synergy from network level
3.3.1. C-T network
Generally speaking, the therapeutic effect of herbal medicine is related to its protein targets, and drug reactions (treatment and adverse reactions) are affected by the network topology. System-level network analysis can provide useful information for understanding and gaining insights into different types of molecular relationships, such as how genes are associated with various diseases or the interaction between drugs and their cellular targets.
A total of 80 targets were predicted from the SysDT model. Presently, in order to investigate how a “multi-components, multi-targets” treatment system such as SFJDC exerts therapeutic effects on SARS-CoV-2, a static C-T network was constructed by using the obtained 94 active compounds and their corresponding 80 target proteins. In this net, the active compounds and targets are denoted by the circles and octagons, respectively. Additionally, the degree, an important quantitative property from the C-T network was also analyzed, which was defined as the number of edges of the nodes. As shown in Fig. 2 , most active compounds have at least > 1 protein targets. For instance, physovenine (HZ01, degree = 31) possesses the largest number of degree, and the following are wogonin (LQ13, degree = 25), artemetin (MB04, degree = 24), eugenol (LG06, degree = 23), showing that these compounds play key roles in the treatment of diseases. Further observations present that the mean number of protein target for each compound is 13, revealing the multi-target features of components in SFJDC. At the same time, at least more than one active compound were hit by the most potential targets. During them, the target with the highest degree (Degree = 59) is ESR1, and the followings are NOS2 (Degree = 48), PPARG (Degree = 42), PIMI (Degree = 40), respectively, demonstrating that they are the potential targets of SFJDC for the treatment of SARS-CoV-2.
Fig. 2.

The C-T network, the active compounds and targets are denoted by the circles and octagons, respectively.
To sum up, the C-T network analysis provides a well understanding of the multicomponent synergy and the biologically relevant connectivity patterns, giving an insight information for the action mechanism of the SFJDC.
3.3.2. T-F Network
It has been well documented that SARS-CoV-2 is characterized by an over exuberant inflammatory response and is the leading cause of lung damage and subsequent death (Stebbing et al., 2020). Additionally, host immune and inflammatory responses are also important factors leading to life-threatening in SARS-CoV-2 patients (Chen et al., 2020). Although researchers are racing against time to develop vaccines and test antiviral drugs in clinical trials, there is no effective prophylactic and clinical treatment for COVID −19 yet (Liao et al., 2020). Therefore, at present, the primary therapies of SARS-CoV-2 are immunomodulatory and anti-inflammatory strategy. Interestingly, most of our predicted targets from the SFJDC are related to inflammation and immune system. Consequently, to further investigate the pharmacology synergistic mechanism of SFJDC against novel coronavirus, the T-F network employing 80 targets are associated with regulating immunity and anti-inflammatory effect was constructed (Fig. 3 ).
Fig. 3.

T-F function network, which the predicted targets related to immunomodulation and inflammation.
3.3.3. Components acting on immunomodulation related targets
As the stimulation of T-cell apoptosis, SARS-CoV-2 can cause abnormal response of antiviral T-cell, and thus the immune system was collapsed (Liao et al., 2020). Therefore, immunomodulation is a critical factor for the treatment of SARS-COV-2. As shown in Fig. 4 , many active compounds and corresponding targets have significant relationships with immunization including ESR1, ESR2, MAPK14, F10, etc. Among them, ESR1 was hit by the largest number of candidate compounds (Degree = 60), indicating its crucial role in regulating immune response. Indeed, a variety of epidemiological data suggest the major role of estrogen (ER) receptors in the etiology and pathophysiology of chronic immune disease (Cutolo et al., 2010). For example, ER receptors regulate the development of innate and adaptive immune system cells and immune cells (Cutolo et al., 2010). In addition, HSP90AA1 reportedly affects the response of systemic lupus erythematosus patients to glucocorticoid therapy and lipopolysaccharide-induced inflammation and immunomodulatory responses (Wang et al., 2020). Besides, ADRB2 and HSP90AA1 may play important roles in chronic obstructive pulmonary disease (COPD)-associated and squamous cell lung carcinoma (SCC) (Wang et al., 2020). All the findings provide an insight evidence that the immunomodulation is probably one of the therapeutic mechanisms of SFJDC against SARS-COV-2.
Fig. 4.

C-T-F function network, which circles and hexagons denote active compounds and corresponding immunomodulatory-related targets.
Collectively, these results provide an insight evidence that the immunomodulation contained in the C-T-F network has important roles in fighting SARS-COV-2, further validating the drug targeting approach.
3.3.4. Components acting on inflammatory related targets
Emerging evidence suggests that more and more inflammation-related biomarkers were found in patients with SARS-COV-2 pneumonia, including erythrocyte sedimentation rate and IL-6 (Zhang & Liu, 2020). Thus, inflammatory responses is the other important predisposing factor for in SARS-COV-2 patients (Zhang & Liu, 2020).
Actually, with further observation of the C-T-F network (Fig. 5 ), we found that many targets with high-degree are related to inflammatory mediators like PTGS2 (Prostaglandin G/Hsynthase 2), NOS2 (Nitric oxide synthase), F2 (Prothrombin), PPARG and PTGS1 (Prostaglandin G/H synthase1). For instance, PTGS2 with the highest number of C-T interactions, is more important in the prostaglandin synthesis associated with inflammation. It is proposed to influence prostate cancer progression and development via neoangiogenesis and increased resistance to apoptosis (Kirschenbaum et al., 2001, Liu et al., 2000, Song, 2002). In addition, current studies also suggest that PPARG can modulate the inflammatory response, showing potential therapeutic applications in SARS-COV-2-caused “cytokine storm” (Cohen et al., 2006). Accordingly, the potential for application of anti-inflammatory drugs has drawn tremendous attention. Remarkably, we observed that many chemicals interacted with the targets associated with inflammation, and all these targets may synergistically regulate the body for the prevention and treatment of SARS-COV-2 pneumonia.
Fig. 5.

C-T-F function network, which circles and hexagons denote active compounds and corresponding inflammatory-related targets.
Overall, the results obtained from this section indicate that multiple target proteins of SFJDC are closely related to immunomodulatory and inflammatory effects, revealing that the therapeutic mechanism of SFJDC against SARS-COV-2 pneumonia is to hit multiple related target proteins through multiple components.
3.4. Gene Ontology analysis and PPI network construction
To further explore whether the targets fished by the active compound of SFJDC are closely associated with immunomodulation and inflammation, we performed the functional bioinformatics analysis. Fig. 6 A and 3B depict the representation of the mutual relationships between target which belong immunomodulation and inflammation, respectively. Fig. 6C, D, E and F show the significantly enriched GO terms form targets related to immunomodulation and inflammation.
Fig. 6.

Gene Ontology (GO) analysis of the SFJDC targets. (A-D) GOChord and Mulberry plots to donate the relationship between GO terms and potential targets which are related to immunomodulatory and inflammatory effects, respectively (E-F) Bubble chart of the GO analysis from the targets.
Obviously, these target proteins are critically involved in various biological processes, which can mainly be grouped into modulating the immune system and the response of inflammation (Fig. 6A and 6B). For example, targets PIK3CG, TNF, ADRA2A and DPP4 involved in the positive regulation of MAP kinase activity (GO: 0043406) and nitric oxide biosynthetic process (GO: 0045429), as well as T cell activation (GO: 0042110) play fundamental roles in the regulation of innate immune responses. In addition, MAPK14, NOS2 participated in the regulation of cytokine production involved in inflammatory response (GO: 1900015), and PTGS2, PTGS1 associated with oxidative stress (GO: 0006979), as well as AR, F2, CDK2 involved in regulation of cell proliferation (GO: 0008284), are all related to inflammatory response. Apart from the regulatory role in response to immunity and inflammation, the predicted targets like HSP90AA1, MAPK14, TOP2A, KDR involved in the ATP binding (GO: 0005524), and integral component of plasma membrane (GO: 0005887) may be directly connected to antiviral response. All these results suggest the probably immunomodulatory and anti-inflammatory mechanism of SFJDC on the treatment of SARS-COV-2 pneumonia.
To further identify the obtained targets related with immunomodulation and inflammation, we also constructed the protein–protein interactions (PPI) network (Fig. 7 ). The PPI network analyses can give us a network of molecular interactions formed between the proteins products of the genes studied. As depicted in Fig. 7A and 7B, ESR1, ESR2, GSK3β, PPARG, PTGS2 and NOS2 present the highest degree centrality values among the obtained targets, indicating the importance of these proteins in the resulting PPI network. Notably, these observations are correlated well with the results obtained from the C-T net, showing the potential hub targets associated with the immunomodulation and inflammation. Fig. 7C and 7D show the similarities which target families occur patterns across genomes. The findings obtained provide a new possibility for revealing the molecular mechanisms of SFJDC against COVID-19 from a systematic point of view.
Fig. 7.

Protein-protein interactions (PPI) PPI networks analysis. (A-B) STRING-generated PPI of targets which are belonging to the immunomodulatory and inflammatory effects, respectively. (C-D) The similarities which target families occur patterns across genomes.
3.5. Target-Pathway analysis
In addition to the multi-components and multi-targets features, herbal medicines also have the characteristic of multiple biological pathways when they present the favorable therapeutic effects in the treatment of diseases. Analysis of biological pathways and interactions networks can uncover the robustness of biological systems, promoting a more comprehensive understanding of SFJDC in the treatment of SARS-COV-2 pneumonia. Herein, all corresponding biological pathways of targets in herbal compounds were obtained from the database of KEGG (http://www.genome.jp/kegg/). Fig. 8 A and 8B display the most significantly enriched GO terms of the pathways. Subsequently, to further investigate the synergistic mechanism of SFJDC against SARS-COV-2 pneumonia, an integrated pathway network including TNF, NF-kappa B, PI3K-Akt, VEGF, JAK-STAT and Estrogen signaling pathways were assembled on the basis of the disease pathology (Fig. 8C).
Fig. 8.

SARS-COV-2 pathway. (A, B) KEGG enrichment analysis from the potential targets. (C) Distribution of SFJDC targets which related to immunomodulatory and anti-inflammatory pathways.
As shown in Fig. 8C, the active ingredients of SFJDC exert the therapeutic effects by regulating the multiple proteins in these six major pathways, most of which revealed associations with immunomodulation and inflammation. For example, PI3K signaling regulates many processes such as autophagy, cell survival, oxidative burst, phagocytosis, and inflammatory cytokine production (Cremer, Butchar, & Tridandapani, 2011). Additionally, when Akt enhances the level of pro-inflammatory cytokines like TNFα, IL-6 and IL-12, it can also reduce anti-inflammatory cytokines IL-10 production (Cremer et al., 2011). Since TNF is a major mediator of apoptosis as well as immunity and inflammation (Chen, 2002), TNF signaling pathway has been reported to largely participate in the pathogenesis of various human diseases and may serve as a therapeutic pathway target in preventing and treating diabetes, rheumatoid arthritis, and inflammatory bowel disease (Chen, 2002). Besides, multiple literatures have demonstrated that Estrogen signaling pathway can affect innate immune cell and regulate CD4+ and CD8+ cells, suggesting the key role in downregulating immune responses (Khan & Ansar Ahmed, 2016).
Overall, the active compounds of SFJDC can act on the same protein in different pathways and are connected to one or more signaling systems. Through the interaction between the potential targets and compounds, these pathways show an interdependent relationship with each other, suggesting that herbs may work synergistically through these different pathways. Also, a candidate compound in herbal medicine may act on different proteins in the same or different pathways, showing the mechanism of multiple targets of SFJDC.
3.6. In silico validation of the C-T interactions
3.6.1. Molecular dynamics simulations
In order to explore the binding information and binding stability of TCM candidates at the binding pocket of the targets, six compounds with their corresponding potential targets were performed for MD simulations. All docking complexes were simulated for 100 ns to extract the kinetic conformational changes of the compounds and the target in aqueous solution. Root-mean-squared deviation (RMSD) was used to study atomic fluctuations during simulations. As depicted in Fig. 9 , whole-molecule RMSDs values for candidates-receptor complexes reveal that each ligand reached stability at approximately 3 ~ 4 Å. This trajectory data confirms that all C-T interactions stabilized after approximately 100 ns and the binding site of the targets are able to accommodate candidate compounds with no major conformational readjustments.
Fig. 9.

The trajectories of the RMSD for six complexes from C-T interactions over long MD simulations.
Fig. 10, Fig. 11 show the surface structure and the cavity depth of the binding sites in each complex. Moreover, the snapshots of the binding mode for compounds saikosaponin D, isaindigodione, villosol, stigmasterol, glycyrol and phillyrin with corresponding their corresponding targets ESR2, GSK3β, NOS2, NR3C1, PPARG and TOP2A with the average structure binding information of the last 1 ns are shown in Fig. 12 .
Fig. 10.

The binding modes of three complexes from C-T interactions. (A) ESR2- saikosaponin D. (B) GSK3-isaindigodione. (C) NOS2-villosol.
Fig. 11.

The binding modes of three complexes from C-T interactions. (A) NR3C1-stigmasterol D. (B) PPARG-glycyrol. (C) TOP2A-phillyrin.
Fig. 12.

H-bonding networks within the accurate binding model of the C-T complexes obtained from the MD simulations. (A) ESR2-saikosaponin D. (B) GSK3-isaindigodione. (C) NOS2-villosol. (D) NR3C1-stigmasterol D. (E) PPARG-glycyrol. (F) TOP2A-phillyrin.
Fig. 12 depicts that six candidates of SFJDC extend deeply into the binding cavity of the targets, in which the key amino acids via the H-bonds and hydrophobic interactions are also observed. Obviously, the saikosaponin D adopts an appropriate orientation that making a strong two hydrogen bonds interactions with the Met494 and Tyr397 (Fig. 12A). Additionally, we can see that the isaindigodione (Fig. 12B) is stabilized in the binding site of the GSK3 by three hydrogen bonds between the saikosaponin D and Ser66, Tyr134 and Cys199, indicating the good inhibitory activity of compound isaindigodione. Fig. 12C indicates that villosol is directly interacted with a deep active site, establishing hydrogen bonds network interaction with residues Tyr491. As shown in Fig. 12D, the conformation of stigmasterol was stabilized by hydrophobic interaction with Ile608, Ile629, Met634, Phe623, Leu563, Leu636, Leu563, Leu608 and Leu733. Besides, Fig. 12E shows that glycyrol can form more hydrogen bond interactions with His323, which was different from the other compounds. Moreover, four hydrogen bonds with residues Asn91, Asn120, Asn95 and Ile125 were observed between the compound and the target (Fig. 12F), demonstrating that phillyrin is directed to the binding site in the entrance cavity of TOP2A. These results indicate that the stabilization of the C-T complexes are mainly via a network of hydrogen bonds interactions.
3.6.2. Binding free energy analysis and bioactivity prediction
From the Table 3 we found that six compounds show low binding free energies (-8.24 ~ -60.40 kcal/mol) by hitting the corresponding targets, demonstrating the good binding affinities to their targets. Additionally, we observe that the contribution of the ΔEelectrostatic energy is higher than the ΔEvdwz energy, indicating that Van der Waals energy plays an important role in the contribution of these six C-T complexes interactions. Moreover, the binding energy component shows that non-polar properties (ΔGSA) provides a larger beneficial contribution than the polar solvation. As a result, a conclusion can be drawn that ΔEvdwz energy and the polar solvation free energy may exert significant contributions in the binding affinities. In addition, the low Ki values of all compounds strongly support the high potential of becoming a lead compound (Reynolds and Reynolds, 2017, Hughes et al., 2011).
Table 3.
Contributions of various energy components to the binding free energy (kcal/mol) and the predicted bioactivities of the compounds.
| Complexes | ΔEvdwz | ΔEelectrostatic | ΔGPB/GB | ΔGSA | ΔGbind | Ki (μM) |
|---|---|---|---|---|---|---|
| ESR2-saikosaponin D | −102.52 | −6.47 | 56.66 | −8.16 | −60.40 | 5.26E-39 |
| GSK3β-quercetin | −31.44 | −10.69 | 37.17 | −3.29 | −8.24 | 0.91 |
| NOS2-eugenol | −20.02 | −11.27 | 22.03 | −2.73 | −11.98 | 1.66E-03 |
| NR3C1-rutin | −62.07 | −3.59 | 19.65 | −5.70 | −51.64 | 1.39E-32 |
| PPARG -isorhamnetin | −40.62 | −2.52 | 28.03 | −4.11 | −19.19 | 8.54E-09 |
| TOP2A −1-quercetin | −65.84 | −24.48 | 70.06 | −6.71 | −26.92 | 1.84E-14 |
ΔEvdw, van der Waals energy; ΔEelectrostatic, electrostatic energy; ΔGPB, the polar solvation energy with the PB model; ΔGbind is the sum of ΔEvdw + ΔEelectrostatic + ΔGPB/GB + ΔGSA.
In summary, analysis of ligand-target complex structures helps visualize structural changes elicited by the test SFJDC candidates during MD simulations, and may provide insights to the possible mechanisms. Besides, the MD simulations, MM-PBSA analysis and bioactivity prediction offer indirect information on biological efficacies of the candidate ligands which may be through their relevant targets for anti-SARS-COV-2, thus further validating our C-T interactions.
3.7. The inhibition of SFDC on SARS-COV-2
To evaluate whether the active molecule can directly inhibit the SARS-COV-2 receptor, the MD simulations were also performed. Based on a recently solved crystal structure (ID: 6LU7) of the SARS-CoV-2, six active compounds were randomly selected from the SFJDC were docked into the SARS-CoV-2 receptor, and then the MD simulations for these six complexes from docking results were performed for further analyzing the dynamics and the binding affinities of active ingredients and the corresponding targets.
Fig. 13 shows the RMSD diagrams of the compounds targets complexes. In this figure, the RMSD value for the six complexes rises to 0.15 ~ 4.0 Å in the initial 10 ns, and keeps this rate over the simulations process, demonstrating the system stability, and thus, the overall accuracy of the topology. Fig. 14, Fig. 15 show the binding modes of compounds isorhamnetin, kaempferol, polydatin, quercetin, taxifolin and wogonin with SARS-COV-2. It can be noted that the interaction modes of six compound exhibits similar features including key residues and hydrogen bonding interactions with SARS-COV-2 in the binding site.
Fig. 13.

The trajectories of the RMSD for targeting SARS-CoV-2 with chrysin, isoliquiritigenin, liquiritigenin, and quercetin over 100 ns MD simulations.
Fig. 14.

The stereoview of binding mode for six active compounds with SARS-COV-2. (A) Isorhamnetin. (B) Kaempferol. (C) Polydatin. (D) Quercetin. (E) Taxifolin. (F) Wogonin.
Fig. 15.

The accurate binding interactions of SARS-COV-2 with active compounds from SFJDC. (A) Isorhamnetin, (B) Kaempferol, (C) Polydatin, (D) Quercetin, (E) Taxifolin, and (F) wogonin.
In addition, the binding free energy were also computed after the 100 ns MD simulations for six active ingredients. The binding free energies (Table 4 ) of isorhamnetin, kaempferol, polydatin, quercetin, taxifolin and wogonin for the SARS-COV-2 model were −9.41, −18.57, −13.21, −18.77, −15.17 and −12.18 kcal/mol respectively, indicating the good binding affinities of these compounds against SARS-COV-2. Specifically, for the compounds Tipranavir, Tegobuvir and Lonafarnib, ΔE vdw contributions are greater than the ΔE electrostatic interaction, suggesting that ΔE vdw contributes the mainly driving force for the combination of these compounds.
Table 4.
Contributions of various energy components of ligand to the SARS-COV-2 (kcal/mol).
| Compounds | ΔEvdwz | ΔEelectrostatic | ΔGPB/GB | ΔGSA | ΔGbind | Ki (μM) |
|---|---|---|---|---|---|---|
| Isorhamnetin | −6.85 | −1.34 | −0.64 | −0.560 | −9.41 | 0.13 |
| Kaempferol | –32.02 | −6.19 | 22.70 | −3.09 | −18.57 | 2.42E-08 |
| Polydatin | −26.96 | −10.45 | 27.40 | −3.28 | −13.21 | 2.08E-04 |
| Quercetin | –33.07 | −12.96 | 30.42 | −3.24 | −18.77 | 1.73E-08 |
| Taxifolin | −31.78 | −7.76 | 27.70 | −3.35 | −15.17 | 7.63E-04 |
| Wogonin | −27.68 | −2.74 | 20.65 | −2.42 | −12.18 | 1.17E-03 |
ΔEvdw, van der Waals energy; ΔEelectrostatic, electrostatic energy; ΔGPB, the polar solvation energy with the PB model; ΔGbind is the sum of ΔEvdw + ΔEelectrostatic + ΔGPB/GB + ΔGSA.
In detail, Table 4 showed that six compound could bind to the binding pockets with SARS-CoV-2 through van der Waals energy and hydrophobic interactions, involving Gln192, Glu166, Cys44, Asn142, Tyr54, His41, Thr190, Thr26, Cys145 and His163. The high binding affinities of the low Ki and good inhibitions of these compounds indicate that the ligands bind well to their receptors, which may suggest the good binding activities of the core active compounds to SARS-CoV-2 receptor. Therefore, we speculate that active compounds from SFJDC have potential activity against SARS-Cov-2.
4. Conclusion
Currently, to elucidate the effect of anti-SARS-CoV-2 from the SFJDC and further to explore its therapeutic mechanism, a systems analyses by a large scale combining the ADME prescreening, targets fishing, network construction, functional enrichment analyses, docking and MD simulations, were carried out. The main findings are as follows:
-
(1)
94 compounds in eight herbs from SFJDC are identified as candidate compounds through ADME screening and their 80 corresponding targets are mostly associated with immunomodulation and inflammation.
-
(2)
The networks, GO and target-pathway analysis provides a better understanding of the synergistic mechanism of SFJDC at the systems molecular level.
-
(3)
The MD results confirm the reliability of the drug-target interactions obtained in this study and the binding free energy analyses demonstrate that the active compounds from SFJDC have potential activity against SARS-Cov-2 receptor.
In summary, all the work suggest that SFJDC may interact with various targets related to immunomodulation and inflammation, uncovering that the potential therapeutic effect of SFJDC against SARS-Cov-2 may be through regulating these target proteins. This study not only offers new insights for understanding the molecular mechanism of SFJDC on anti-SARS-CoV-2, but also provides some valuable clues for developing novel pharmaceutical strategies to control the current coronavirus.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This project was supported in part by National Natural Science Foundation of China (NO. 81773967); The Open Research Fund of Anhui Provincial Cardiovascular Institute (NO. KF2018015); Anhui Key Research and Development Program (NO. 201904a07020092)
Author contributions
Zhengang Tao, Lei Zhang and Thomas Friedemann collected, organized and compiled the Guangshan Yang, Jinhu Li and Yaocai Wen scrutinized the data. Zhengang Tao, Lei Zhang and Thomas Friedemann wrote the manuscript and prepared figures. Jinghui Wang and Aizong Shen conceived the idea and coordinated the project.
References
- CDC COVID-19 Response Team, CDC COVID-19 Response Team, Bialek, S., Boundy, E., Bowen, V., Chow, N., … Sauber-Schatz, E. (2020). Severe Outcomes Among Patients with Coronavirus Disease 2019 (COVID-19) -United States, February 12-March 16, 2020. MMWR. Morbidity and Mortality Weekly Report, 69(12), 343-346. [DOI] [PMC free article] [PubMed]
- Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology. 2018;17(3):181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P.…Jiang T. Genome composition and divergence of the novel coronavirus (2019-ncov) originating in China. Cell Host & Microbe. 2020;27(3):325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C. Traditional Chinese Medicine is a resource for drug discovery against 2019 novel coronavirus (SARS-CoV-2) Journal of Integrative Medicine. 2020;18(2):87–88. doi: 10.1016/j.joim.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Chang N., Han Y., Zhou M., Gao J., Hou Y.…Bai G. Anti-inflammatory effects of Shufengjiedu capsule for upper respiratory infection via the ERK pathway. Biomedicine & Pharmacotherapy. 2017;94:758–766. doi: 10.1016/j.biopha.2017.07.118. [DOI] [PubMed] [Google Scholar]
- Ji S., Bai Q., Wu X., Zhang D.-W., Wang S., Shen J.-L., Fei G.-H. Unique synergistic antiviral effects of Shufeng Jiedu Capsule and oseltamivir in influenza A viral-induced acute exacerbation of chronic obstructive pulmonary disease. Biomedicine & Pharmacotherapy. 2020;121 doi: 10.1016/j.biopha.2019.109652. [DOI] [PubMed] [Google Scholar]
- Zhang L., Liu Y. Potential interventions for novel coronavirus in China: A systematic review. Journal of Medical Virology. 2020;92(5):479–490. doi: 10.1002/jmv.25707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Wang Y., Lu Y., Li J., Song Y., Nyamgerelt M., Wang X. Diagnosis and treatment of novel coronavirus pneumonia based on the theory of traditional Chinese medicine. Journal of Integrative Medicine. 2020;18(4):275–283. doi: 10.1016/j.joim.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV) BioScience Trends. 2020;14(1):69–71. doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- Qu X.K., Hao S.L., Ma J.H., Wei G.Y., Song K.Y., Tang C.…Gao Y.F. Observation on clinical effect of Shufeng Jiedu Capsule combined with Arbidol Hydrochloride Capsule in treatment of COVID-19. Chinese Traditional and Herbal Drugs. 2020;5(51):1167–1170. [Google Scholar]
- Yu H., Chen J., Xu X., Li Y., Zhao H., Fang Y.…Wang Y. A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Zhang W., Huang C., Li Y., Yu H., Wang Y.…Ling Y. A Novel Chemometric Method for the Prediction of Human Oral Bioavailability. International Journal of Molecular Sciences. 2012;13(6):6964–6982. doi: 10.3390/ijms13066964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett P., Barnard J.M., Downs G.M. Chemical Similarity Searching. Journal of Chemical Information and Computer Sciences. 1998;38(6):983–996. [Google Scholar]
- Pham The H., González-Álvarez I., Bermejo M., MangasSanjuan V., Centelles I., Garrigues T.M., Cabrera-Pérez M.Á. In silico prediction of caco-2 cell permeability by a classification QSAR approach. Molecular Informatics. 2011;30(4):376–385. doi: 10.1002/minf.201000118. [DOI] [PubMed] [Google Scholar]
- Shannon P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arooj M., Sakkiah S., Kim S., Arulalapperumal V., Lee K.W. A combination of receptor-based pharmacophore modeling & qm techniques for identification of human chymase inhibitors. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0063030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91(1–3):43–56. [Google Scholar]
- Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Structure, Function, and Bioinformatics. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L.…Cheatham T.E. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts of Chemical Research. 2000;33(12):889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Li Y., Yao J., Han C., Yang J., Chaudhry M., Wang S.…Yin Y. Quercetin, inflammation andimmunity. Nutrients. 2016;8(3):167. doi: 10.3390/nu8030167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand David A., Arulmoli R., Parasuraman S. Overviews of biological importance of quercetin: A bioactive flavonoid. Pharmacognosy Reviews. 2016;10(20):84. doi: 10.4103/0973-7847.194044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W., Wu Y., Xie X., Zhou X., Wei M., Soromou L.-W.…Wang D. Phillyrin attenuates LPS-induced pulmonary inflammation via suppression of MAPK and NF-κB activation in acute lung injury mice. Fitoterapia. 2013;90:132–139. doi: 10.1016/j.fitote.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Wang J., Li Y., Yang Y., Du J., Zhao M., Lin F.…Wang B. Systems pharmacology dissection of multiscale mechanisms of action for herbal medicines in treating rheumatoid arthritis. Molecular Pharmaceutics. 2017;14(9):3201–3217. doi: 10.1021/acs.molpharmaceut.7b00505. [DOI] [PubMed] [Google Scholar]
- Hassanin M., Tolba M., Tadros M., Elmazar M., Singab A.-N. Wogonin a promising component of scutellaria baicalensis: A review on its chemistry, pharmacokinetics and biological activities. Archives of Pharmaceutical Sciences Ain Shams University. 2019;3(2):170–179. [Google Scholar]
- Błach-Olszewska Z., Jatczak B., Rak A., Lorenc M., Gulanowski B., Drobna A., Lamer-Zarawska E. Production of cytokines and stimulation of resistance to viral infection in human leukocytes by scutellaria baicalensis flavones. Journal of Interferon & Cytokine Research. 2008;28(9):571–582. doi: 10.1089/jir.2008.0125. [DOI] [PubMed] [Google Scholar]
- Choi E.-J., Lee C.-H., Kim Y.-C., Shin O.S. Wogonin inhibits Varicella-Zoster (shingles) virus replication via modulation of type I interferon signaling and adenosine monophosphate-activated protein kinase activity. Journal of Functional Foods. 2015;17:399–409. [Google Scholar]
- Ye J., Piao H., Jiang J., Jin G., Zheng M., Yang J.…Yan G. Polydatininhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-κB and Nrf2/HO-1 pathways. Scientific Reports. 2017;7(1) doi: 10.1038/s41598-017-12252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Wu M., Xiong W., Li J., An Y., Ren J.…Zhong G. Saikosaponin-d ameliorates dextran sulfate sodium-induced colitis by suppressing NF-κB activation and modulating the gut microbiota in mice. International Immunopharmacology. 2020;81 doi: 10.1016/j.intimp.2020.106288. [DOI] [PubMed] [Google Scholar]
- Stebbing J., Phelan A., Griffin I., Tucker C., Oechsle O., Smith D., Richardson P. COVID-19: Combining antiviral and anti-inflammatory treatments. The Lancet Infectious Diseases. 2020;20(4):400–402. doi: 10.1016/S1473-3099(20)30132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Qi F., Shi K., Li Y., Li J., Chen Y.…Xia J. Thalidomide combined with low-dose short-term glucocorticoid in the treatment of critical Coronavirus Disease 2019. Clinical and Translational Medicine. 2020;10(2) doi: 10.1002/ctm2.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J.…Zhang Z. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nature Medicine. 2020;26(6):842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Cutolo M., Brizzolara R., Atzeni F., Capellino S., Straub R.H., Puttini P.C.S. The immunomodulatory effects of estrogens. Annals of the New York Academy of Sciences. 2010;1193(1):36–42. doi: 10.1111/j.1749-6632.2009.05383.x. [DOI] [PubMed] [Google Scholar]
- Yang Y., Li Y., Wang J., Sun K., Tao W., Wang Z.…Wang Y. Systematic Investigation of Ginkgo Biloba Leaves for Treating Cardio-cerebrovascular Diseases in an Animal Model. ACS Chemical Biology. 2017;12(5):1363–1372. doi: 10.1021/acschembio.6b00762. [DOI] [PubMed] [Google Scholar]
- Kirschenbaum A., Liu X.-H., Yao S., Levine A.C. The role of cyclooxygenase-2 in prostate cancer. Urology. 2001;58(2):127–131. doi: 10.1016/s0090-4295(01)01255-9. [DOI] [PubMed] [Google Scholar]
- Liu X.H., Kirschenbaum A., Yao S., Lee R., Holland J.F., Levine A.C. Inhibition of cyclooxygenase-2 suppresses angiogenesis and the growth of prostate cancer in vivo. Journal of Urology. 2000;164(3 Part 1):820–825. doi: 10.1097/00005392-200009010-00056. [DOI] [PubMed] [Google Scholar]
- Song X. Cyclooxygenase-2, player or spectator in cyclooxygenase-2 inhibitor-induced apoptosis in prostate cancer cells. CancerSpectrum Knowledge Environment. 2002;94(8):585–591. doi: 10.1093/jnci/94.8.585. [DOI] [PubMed] [Google Scholar]
- Cohen B.L., Gomez P., Omori Y., Duncan R.C., Civantos F., Soloway M.S.…Lokeshwar B.L. Cyclooxygenase-2 (cox-2) expression is an independent predictor of prostate cancer recurrence. International Journal of Cancer. 2006;119(5):1082–1087. doi: 10.1002/ijc.21749. [DOI] [PubMed] [Google Scholar]
- Cremer T.J., Butchar J.P., Tridandapani S. Francisella subverts innate immune signaling: Focus on PI3K/Akt. Frontiers in Microbiology. 2011;2:13. doi: 10.3389/fmicb.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. TNF-R1 signaling: A beautiful pathway. Science. 2002;296(5573):1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Khan D., Ansar Ahmed S. The immune system is a natural target for estrogen action: Opposing effects of estrogen in two prototypical autoimmune diseases. Frontiers in Immunology. 2016;6:635. doi: 10.3389/fimmu.2015.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel M.D., Jurs P.C., Tolan J.W., Muskal S.M. Prediction of human intestinal absorption of drug compounds from molecularstructure. Journal of Chemical Information and Computer Sciences. 1998;38(4):726–735. doi: 10.1021/ci980029a. [DOI] [PubMed] [Google Scholar]
- Lin J.-H., Perryman A.L., Schames J.R., McCammon J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. Journal of the American Chemical Society. 2002;124(20):5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Lynn A. g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Yang Y., Li Y., Zhou W., Chen Y., Wu Q., Pan Y.…Yang L. Exploring the structural determinants of novel xanthine derivatives as A2B adenosine receptor antagonists: A computational study. Journal of Biomolecular Structure and Dynamics. 2018;37(13):3467–3481. doi: 10.1080/07391102.2018.1517612. [DOI] [PubMed] [Google Scholar]
- Jedrzejas M.J., Singh S., Brouillette W.J., Air G.M., Luo M. A strategy for theoretical binding constant, Ki, calculations for neuraminidase aromatic inhibitors designed on the basis of the active site structure of influenza virus neuraminidase. Proteins: Structure, Function, and Genetics. 1995;23(2):264–277. doi: 10.1002/prot.340230215. [DOI] [PubMed] [Google Scholar]
- Jangam C.S., Bhowmick S., Chorge R.D., Bharatrao L.D., Patil P.C., Chikhale R.V.…Islam M.A. Pharmacoinformatics-based identification of anti-bacterial catalase-peroxidase enzyme inhibitors. Computational Biology and Chemistry. 2019;83 doi: 10.1016/j.compbiolchem.2019.107136. [DOI] [PubMed] [Google Scholar]
- Reynolds C.H., Reynolds R.C. Group Additivity in Ligand Binding Affinity: An Alternative Approach to Ligand Efficiency. Journal of Chemical Information and Modeling. 2017;57(12):3086–3093. doi: 10.1021/acs.jcim.7b00381. [DOI] [PubMed] [Google Scholar]
- Hughes J., Rees S., Kalindjian S., Philpott K. Principles of early drug discovery. British Journal of Pharmacology. 2011;162(6):1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]