

Graphical abstract
Keywords: Cytokines, Cytokine storm, Innate Immunity, PAMPs, Sepsis, TLRs
Highlights
-
•
Sepsis infects more than 48.9 million people world-wide, with 19.7 million deaths.
-
•
Cytokine storm plays a significant role in sepsis, along with severe COVID-19.
-
•
TLR signaling pathways plays a crucial role in generating the cytokine storm.
-
•
Endogenous negative regulators of TLR signaling are crucial to regulate cytokine storm.
Abstract
Cytokine storm generates during various systemic acute infections, including sepsis and current pandemic called COVID-19 (severe) causing devastating inflammatory conditions, which include multi-organ failure or multi-organ dysfunction syndrome (MODS) and death of the patient. Toll-like receptors (TLRs) are one of the major pattern recognition receptors (PRRs) expressed by immune cells as well as non-immune cells, including neurons, which play a crucial role in generating cytokine storm. They recognize microbial-associated molecular patterns (MAMPs, expressed by pathogens) and damage or death-associate molecular patterns (DAMPs; released and/expressed by damaged/killed host cells). Upon recognition of MAMPs and DAMPs, TLRs activate downstream signaling pathways releasing several pro-inflammatory mediators [cytokines, chemokines, interferons, and reactive oxygen and nitrogen species (ROS or RNS)], which cause acute inflammation meant to control the pathogen and repair the damage. Induction of an exaggerated response due to genetic makeup of the host and/or persistence of the pathogen due to its evasion mechanisms may lead to severe systemic inflammatory condition called sepsis in response to the generation of cytokine storm and organ dysfunction. The activation of TLR-induced inflammatory response is hardwired to the induction of several negative feedback mechanisms that come into play to conclude the response and maintain immune homeostasis. This state-of-the-art review describes the importance of TLR signaling in the onset of the sepsis-associated cytokine storm and discusses various host-derived endogenous negative regulators of TLR signaling pathways. The subject is very important as there is a vast array of genes and processes implicated in these negative feedback mechanisms. These molecules and mechanisms can be targeted for developing novel therapeutic drugs for cytokine storm-associated diseases, including sepsis, severe COVID-19, and other inflammatory diseases, where TLR-signaling plays a significant role.
1. Introduction
Sepsis leading to multiple organ failure and shock-associated deaths is a very common life threatening condition throughout the world. The annual incidence of sepsis was 31.5 million, with 19.4 million cases of severe sepsis, causing death of 5.3 million people in the high income countries throughout the world in 2015–2016 [1]. This has now increased to 48.9 million cases of sepsis with 11.0 million sepsis-related deaths (represents 19.7% of all global deaths) in 2020 [2] The situation seems worse as these figures are based upon data collected only from high income countries, as there is scarcity of data on its incidence in low- and middle income countries (LMICs). It is important to note that the pattern of origin of sepsis and associated mortality is quite different in LMICs. The major causal factors causing an increased risk for sepsis in the population from LMICs are poverty, overcrowding, high incidence of nosocomial infections, low access to basic healthcare facility, improper hygiene with few public health programs [3], [4]. In addition, in some countries, a very high prevalence of HIV infection (South African, South American and Asian countries etc.) makes sepsis worse [5]. For example, in patients suffering from HIV infection, the incidence of sepsis goes up to 1,000 cases per 100,000 persons [6] and the outcome is worse [7]. Otherwise, its incidence in normal population without AIDS is 150–300 cases per 100,000 persons, while among patients with chronic diseases (diabetes, obesity, hypertension, and cancer) the rate increases to 700 cases per 100,000 patients [8], [9]. Also, COVID-19 is more severe and develops into sepsis in patients with comorbidities (hypertension, diabetes, heart diseases, and obesity etc.) and old age people than younger healthy people [10], [11].
Increasing aged population in developed and high income countries contributes as an additional factor for an increasing incidence of sepsis [12], whereas in LMICs, this is not a major contributing factor for sepsis-related deaths [12]. Despite advances in drug discovery and medicine, the incidence of sepsis has dramatically increased by 8–13% every year over the last decade [13]. It has claimed more lives as compared to combined deaths caused by bowel and breast cancer (World Sepsis Declaration). In addition to high mortality associated with sepsis, its management is a major economic burden, for example, its cost has increased from 14 to 24 billion dollars annually in the United States alone [13], [14]. Thus, sepsis is a major health problem for both developing as well as developed world, and it needs a great attention. A recent step in this direction has been taken by WHO and on Friday, May 26th, 2017, the World Health Assembly (WHA) and the WHO made sepsis a global health priority, by adopting a resolution to improve, prevent, diagnose, and manage sepsis [15]. This decision by WHA and WHO marks a quantum leap in the global fight against sepsis. At this juncture, it has become a very appropriate task to understand pathogenesis of sepsis to design newer therapeutics for its treatment.
Cytokine storm (a profound increase in cytokines, chemokines, and interferons causing severe inflammation and tissue damage, including acute lung injury or ALI) plays a crucial role in sepsis, influenza, and severe COVID-19 pathogenesis [16], [17], [18], [19], [20], [21]. The term cytokine storm was first used in 1993 in an article published on the graft-versus host disease (GVHD) and it popularized in infectious diseases in 2005 due to its use in H5N1 influenza infection [22], [23]. The details of cytokine storm are discussed somewhere else [24]. The current article focuses on Toll-like receptors (TLRs) signaling pathways in sepsis generating cytokine storm and host-derived endogenous molecules and factors involved in the negative regulation of TLR signaling with a potential for future therapeutics .
2. Sepsis as a clinical disease and syndrome
People with immature (neonates and young children) or dysfunctional immune response (old age people) are unable to mount a robust but regulated immune response to clear the pathogen to prevent from developing severe infection and its dissemination to the circulation (bacteremia, viremia, or fungemia) that may lead to sepsis (Fig. 1 ) [25]. For example, in 2017 half of the sepsis cases in the world were detected in children with an estimation of 20 million cases and 2.9 million deaths among children below five years of age [2]. Also, diarrheal diseases (9.2 to 15 million cases/year) and lower respiratory tract infections (1.8 to 2.8 million cases/year) are the largest contributors for sepsis and associated mortality [2]. According to the quick sequential organ failure assessment (qSOFA) in the presence of the suspected infection the increased respiratory rate that is greater than 22 breaths/min (or arterial pCO2 < 32 mmHg, indicating hyperventilation), change in blood pressure (systolic blood pressure ≥ 100 mm Hg or less), and altered mentation (Glasgow Coma Scale score < 15) provide simple bedside criteria for identifying development of sepsis or the poor outcome [26]. Thus according to the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) held in 2016, sepsis is defined as a life-threatening organ dysfunction originated due to the dysregulated host immune response to the infection [26]. Also, Sepsis-3 has recommended that septic shock should now be defined as subset of sepsis, where exaggerated and abnormal response in circulatory, cellular, and metabolic system are major factors for greater risk of mortality among patients as compared to sepsis alone [26]. For example, clinically patients with septic shock should be identified by their requirement to vasopressors to maintain their mean arterial pressure of 65 mm Hg or higher and serum lactate levels more than 2 mmol/L or greater than 18 mg/dL in the absence of hypovolemia [26]. This combination is associated with hospital mortality rates greater than 40% [26].
Fig. 1.

Schematic representation of the breach in the host innate immunity due to different pathogenic infections and the development of sepsis. The dysregualted immune response, including TLR signaling plays a crucial role in the cytokine storm generation. The cytokine storm and exaggerated innate immune response plays a crucial role in the multi-organ damage (MOD) or dysfunction syndrome (MODS). The development of MODS leads to the ultimate death of the patients.
3. TLRs in sepsis
Innate immune system is the primary defence mechanism of the host to protect against invading pathogens. In humans, the innate immune system mainly comprise innate immune cells (i.e. monocytes/macrophages, neutrophils, dendritic cells (DCs), natural killer (NK) cells, mast cells (MCs), eosinophils, basophils along with newly identified innate lymphoid cells (ILCs) and mucosa-associated invariant T (MAIT) cells etc.) [27], [28], [29], [30], [31], [32]. It also includes innate humoral components such as complement system, cytokines, chemokines and antimicrobial peptides (AMPs; LL37 and Bactericidal/permeability increasing protein (BPI) etc.) secreted by the innate immune cells [33], [34], [35], [36]. The innate immune cells express various pattern recognition receptors (PRRs) such as TLRs, nod-like receptors (NLRs; NOD1 and NOD2), RIG-like helicases (RLH) such as melanoma differentiation-associated protein 5 (MDA-5), C-type lectin receptors (CLRs) [Dectin 1 or C-type lectin domain containing 7A (CLEC7A), dectin 2 or CLEC6A, DC-specific ICAM3-grabbing non-integrin (DC-SIGN)], complement receptor 3 (CR3), Triggering receptors expressed on myeloid cells (TREM-1), and myeloid DNAX-activating protein of 12 kDa (DAP-12) associated lectin (MDL-1) [37], [38], [39], [40], [41]. These PRRs recognize pathogen-associated molecular patterns (PAMPs) or MAMPs expressed/released by pathogens and transmit intracellular signaling. The recognition of PAMPs by PRRs is the first step in the development of effective innate immune responses against pathogens and induction of sepsis. Of these PRRs, TLRs are the most studied and relevant ones in context to the cytokine storm and sepsis, and are the focus of this article. The author has discussed TLRs in different inflammatory diseases and their ligands somewhere else [42], [43]. Table 1 shows the complete list of TLRs, their adaptors, and potential ligands. The immune system plays a crucial role in the pathogenesis of sepsis and its outcome, which are described somewhere else in detail [37], [44], [45]. The goal of the present article is to describe the role of TLRs in the sepsis-associated cytokine storm and the endogenous negative regulators of TLR signaling that may serve as molecular targets for developing novel anti-sepsis therapeutics.
Table 1.
Mammalian TLRs, their cellular location, specific ligands and their sources.
| TLRs | TLR Localization | PAMPs or MAMPs | DAMPs | Signal Adaptors and Role in cytokine storm |
|---|---|---|---|---|
| TLR1 | Plasma membrane | Triacyl lipopeptide Soluble factors [362] | Unknown | MAL or TIRAP/MyD88 adaptors and Pro-inflammatory cytokines |
| TLR2 | Plasma membrane, Phagosomes [363] | PGN, LTA, Lipoproteins, zymosan, atypical LPS, [362] | Hsp60, 70, and Gp96, and Hyaluronic acid (HA), serum amyloid A (SAA), HMG-B1, Versican, Matrix metalloproteinase 2 (MMP2), Pancreatic adenocarcinoma upregulated factor (PAUF) [364] | MAL or TIRAP/MyD88 adaptors and Pro-inflammatory cytokines |
| TLR3 | Endosomes and Endolysosomes [365] | dsRNA [362] | mRNA | TRIF adaptor, and pro-inflammatory cytokines and type 1 IFNs |
| TLR4 | Plasma membrane, phagosomes | LPS, Taxol, Fusion protein, Envelope proteins, [362], [366] | Hsp60, Hsp70, Type III fibronectin, glycoprotein 96 (gp96), HA, heparin sulphate, Fibrinogen, SAA, HMG-B1 [364] | MAL or TIRAP/MyD88 and TRIF/TRAM adaptors, Pro-inflammatory cytokines and type 1 IFNs |
| TLR5 | Plasma membrane | Flagellin [362] | HMG-B1 [367] | MyD88, pro-inflammatory cytokines |
| TLR6/TLR4 | Plasma membrane and Phagosomes | Di-acyl lipopeptides From mycoplasma [366] | Amyloid β and oxidized LDL | MyD88 and TRIF, Pro-inflammatory cytokines |
| TLR7 | Endosomes and Endolysosomes Lysosomes | ssRNA from viruses [366] | ssRNA (Immune complex) | MyD88, Pro-inflammatory cytokines |
| TLR8 | Endolysosome [365] | ssRNA [77], [362] | ssRNA | MyD88, Pro-inflammatory cytokines |
| TLR9 | Endosomes, Endolysosomes, Lysosomes, and Phagosomes [368] | CpG ODN, dsDNA, Hemozoin pigment [362], [366] | Host DNA from dying cells, mitochondrial DNA (mtDNA), HMG-B1-DNA complex [369] | MyD88, Proinflammatory cytokines |
| TLR10 | Endolysosomes | HIV-1 gp41 [370], [371] | Unknown | MyD88 adaptor, Anti-inflammatory action [371], [326] |
| TLR11 | Endolysosomes | Profilin-like protein From Toxoplasma gondii[77], [372] | Unknown | MyD88, dependent IRF8 activation, but not NF-κB activation, Pro-inflammatory cytokines [372] |
| TLR12 | Endolysosomes | Profilin-like protein [366], [372], [373] | Unknown | MyD88, dependent IRF8 activation, but not NF-κB activation, Pro-inflammatory cytokines [372] |
| TLR13 | Endosomes and Endolysosomes | 23 s ribosomal RNA [366], [374] | Unknown | MyD88, Pro-inflammatory cytokines |
TLRs are the crucial PRR expressed, both on the outer cell membrane (TLR1, TLR2, TLR4, and TLR6) and in the cytosolic organelles of the cells, including lysosomes, endosomes, phagosomes, phagolysosomes, and endolysosomes (TLR3, TLR7, TLR8, and TLR9). However, TLR2 and TLR4 are also present inside the cells (human DCs) and are essential for IL-12 production in response to the internalized bacteria [46]. The cell-specific expression of TLR4 controls bacterial clearance, induction of pro-inflammatory immune response during bacterial sepsis [47]. The TLR2 and TLR4 expression on the neutrophils of sepsis patients are more dynamic than on monocytes [48]. The TLR4 activation in hepatocytes during sepsis increases caspase 11 (CASP11, a cytosolic LPS receptor mediates pyroptosis) expression and thus, the hepatocyte pyroptosis and exosome release [49], [50]. The CASP11/gasdermin-D (GsdmD) activation/cleavage induces the of high mobility group box-1 protein (HMGB1) accumulation in the cytoplasm via calcium-induced phosphorylation of calcium-calmodulin kinase kinase (Camkk)-β. Hence, HMGB1 release in the circulation depends on TLR4 signaling. The circulating extracellular HMGB1 proves lethal to the host during sepsis and preventing its release in the cytosol and circulation decreases the severity and mortality associated with the sepsis [50], [51]. The platelets isolated from sepsis patients show unaltered TLR expression, although they exhibit active phenotype [52]. The stimulation of platelets isolated from healthy adult humans and mice with LPS also do not alter TLR expression, but it alters macrophages respiration in a TLR4 activation-dependent manner [52]. However, these platelets do not show a classical activation (as indicated by P-selectin, CD63 and phosphatidylserine expression) and platelet-leukocyte complex formation. The details of TLR expression in different immune cells and their role in immunity and inflammation have been discussed by the author somewhere else [42], [43].
The DC-specific intrinsic TLRs signaling also plays a crucial role in the pathogenesis of polymicrobial sepsis via regulating the production of pro-inflammatory TNF-α and survival [53]. The study indicates that the DC-specific gp96-/- mice (representing DC-specific pan TLR knockout (KO) mouse model) subjected to polymicrobial sepsis show an increased survival rate (33%) as compared to wild-type (WT) animals (5% survival rate) and decreased serum pro-inflammatory cytokines (TNF-α). The heat shock protein (Hsp) gp96 (also known as grp94 or Erp99 or endoplasmin, which is an endoplasmic reticulum paralog of Hsp90) is a master chaperone of TLRs and regulates the expression of TLR2, TLR4, TLR5, TLR7, and TLR9, and mice deficient in gp96 are resistant to endotoxic or septic shock [54]. This gp96-mediated regulation of TLRs in macrophages also controls macrophage polarization to pro-inflammatory M1 phenotype following stimulation with lipopolysaccharide (LPS) [55]. However, gp96-/- macrophages are still able to upregulate TLR expression upon stimulation with LPS indicating other chaperone proteins also regulate TLR expression [56]. However, gp96 deficient macrophages are defective in producing TLR4-dependent pro-inflammatory cytokine release indicating its role in TLR4-downstream signaling. The gp96 deficiency also impairs ERK and p38MAP kinase phosphorylation upon colony stimulating factor 1 (CSF1) signaling [56]. The initial TLR signaling during a low grade polymicrobial sepsis protects from severe cardiac dysfunction, kidney and liver injuries, and increase the survival [57].
The TLR2 and TLR4 expression in the lungs of mice subjected to the cecal-ligation and puncture (CLP)-induced polymicrobial sepsis increases within 24 h as compared to the sham group [58]. Whereas, TLR3 and TLR7 expression increases in their intestine within 24 h. This can be speculated as gut epithelial cells normally express a lower level of TLR2 (at apical surface) and TLR4, but a higher level of TLR3 and TLR5 [59], [60], [61]. The TLR2 and TLR4 are expressed on the basolateral surface of enterocytes of the small intestine, whereas TLR5 on the basolateral surface of enterocytes of the colon [59], [61], [62]. The details of TLR expression in the human gut are explained somewhere else [63]. Thus, it would be interesting to observe the kinetics and dynamics of TLR expression in different organs during sepsis.
The newborns with sepsis also show an increased TLR4 expression on monocytes and have high circulating pro-inflammatory cytokines (IL-8, IL-6, and IL-1β) [64]. Another study has shown the increase in the TLR4 and TLR2 expression in peripheral blood monocytes (PBMCs) during the Gram-negative and Gram-positive bacterial sepsis in both adults and children in Iraq [65]. Even the population with TLR9-1237 T/C polymorphism are more prone to progression of an infection-induced sepsis [66]. However, TLR9 (-1486 T > C) and TLR-9 (C > T) gene polymorphisms do not show any correlation with sepsis [67]. The serum TLR9 levels are low in sepsis patients but their circulating levels increases at later immunosuppressive stage of sepsis, including septic shock in patients with a higher (greater than 5 mmol/l) lactate level [68]. This indicates that the profound immune cell death may have caused the increase in the circulating TLR9 (recognizes bacterial and viral DNA). Further studies are required in the direction.
The septic mice along with the increased TLR expression also show an increased level of circulating creatine, IL-6, and IL-10. Thus a higher TLR2 and TLR4 expression in the lungs of septic mice plays a crucial role in the sepsis-induced acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) via controlling the secretion of pro-inflammatory cytokines and other cytotoxic mediators [reactive oxygen or nitrogen species (ROS or RNS)] [69]. Additionally, mice with sepsis also show an increased TLR2, TLR3, TLR4, and TLR7 expression in kidneys and intestine indicating their pro-inflammatory role increases with the severity of the infection causing sepsis-induced organ damage [70]. Also, CD38-/- mice subjected to sepsis show an increased TLR4 expression in kidneys and severe or acute kidney injury (AKI) due to the increased pro-inflammatory cytokine levels (IFN-γ, TNF-α, IL-1β, and IL-6) as compared to the WT mice with sepsis [71]. However, a high IFN-γ level was present only in serum only. Hence, sepsis increases the TLR expression on different organs and tissues that aggravates the inflammatory signaling, and thus the cytokine storm and organ damage. For example, TLR2-/-, TLR4-/-, and MyD88-/- mice subjected to the CLP-induced polymicrobial sepsis show preserved renal or kidney morphology, and fewer areas of hypoxia and apoptosis as compared to the WT mice [72]. MyD88-/- mice are completely protected from sepsis-induced AKI in comparison to the WT mice. MyD88-/- mice have very low levels of HMG-B1 and heat shock protein 70 (Hsp70) [72].
The TLR2-/-, TLR4-/-, and MyD88-/- mice show reduced levels of pro-inflammatory cytokines [IL-1β, TNF-α, IL-6, IL-8, IL-17, and induced nitric oxide synthase (iNOS)] neutrophil infiltration in kidneys, and vascular permeability than WT mice subjected to CLP-induced polymicrobial sepsis [72]. Of note, MyD88-/- mice with CLP-induced sepsis had least levels of these pro-inflammatory cytokines and mediators in kidneys [72]. The prevention of neutrophil infiltration in the kidneys in WT mice protects them from sepsis-induced AKI. The study also shows the importance of MyD88-dependent pathway in the generation of innate immune response that causes an increased neutrophil infiltration in kidneys, increased pro-inflammatory cytokines, vascular permeability, hypoxia, and apoptosis of tubular cells [72]. The MyD88-/- mice show increased susceptibility to the polymicrobial sepsis due to the impaired local inflammatory immune response (neutrophil infiltration) and chemokine production [73]. However, MyD88-/- mice are protected from developing sepsis during Neisseria meningitidis infection as compared to the WT mice, who develop sepsis and septic-shock associated 100% fatality [74]. Hence, both human and animal studies have shown the strong the association of TLRs in the sepsis pathogenesis and their pattern of expression during sepsis.
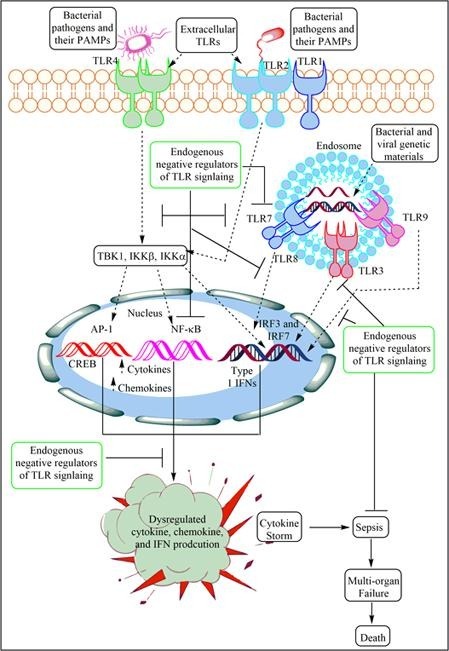
4. TLR signaling causing cytokine storm during sepsis
The recognition of bacterial PAMPs such as lipopolysaccharide (LPS), lipoteichoic acid (LTA), peptidoglycan (PGN), porins, flagellin and CpG-DNA by their corresponding TLRs (TLR4, TLR2, TLR5, TLR9) induces the events, which converge at the activation of nuclear factor κ-light-chain-enhancer of activated B cell (NF-κB) inducing the gene expression responsible for the synthesis and release of pro-inflammatory mediators, including cytokines, chemokines, ROS and RNS (Fig. 2 ) [75], [76], [77]. Salmonella and E. coli flagellin FliC and Toxoplasma gondii profilin-like protein act as PAMPs for TLR11, which is present and functional in mice but not in humans [78], [79]. TLR5 physically interacts with TLR4 that biases the signaling pathway towards myeloid differentiation factor 88 (MyD88)-dependent by forming Myddosome (a complex formed of MyD88 and interleukin 1 receptor-associated kianse 4 or IRAK4) to activate NF-κB and associated downstream pro-inflammatory genes expression, including cytokines to cause ALI during endotoxemia [80]. The deficiency of TLR5 alters the response to the TLR4 stimulation. For example, people with a dominant negative TLR5 single nucleotide polymorphism (SNP) (rs5744168) for the common stop codon polymorphism in the ligand-binding domain of the TLR5 (TLR5392STOP) do not respond to the flagellin and are more prone to Legionella pneumophila pneumonia [81]. Macrophages isolated form rs5744168 minor-allele carriers also show a defective response to TLR4 activation, but not to TLR2, TLR3, and TLR9 [80].
Fig. 2.

Schematic representation of the activation of various TLR signaling pathways leading to the development of cytokine storm and sepsis. The stimulation of TLR4 and TLR2 via their corresponding PAMPs activate a cascade of events, which results in the activation of NF-κB, p38 and JNK, CREB, and AP-1 etc. which regulates the synthesis and release of various pro-inflammatory cytokines (IL-1, IL-6, IL-12, IL-8, and TNF-α), anti-inflammatory cytokine (IL-10), and type 1 IFNs. The exaggerated levels of these inflammatory mediators develop cytokine storm during sepsis and other severe infections, including COVID-19. The patient develops multi-organ failure, including ALI and dies.
The activation of TLR signaling in response to pathogens, their PAMPs, and DAMPs depends on the MyD88-dependent and MyD88-independent (TIR-domain containing Adaptor inducing interferon-β or TRIF-dependent) signaling events activating NF-κB as shown in the Fig. 2. The NF-κB activation induces expression and release of several pro-inflammatory cytokines (TNF-α, IL-1α, IL-6, and IL-8) and other molecules responsible for the generation of so-called cytokine storm, systemic inflammation and development of sepsis (Fig. 2). The details of TLR signaling responsible for cytokine and other pro-inflammatory mediators generation to cause sepsis-associated cytokine storm is beyond the scope of current article and has been discussed somewhere else [76], [82], [83]. However, the TLR signaling is controlled by its endogenous regulators that are described in following sections.
5. Host-derived negative regulators of TLR signaling
There are a variety of molecules/mechanisms that negatively regulate TLR-induced signaling. They are usually induced by the ligands, which primarily activate TLRs, and serve as built-in negative feedback mechanisms to maintain homoeostasis and prevent excessive and infinite activation of TLR-activated pro-inflammatory pathway. Depending upon their mode of action, these regulatory molecules/mechanisms have mainly been classified into 3 types: 1) Dissociation of adaptor molecule complex; 2) Degradation of proteins involved in TLR signaling causing inflammation and; 3) Transcriptional regulation of TLR signaling [84]. Table 2 describes the list of endogenous negative regulators of TLR signaling pathways, their category, and mode of action.
Table 2.
Host-derived endogenous negative regulators of TLR-signalling-mediated inflammatory pathways generating cytokine storm and sepsis. See text for details.
| Host-derived negative regulators of TLR signalling | Category/Classification | Target/Mode of action |
|---|---|---|
|
Splice variant of TRAM | Competes with TRAM for binding to TRIF. |
|
TIR-domain containing adaptor molecule | Binds and inhibits TRIF-dependent TLR signalling |
|
IFN regulatory factor (IRF) family of transcription factors | Inhibits IRF5 binding to MyD88 and TRAF6 and its translocation to nucleus |
|
Member of TNF-α-induced protein-8 (TNFAIP8) family | Inhibits TLR signalling via binding to CASP8 and inhibiting NF-κB and AP-1 activation |
|
Transmembrane adaptor protein | DAP12 mediated inhibition of TLR signalling involves another adaptor protein called DOK-3 |
|
Adaptor Proteins | Inhibit Ras-Erk dependent signalling |
|
TAM family of receptor kinases | Inhibits NF-κB mediated production of TNF-α |
|
Member of serine/threonine kinase family | Inhibits dissociation of IRAK1 and IRAK-4 form MyD88 and formation of IRAK-TRAF6 complex |
|
Adaptor protein that interacts with cytoplasmic TIR domain of IL-1Rs | Inhibits phosphorylation and kinase activity of IRAK1 |
|
Intracellular tyrosine phosphatase | Inhibits MAP kinase and NF-κB activation through binding to IRAK1 |
|
A serine/threonine phosphatase | Specific pathway unknown |
|
Intracellular tyrosine phosphatase | Inhibits MAPKs, NF-κB and IRF3 |
|
Ubiquitin modifying enzyme | Inhibits NF-κB signalling as a negative feedback via removing ubiquitin moieties from TRAF6 |
|
Tumour suppressor deubiquitinase | Inhibits TLR2 signalling via inhibiting MyD88, TRAF2, TRAF6 TRAF7 and NEMO |
|
Deubiquitinase | Inhibits TRL4 signalling via deubiquinating TRAF6 and inhibiting its adaptor function |
|
Isopeptidase | Cleaves the K63-linked polyubiquitin chains of TAK1 and also targets NEMO |
|
Cysteine protease | Removes K63-ubiquitin chain from TRAF3 to inhibit NF-κB and IRF3 activation |
|
Member of nuclear receptor 4A receptor subfamily | Prevent auto-ubiquitination of TRAF6 via binding to TRAF6 |
|
Member of nuclear receptor 4A receptor subfamily | Inhibits NF-κB activation downstream to TLR4 signalling |
|
Orphan nuclear receptor | Prevents Lys63-linked polyubiquitination of TRAF6 and subsequent activation of NF-κB |
|
Nuclear kinase sharing homology with ribosomal S6 kinase (p90rsk) family | Phosphorylate histone H3 and CREB that negatively regulates TLR signalling and induces several anti-inflammatory genes |
|
TRAF binding protein | Binding of TANK to TRAF6 inhibits NF-κB and AP-1 activation |
|
PDZ and LIM domain containing Alkaline Phosphatase | Inhibits TLR signalling by acting as a nuclear E3 ubiquitin ligase and inhibits NF-κB activation |
|
Zinc finger and RING finger domain containing nuclear protein | Binds to PDLIM2 and inhibits activation of NF-κB downstream to TLR signalling |
|
PDZ and LIM domain containing protein of APL subfamily | Inhibits NF-κB activation by sequestering p65 into cytosol |
|
A member of tripartite-motif (TRIM) protein family | Inhibits TRAF6 autoubiquitination by degrading TAB2 and TAB3and suppresses NF-κB activation |
|
Acts as ubiquitin E3 ligase | Polyubiquitinates TRIF and inhibits TRIF-TBK1 interaction |
|
RING finger type E3 ubiquitin ligase | Degrades TLR proteins |
|
Member of NLR family | Binds to IKK complex causing an inhibition of IKKα and IKKβ phosphorylation and NF-κB activation |
|
Member of NLR family | It interacts with TRAF6 to attenuate its K63-linked ubiquitination to inhibit NF-κB activation |
|
Member of NLR family | Inhibits IKK complexes to inhibit NF-κB activation and type 1 IFN signalling pathways |
|
Serves as a part of IL-33 receptor | Binds to sequester MyD88 and MAL without affecting TRIF and IRAK to inhibit TLR-induced NF-κB activation |
|
Member of TLR/IL-1R superfamily | Inhibits TLR-mediated NF-κB and JNK activation via blocking the IRAK and TRAF6 recruitment to MyD88 |
|
Member of TLR family | Activates PI3K/Akt/IL-1R antagonist pathway and also inhibits MyD88 and TRIF-dependent signaling pathways |
|
Member of activating transcription factor/cAMP response element family of bZip transcription factors | Bind to consensus c-AMP response element (CRE) sequences |
|
Member of IL-1 cytokine family | Inhibits NF-κB and MAPK activation |
5.1. Negative regulators that dissociate of adaptor molecule complexes
TAG (TRAM (Translocating chain-associated membrane protein) adaptor with Golgi dynamics (GOLD) domain) is a splice variant of TRAM, having a GOLD domain coupled to TRAM's Toll-interleukin 1 receptor (TIR) domain [85]. The GOLD domain containing proteins are involved in Golgi dynamics, secretion, and localization to membranous vesicles. The LPS-mediated stimulation of TLR4 localizes both TRAM and TAG in the late endosomes, positive for Rab7a GTPase, where TAG competes with TRAM to bind to TRIF (Table 2). The competitive binding results in TLR4 degradation and TRIF-induced activation of interferon regulatory factor 3 (IRF3) and production of type I interferons inhibition [85], [86]. Also, macrophages lacking TAG release CCL5 or regulated on activation, normal T cells expressed and secreted (RANTES) upon stimulation with LPS but no IL-8 [85], a major chemoattractant for neutrophils and also a biomarker for sepsis [87], [88]. Thus, TAG could serve as a molecular target for developing anti-sepsis drugs as neutrophils serve as major immune cells for organ damage associated with sepsis, including sepsis- and severe COVID-19-induced acute lung injury (ALI) [11], [69].
Sterile alpha- and armadillo-motif-containing protein (SARM) is one of the five TIR-domain-containing adaptor molecules. However, it directly binds to TRIF and inhibits TRIF-induced signaling in response to TLR stimulation (Table 2) [89]. SARM specifically inhibits TRIF-dependent activation of IRF3 and IRF7 (master regulators of IFN production), but not tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) and MyD88-dependent pathways. Thus it inhibits the release of RANTES (CCL5) and IFN-β in response to the LPS-induced TLR stimulation [89], [90]. The SARM-mediated TRIF inhibition requires sterile α-motif and TIR domains of SARM [89]. Thus TRIF inhibition during TLR signaling does not affect IL-1-MyD88-mediated IL-8 release. However, SARM also inhibits MAPK activation directly in human embryonic kidney (HEK) 293 cells and U937 cells (a human macrophage cell line, used to study the behaviour and differentiation of human monocytes), and reduces the p38 phosphorylation [91]. SARM also inhibits both TRIF and MyD88-depdendent activation of both endogenous and induced AP-1 [91]. Thus, the inhibitory effects of this negative regulator of TLR-mediated signaling appear to be broader. Recently, in a mouse model of polymicrobial sepsis a decreased expression of SARM has been observed in their spleen that well correlates with the apoptosis of splenocytes [92]. The increased caspase 3 (CASP3) activation and ATP depletion in splenocytes indicates their apoptotic death. Hence, SARM is essential to prevent sepsis-induced apoptotic death of splenocytes and associated immunosuppression at later stages. Further, studies are required in the field. SARM does not act only as a negative regulator of TLR signaling but also negatively affects NLRP3 activation and IL-1β production [93]. Thus it becomes crucial to study SARM in context to the bacterial sepsis and severe COVID-19-associated sepsis as lymphopenia is seen in these patients.
Interferon regulatory factor 4 or IRF4 (MUM1) is another negative regulator of TLR signaling that acts via inhibiting the binding of IRF5 to MyD88 and TRAF6 in a competitive manner and inhibits nuclear translocation of IRF5 (Table 2) [94]. IRF5 and IRF7 bind to MyD88 upon stimulation of TLRs to induce pro-inflammatory cytokines and type 1 IFNs [95]. However, IRF4 only competes with IRF5 (and not with IRF7) to interact with MyD88 [95]. The IRF5 inhibition by IRF4 during TLR signaling inhibits synthesis and release of pro-inflammatory cytokines (IL-6, IL-12, and TNF-α), which are responsible for the induction of sepsis-associated cytokine storm and multi-organ damage, failure, and mortality. Not surprisingly, IRF5-/- mice are resistant to lethal septic shock induced by unmethylated DNA and endotoxin/LPS as well [94]. Consistent with these observations, peritoneal macrophages isolated from IRF4 KO mice release higher amounts of pro-inflammatory cytokines upon stimulation with LPS [95]. Metformin (an antidiabetic drug) protects against sepsis-induced myocarditis through increasing the IRF4 expression in the cytosol and mitochondria [96], [97]. The mitochondrial IRF4 interacts with protein kinase C ε (PKCε). The protective action of IRF4 on sepsis-associated organ damage should be further explored.
Tumour Necrosis Factor α-induced Protein 8-like 2 (TIPE2) is a member of tumor necrosis factor-α-induced protein-8 (TNFAIP8) family, and is expressed mainly in lymphoid organs (spleen and lymph nodes). TIPE2-/- mice develop multi organ inflammation, splenomegaly, and premature death due to over-activated TLR signaling and pro-inflammatory cytokine generation [98]. These mice are more prone to develop sepsis and septic shock [98]. TIPE2 inhibits TLR signaling via binding to the caspase 8 (CASP8), and inhibiting NF-κB and AP-1 activation (Table 2) [98]. Also, TIPE2 inhibits TAK1 in murine macrophages and this inhibition involves the interaction between TIPE2 amino acid 101–140 region and amino acid 200–291 region of the internal kinase domain of transforming growth factor-β-activated kinase 1 (TAK1) or mitogen-activated protein kinase kinase kinase 7 (MAP3K7) [99]. Thus, TIPE2 interferes with the formation of the TAK1-TGF-beta-activated kinase 1 and MAP3K7-binding protein 1 (TAB1)-TAB2 complex, and inhibits TAK1 activation and its downstream signaling events resulting in the inhibition of the synthesis and release of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α) [99], important components of cytokine storm associated with development of sepsis. Further study has shown that TIPE2 promote the host resistance to Pseudomonas aeruginosa infection via inhibiting TAK1-mediated TLR signaling and inflammatory immune cell infiltration [100]. Along with inhibiting TLR signaling, TIPE2 also inhibits NOD-2 (Nucleotide-binding oligomerization domain-containing protein 2 or CARD-15 or caspase recruitment domain containing protein 15) induced MAPK and NF-κB signaling pathways to reduce pro-inflammatory cytokine production [101]. TIPE2 also regulates the immunosuppressive action of CD4+CD25+FoxP3+T regulatory cells (Tregs) via regulating the IL-10 and transforming growth factor-β (TGF-β) production, which play a crucial role in immunoparalysis or immunosuppression during later stages of sepsis [102]. Xuebijing (an ancient Chinese traditional medicine) injection (XBJ) protects from sepsis-induced lung and liver injury, and vascular leakage via increasing TIPE2 and Tollip expression that inhibit TLR4-induced MAPK and NF-κB signaling pathways, IRAK1, and TRAF6 activation to inhibit the pro-inflammatory cytokine release [103], [104]. The TIPE2-based gene therapy has shown a protective anti-inflammatory and immunomodulatory effects in a mouse model of collagen-induced arthritis (CIA) analogous to human rheumatoid arthritis (RA) [105]. Hence, TIPE2-based therapies should also be explored in sepsis and severe COVID-19-induced cytokine storm.
5.2. Tyrosine kinases and phosphatases as negative regulators of TLR signaling
Transmembrane proteins having intracytoplasmic immunoreceptor tyrosine-based activation motif (ITAM) also regulate TLR signaling in a negative manner. For example, macrophages isolated from DAP-12 deficient mice secrete a large amount of pro-inflammatory cytokines (IL-6, TNF-α, and IL-12p40) upon stimulation of different TLRs (TLR4, TLR2, TLR3, and TLR9) [106]. This adaptor protein was first identified in NK cells [107]. Also, named as KARAP (Killer cell activating receptor-associated protein) or TYROBP (Tyrosine kinase-binding protein), it is used to propagate intracellular inhibitory and activating signals initiated by a variety of receptors in DC, osteoclasts, NK, and myeloid cells. DAP12 deficient mice are more prone to develop endotoxemia and sepsis [106]. They produce more cytokines and show enhanced ERK1/2 activation in response to low dose LPS. The DAP-12-mediated negative regulation of TLR signaling involves another adaptor protein called DOK (downstream of kinase)-3, which physically associates with the ITAM of DAP-12 via its phosphotyrosine-binding domain [108]. TLR4 activation by LPS phosphorylates DOK3 in a DAP12 and Syk (spleen tyrosine kinase)-dependent manner causing translocation of DOK3 towards plasma membrane, where it binds to TREM (triggering receptor expressed on myeloid cells)-2 and inhibits downstream TLR signaling [108], [109]. The inhibitory effects of the TREM2-DAP12 interaction has also been demonstrated in the D-galactosamine-potentiated endotoxaemia model, where mice are sensitized by D-galactosamine and challenged with low doses of LPS (1–5 µg per kg or 20–100 ng for a 20 g mouse) [103], [105]. To the contrary, the TREM1–DAP12 interaction was shown to exacerbate inflammation in the endotoxaemia model in which mice are injected with the LPS at doses of 5–10 mg/kg or approximately 100–200 µg of LPS for a 20 gm animal [110], [111]. Hence, the positive and negative impact of DAP12 on TLR signaling depends on intensity of TLR signaling stimulation. Further studies are required in the direction.
Downstream of kinase 1 and 2 (DOK1 and DOK2) are other adaptor proteins that regulate LPS-stimulated TLR4 signaling in a negative manner via inhibiting Ras-Erk-dependent signaling downstream of protein tyrosine kinases (PTKs) [112], [113]. The LPS-mediated TLR4 stimulation induces tyrosine phosphorylation of DOK1 and DOK2, which inhibit ERK and decrease the release of TNF-α and nitric oxide (NO.) (Table 2). This pathway, however, is not operative when other TLRs are stimulated [112]. Thus, the pathway is a specific inhibitor of TLR4 stimulation and can be used as a therapeutic approach for the Gram-negative bacterial sepsis and associated cytokine storm. Of note, the expression of DOKI in mouse bone marrow-derived macrophages (BMDMs) decreases upon LPS-mediated TLR4 stimulation [112], while DOK2 expression in NFS-60 myeloid leukaemia cells increases in response to M−CSF, GM-CSF, and (IL-3) [114].
The TAM family of receptor tyrosine kinases (RTKs) comprises Tyro-3, Axl, and Mer. Each of these kinases is characterized by a conserved sequence within the kinase domain and adhesion molecule-like extracellular domains. These kinases act as important homeostatic regulators in adult tissues and organ systems, and mainly mature cells in the immune, nervous, vascular, and reproductive system express them [115], [116]. Genetic deficiencies in TAM signaling components are associated with chronic inflammatory and autoimmune diseases. The TAM family members play important roles in immune homeostasis independently or collectively by three mechanisms: 1) negative regulation of innate immune activation, 2) phagocytosis of apoptotic cells, and 3) restoration of vascular integrity [117]. Negative regulation of TLR signaling by TAMs has been observed in macrophages and DCs [118]. In these cells, Axl gene is modestly expressed in their steady-state. The activation of TLRs increases the Axl expression and activation [118]. The up-regulated and activated Axl protein forms a complex with R1 chain of type I IFN receptor (IFNAR). The Axl-IFNAR complex switches pro-inflammatory action of the IFNAR signaling into an anti-inflammatory or more specifically into an immunosuppressive one. It does so by activating a STAT1 homodimer, which activates suppressor of cytokine signaling-1 (SOCS)-1 and SOCS-3 [118], [119]. The later proteins recognize and degrade number of TLR signaling molecules such as IRAKs, MyD88-adaptor like (MAL), TRAF3, TRAF6, apoptosis signal-regulated kinase 1 (ASK1) and Janus-associated kinases (JAKs) [117], [120].
An SH2 domain present in the central regions of SOCS1 and SOCS3 proteins binds to JAKs and IFNAR via their phosphotyrosine residues. The SOCS proteins inhibit catalytic activity of JAKs and their carboxy-terminal SOCS box degrades associated proteins (MAL, TRAFs, and JAKs) via activating ubiquitin–proteasome pathway [109]. The amino-terminal regions of SOCS1 and SOCS3 also contain a kinase-inhibitory region (KIR) that acts as a pseudosubstrate for JAKs [113], [115]. Thus, SOCS1 and SOCS3 induction via TAM kinase signaling pathway activation due to TAM-IFNAR multimeric complex formation and activation in myeloid cells (macrophages and DCs) terminates the pro-inflammatory innate immune response [112]. Hence, TAMs-mediated TLR signaling inhibition is not only dependent on the JAK-STAT pathway inhibition, but also involves degradation of several other key molecules implicated in the propagation of the TLR-initiated signaling cascades involved in the pathogenesis of sepsis via inducing potent cytokine storm and other potential inflammatory mediators [117].
TAM signaling component Axl also inhibits the TLR-mediated TNF-α production via inducing Twist protein (a basic helix-loop-helix (bHLH) family transcriptional repressor that binds to E box elements in the TNF promoter region and suppress NF-κB-dependent transcription and release of TNF-α gene) [121]. The Gas6 and protein S (ProS) binding to Axl and apoptotic cells induce Twist [121]. Also, TAM receptor ligands Gas6 and ProS1 are very potent negative regulators of TLR3, TLR4 and TLR9 signaling in both macrophages and DCs [118]. As mentioned above, the mechanism of Gas9 and ProS1-mediated TLR signaling inhibition involves activation of a signaling pathway via binding through R1 subunit of type 1 IFNR, which forms a complex with Axl, resulting in STAT1 activation and SOCS proteins induction [122]. Reports have shown a significant early changes in the expression MerTK (another kinase of the TAM family) on the surface of monocytes and neutrophils isolated from patients with sepsis and septic shock. A persistent overexpression of MerTK in patients with septic shock proves detrimental [123], [124]. The receptors get cleaved from the cell surface by ADAM metallopeptidase domain 17 (ADAM-17) or tumor necrosis factor-α-converting enzyme (TACE) into decoy receptors, which sequester the ligands (ProS and Gas-6). A decreased availability of the ligands makes TAMs inefficient in controlling inflammation (1 1 9). Tyro-3 and Axl levels also increase on the surface of monocytes isolated from trauma patients [123]. MerTK expressed on macrophages plays a crucial role in the phagocytosis of apoptotic cells and MerTKY867F mutant, that lacks autophosphorylation site, inhibits phagocytosis of apoptotic cells, but does not inhibit LPS-stimulated TLR signaling-mediated NF-κB activation [125], [126]. Thus, TAMs-mediated negative regulation of TLR signaling does not depend upon the receptors’ ability to promote phagocytosis of apoptotic cells.
IRAK-M (Interleukin-1 receptor-associated kinase-M) or IRAK-3 is a member of serine/threonine kinase family called interleukin-1 receptor-associated kinase (IRAK). The family comprises 4 members, of which only two exhibit kinase activity (IRAK-1 and IRAK-4), while the other two (IRAK-2 and IRAK-M/IRAK-3) do not have any kinase activity [127]. Except IRAK-M, which is only expressed in monocyte and macrophages, all other IRAKs are ubiquitous in nature [127]. The TLR4 and TLR9 activation upregulates IRAK-M expression in monocytes and macrophages. The kinase acts as a negative regulator of TLR signaling by inhibiting the dissociation of IRAK-1 and IRAK-4 from MyD88 and formation of IRAK-TRAF6 complex (Table 2). IRAK-M-/- mice show an enhanced pro-inflammatory immune response upon treatment with LPS, and in response to bacterial infections (E. coli and S. typhimurium) and develop sepsis [127]. Along with inhibiting TLR4 and TLR9 signaling, IRAK-M also inhibits TLR2 signaling in macrophages and induces tolerance among them against PGN [128]. A further study has shown that IRAK-M plays a crucial role in the sepsis-induced suppression of lung innate immunity [129]. For example, IRAK-M-/- mice subjected to peritonitis-induced sepsis show increased release of pro-inflammatory cytokines ex vivo and express higher levels of co-stimulatory molecules in vivo as compared to WT animals. These mice show increased survival and improved bacterial clearance from lungs and systemic circulation following sepsis [129]. However, this exaggerated innate immune response in IRAK-M-/- mice causes an increased neutrophil infiltration in the lungs of the animals and impaired macrophage-mediated innate immune response making them prone to develop secondary infections following sepsis [129]. IRAK-M has also a potential to influence macrophage polarization via inducing their polarization towards anti-inflammatory phenotype called M2 macrophages or alternatively activated macrophages (AAMs) that further has a potential to decrease pro-inflammatory cytokine levels [130]. However, the IRAK-M activation during sepsis-associated immunosuppression (SAIS) can prove harmful to the host as it may aggravate it. Hence, the silencing of IRAK-M in the immune cells of SAIS mice restores the pro-inflammatory cytokine production and reverses the SAIS [131]. Thus, IRAK-M plays a dual role (both harmful and beneficial) for the host in sepsis and further studies are required to learn how it could be targeted for managing sepsis. Humans with IRAK-M + 22148 G > A genetic polymorphism are more susceptible to sepsis than normal population [132]. The G/G genotype of IRAK-M has a more risk for developing sepsis than normal population, whereas A/A genotype exerts a protective action against developing sepsis. Hence, IRAK-M modulators or gene-based therapies should be used with a caution depending on the immune status of the host during sepsis.
TOLLIP-mediated inhibition of TLR signaling
TOLLIP (Toll-interacting protein) was originally identified during the screening of IL-1 receptor accessory proteins and acts, in addition to MyD88, as another adaptor protein, which interacts with the cytoplasmic TIR domain of IL-1Rs upon IL-1 stimulation [133]. TOLLIP is ubiquitously expressed and also associates directly with TLR4 and TLR2, and inhibits TLR signaling through phosphorylation and kinase activity of IRAK1 (Table 2) [134]. However, TOLLIP serves as the first substrate for the IRAK1-mediated phosphorylation [134]. It inhibits IRAK1 by ubiquitinylation and subsequent kinase degradation [135]. Furthermore, the N-terminal conserved-2 (C2) domain of TOLLIP plays a crucial role in the recognition of phosphoinositides in lipid-rich cellular membranes (for example, its localization to endosomes and lysosomes is important for autophagy) [136]. An essential mutation changing vital lysine residue to glutamic acid (TOLLIPK150E) within the C2 domain abolishes the binding of TOLLIP with phosphatidylinositol-3-phosphate and phosphatidylinositol-3,4,5-phosphate (PI3P), and the TOLLIPK150E mutant animals are defective in negatively regulating the LPS/TLR4-mediated NF-κB activation. Thus, TOLLIP serves as a pivotal internal inhibitor of TLR signaling involved in the sepsis-associated cytokine storm. For example, the minor C-allele of TOLLIP carrying the rs5743867 SNP is significantly associated with the decreased risk of sepsis in Chinese Han population [137]. After exposure to LPS, the TOLLIP levels increase significantly in PBMCs from homozygotes (CC) as compared to heterozygotes (CT) and homozygotes (TT) for the rs5743867T/C variation [137]. Also, the TNF-α and IL-6 levels decrease significantly in culture supernatants of these PBMCs isolated from people with CC genotype than CT and TT genotypes [137].
The neutrophils from TOLLIP-deficient mice become dysfunctional and inflammatory in nature [138]. TOLLIP deficient neutrophils exhibit defect in their migration capacity towards fMLF (formyl-methionyl-leucyl phenylalanine, a bacterial peptide that chemoattracts neutrophils) due to reduced activity of AKT and FPR2 (the receptor for fMLF). Their potential to release neutrophil extracellular traps (NETs) or NETosis and kill bacteria decreases [138]. However, they highly express CCR5 (Chemokine receptor 5), which increases their migration towards inflamed tissue [138]. TOLLIP acts as a negative regulator of TLR signaling in acute microbial infections when the host is exposed to high dose LPS and is important for homeostatic clearance of the inflammation. However, it serves to propagate the TLR signaling when the host is exposed to super low doses of LPS (<100 pg/ml) in certain conditions, including aging. Under these conditions, MyD88 is not recruited, no activation of NF-kB occurs and negative regulators of TLR-signaling are not induced. Instead, TOLLIP translocates to mitochondria, promotes ROS production, and induces cytokines release [139], [140]. In agreement with these observations, TOLLIP-KO cells produce less IL-6 and TNF-α and more TGF-β [141]. Thus, TOLLIP plays a dual role and adjusts the cell’s response to changing concentrations of LPS, a major player in the pathogenesis of Gram-negative bacterial sepsis and associated cytokine storm.
SHP (Src homology 2 domain-containing protein tyrosine phosphatase)-1
SHP-1 is an intracellular tyrosine phosphatase having two tandemly linked SH2 domains at its amino terminus followed by a catalytic domain [122]. Myeloid cells, including neutrophils, inflammatory macrophages, and plasmacytoid dendritic cells (pDCs) highly express SHP-1 [142]. SHP1 acts as a negative regulator of TLR signaling due to its inhibitory action on NF-κB and Erk1/2 MAP kinase activation via inhibiting IRAK1 (Table 2) [122], [143], [144], [145]. The binding of SHP1 to IRAK1 completely inhibits its enzymatic activity required for pro-inflammatory functions of myeloid cells, including macrophages upon TLR stimulation [144]. The SHP-1 and IRAK1 binding involves an evolutionarily conserved ITIM-like motif present in the kinase domain of IRAK1 that is called KTIM (Kinase Tyrosyl-based Inhibitory Motif) [144]. The KTIM is only present in the IRAK1 but not in any other IRAK family members [144]. Hence, pathogens responsible for sepsis may exploit SHP-1 to increase their pathogenicity and the infection severity. Along with inhibiting pro-inflammatory TLR signaling, SHP-1 is also involved in increasing the type 1 IFN production upon activation of TLR and RIG-1 like receptors (RLRs) by directly inactivating IRAK1 and IRAK2, which play important role in the NF-κB activation and type 1 IFN inhibition [86], [143].
Quite expectedly, SHP1 KO mice develop severe inflammatory lesions in response to LPS stimulation or endotoxemia due to increased production of pro-inflammatory mediators [142], [143]. Thus SHP-1 is a key regulator of a switch from production of pro-inflammatory (TNF-α) cytokines to anti-inflammatory (IFN-I) upon TLR stimulation. It should be novel to observe the effect of SHP-1 during severe viral infections causing sepsis, including severe COVID-19. This is because SHP-1 can inhibit the release of pro-inflammatory cytokines (IL-6 and TNF-α) responsible for cytokine storm and associated organ damage and mortality in severe COVID-19 but increase the antiviral type-1 IFN secretion in response to both TLR and RLR activation. However, SHP-1 upregulates the IL-12p40 production in BMDMs upon TLR stimulation via interacting with phosphatidylinositol 3-kinase (PI-3 K), without affecting c-Rel activation [146]. The IL-6 production in macrophages depends on SHP-1 as deficiency of SHP-1 in myeloid cells increases its production and expression of SHP-1 inhibits IL-6 production [145]. IL-6 is a major pro-inflammatory cytokine associated with the severe COVID-19 cytokine storm and IL-12p40 activates antiviral action of NK cells and Th1 cells to clear the infection [10], [11]. Hence, SHP-1 levels and functions should be studied in context to severe COVID-19-associated sepsis for designing better immunomodulatory drugs for sepsis and severe COVID-19. In a most recent study the treatment with adenovirus-mediated ectopic expression of SHP-1 (AdSHP1) in the mouse model of hepatitis has ameliorated the production of pro-inflammatory cytokines (IL-6 and TNF-α) [147]. However, the interaction between SHP-1 and TRAF3 during herpes simplex virus-1 (HSV-1) and vesicular stomatitis virus (VSV) infection inhibits the antiviral innate immune response through diminishing the conserved serine-threonine kinase CK1ε recruitment to TRAF3 [148], [149]. The CK1ε-deficient mice are more prone to develop viral infections [149]. Thus, SHP-1-based therapeutics should be used depending on the pathogen-specific sepsis and cytokine storm.
Another tyrosine phosphatase called SHP-2 also acts as a negative regulator of TLR3 and TLR4 signaling. It specifically inhibits TRIF-dependent gene expression and inhibits IFN-β, IL-6, and TNF-α production. The phosphatase partially inhibits TBK1 activation by directly binding to its kinase domain independent of its kinase activity [150]. Thus, SHP-1 and SHP-2 perform opposing roles as the former promotes IFN-β production, while later inhibits IFN-β synthesis upon TLR stimulation. Interestingly, it was recently shown that glycogen synthase kinase (GSK)-3β interacts with SHP-2 and counters the inhibitory effect of the phosphatase and increases IFN-I production upon TLR2 stimulation [151].
Calcineurin is a serine/threonine phosphatase comprising two sub-units: a catalytic subunit called CnA, and a regulatory subunit called CnB [152]. It impacts a diverse array of biological functions, including lymphocyte activation, neuronal development, muscle remodelling, memory, heart valve morphogenesis, and innate immune response via modulating the function of NFAT (nuclear factor of activated T cells) in a Ca2+ calmodulin dependent manner [152], [153]. Although, calcineurin plays a positive role in T cell activation, it inhibits TLR signaling in macrophages (Table 2). The inhibitory role of calcineurin for TLR signaling in macrophages was suggested by the observations that its inhibition by immunosuppressive drugs (cyclosporine and FK506) or siRNA results in the increased production of pro-inflammatory cytokines. Calcineurin physically associates with TLR2, TLR4, MyD88, and TRIF, but not with TLR3 and TLR9 [154]. The association occurs in unstimulated macrophages and fibroblasts.
The calcineurin inhibition by specific drugs such as FK506 or cyclosporine A (CsA) stimulates TLR signaling pathway, and activation of NF-κB since the drugs cause removal of calcineurin from MyD88 and TRIF, and constitutively initiating the TLR signaling [154]. Thus, inhibition of calcineurin-NFAT signaling could prove beneficial in sepsis and drugs such as FK506, which is used by in organ transplant patients by inhibiting T cell-mediated responses, could be repurposed for sepsis patients and severe COVID-19 patients with sepsis. However, caution is warranted as the calcineurin-NFAT pathway is also used by myeloid cells for inducing calcineurin-NFAT pathway-mediated innate immune response [155], [156], [157]. Its loss/inhibition may lead to decreased production of IL-2, IL-12-p40, IL-23 and IFN-I in DC in response to Sendai virus and tolerance to LPS in macrophages [155]. Calcineurin may be required to prevent TLR signaling under steady state constitutive conditions. However, upon TLR stimulation, it dissociates from TLRs and their adaptors, and is required to induce a component of the innate immune system via calcineurin-NFAT pathway. Therefore, its loss or inhibition may lead to tolerance in myeloid cells to a variety of stimuli including LPS. Given the complexity of calcineurin regulated pathways, a careful assessment of calcineurin inhibitors in sepsis patients is required.
Protein tyrosine phosphatase-1B (PTP1B) is an intracellular tyrosine phosphatase and negatively regulate insulin signaling pathway [158]. A recent report has also shown its negative role in TLR signaling through inhibiting the MAPKs, NF-κB, and IRF3 activation along with Tyk2 and STAT1 activation in LPS-induced macrophages (Table 2) [159] The phosphatase inhibits both MyD88 and TRIF-dependent pathways in TLR-stimulated cells. As expected, PTPIB-/- mice are more prone to develop endotoxic shock and associated mortality due to higher levels of pro-inflammatory mediators (TNF-α, IL-6, CCL2 ,and CCL17) [160]. PTP1B regulates the release of pro-inflammatory mediators in Pseudomonas aeruginosa infection [161]. Thus, inhibition of PTP1B, envisaged for diabetes, may also lead to increased production of pro-inflammatory cytokines due to upregulation of TLR-mediated signaling, and hence, increased predisposition to bacterial pathogenesis and sepsis.
5.3. Negative regulation of TLR signaling via deubiquitination
A20, named after its cDNA clone number, is also known as TNF-α induced protein 3 (TNFAIP3). It is an ubiquitin modifying enzyme located in cellular cytosol and is induced by NF-κB dependent signals, which in turn inhibits NF-κB signaling as a negative feedback mechanism (Table 2) [162], [163]. A20 restricts NFκB signaling downstream of TNFR1, CD40, TLRs, NODlike receptors (NLRs) and IL1R [164], [165], [166], [167]. It directly removes ubiquitin moieties (K63-linked polyubiquitin chains) from TRAF6 [122], [167]. During TLR stimulation via LPS, A20 modifies the E3 ligase activity of TRAF6, TRAF2, and cellular inhibitor of apoptosis protein 1 (cIAP1) or BIRC2 through preventing its interaction with the E2 ubiquitin conjugating enzymes Ubc13 and UbcH5c [168]. Further, A20, together with the regulatory molecule TAX1BP1, interacts with Ubc13 and UbcH5c and induces their degradation through ubiquitin and proteasome system (UPS) [168], [169]. A20 phosphorylation during the TLR4-LPS signaling pathway via IκB kinase β increases its NF-κB inhibitory action via an unknown mechanism [170]. In human cases of sepsis, the expression of A20 mRNA and NF-κB was diminished in PBMCs of sepsis patients progressing towards immunosuppression [171]. The inhibition of A20 through downstream regulatory element antagonist modulator (DREAM, a Ca2+-binding protein family member containing 4 Ca2+ binding motifs (“EF-hands”), which interact as a tetramer with downstream regulatory element (DRE) to inhibit transcription) aggravates the sepsis-associated inflammation (acute lung injury) and mortality among animals through strengthening the NF-κB signaling in response to the TLR4 signaling activation [172], [173]. The DREAM-mediated inflammation involves the TAK1-mediated promotion of pro-inflammatory NF-κB signaling. On the other hand, the transcription factor upstream stimulatory factor 1 (USF1) also binds the E-box domain of the DRE of the A20 promoter to increase its transcription [172]. Hence, activating USF1 can increase A20 levels and suppress the pro-inflammatory TLR signaling responsible for cytokine storm during sepsis. Thus, A20 negatively regulates TLR signaling via various mechanisms and may act as a good molecular target for the management of sepsis and cytokine storm.
Tumor suppressor deubiquitinase (DUB), cylindromatosis (CYLD), inhibits TLR2 signaling responsible for the recognition of Gram-positive bacterial PAMPs [PGN, LTA, MALP-2 (Mycoplasma-derived lipopeptide 2), and Pam3CSK4 (a synthetic triacylated lipopeptide recognized by TLR1/TLR2 heterodimer] [174]. TLR2 stimulation upon binding with these ligand induces IKKs-IκBα and MKK3/6-p38 pathways via activating TRAF6 and TRAF7 (a newly discovered TRAF family member, as a signaling transducer upstream of the MEKK3-AP1 pathway), causing synthesis and release of CYLD in addition to pro-inflammatory cytokines (TNF-α, IL-1β, and IL-8) [174], [175]. The TLR2 signaling inhibition by CYLD involves the TRAF6 and TRAF7 inhibition via their deubiquitination (Table 2) [174]. CYLD also inhibits TRAF2 and NEMO (NF-κB essential modulator, a regulatory subunit of IKK), and blocks NF-κB signaling downstream to TLR activation [176], [177], [178]. It also acts as a crucial negative regulator of inflammatory pathway in Escherichia coli (E. coli)-induced pneumonia and sepsis, where it inhibits LPS/TLR4-mediated activation of NF-κB activation [179].
A role for inhibiting TRAF6 and TRAF7 for CYLD has been described in non-type able Haemophilus influenzae (NTHi) infection [174], [180]. However, a more recent study has shown that CYLD inhibits TLR signaling by deubiquitinating NTHi-induced K63-linked polyubiquitination of MyD88 at lysine 231 [181]. NTHi is a Gram-negative bacteria that initiates inflammatory immune response via activating TLR4-dependent TRAF6–IKK–NF-κB and TRAF6–MKK3/6–p38 signaling pathways [182]. Additionally, CYLD also regulates the TLR induced necroptosis of macrophages via inhibiting RIP1 (Receptor interacting protein-1; a serine/threonine kinase), which is activated downstream to TLR activation and integrates inflammation and necroptosis [183]. Thus, CYLD inhibits MyD88, RIP1 (receptor-interacting serine/threonine-protein kinase 1), TRAF2, TRAF6, TRAF7, and NEMO downstream to TLR signaling and regulates exaggerated inflammation which can lead to the development of severe infection causing sepsis and associated organ damage. A recent study has also shown that RIP1 inhibition blocks inflammatory diseases [184]. Hence, it will be worthwhile to study CYLD in context the sepsis-associated cytokine storm.
Ubiquitin-specific Protease 4 (USP4) is another DUB that inhibits TLR4 signaling via deubiquitinating Lys-63-polyubiquitinated TRAF6 and inhibiting its adaptor function (Table 2) [185] (Fig. 3). The TRAF6 inhibition prevents the NF-κB and AP-1 activation, responsible for generating pro-inflammatory immune response and the cytokine storm during sepsis [185]. Animals, lacking USP4 are more prone to develop endotoxemia and sepsis upon LPS challenge as compared to WT animals [185]. Thus, further studies are required to learn more on the role of USP4 as a novel regulator or TLR signaling and sepsis.
Ubiquitin-specific protease 18 (USP18 or UBP43) is also known as ISG15 (Interferon-stimulated gene 15) isopeptidase. It is a well-known negative regulator of type I and type III interferon signaling [186]. It is highly expressed in liver, spleen, and thymus, but at low levels in lungs, bone marrow and adipose tissues [187], [188]. Among immune cell types, USP18 is expressed at high levels in human CD169+ macrophages, bone marrow-derived DCs (BMDCs), monocyte derived and peritoneal macrophages, T cells, and B cells as well as in murine macrophages [187], [188], [189], [190]. However, it is not expressed in lung fibroblasts and bone marrow derive macrophages (BMDMs) [191]. In cells, it is localized to cellular cytosolic environment [191]. Humans have two isoforms of USP18: 1) USP18, located in the cytosol and, 2) USP18-sf isoform, which is an N-terminal truncated isoform, and is equally distributed in the cytosol and in the nucleus [191], [192]. The LPS-mediated TLR4 activation in human macrophages upregulates USP18, which in turn inhibits NF-κB activation, and thus the secretion of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) via interacting with TAK1-TAB1 and IKKα/β-NEMO complexes (Table 2) [193]. Upon binding with the TAK1-TAB1 complex, USP18 cleaves the K63-linked polyubiquitin chains of TAK1 through its proteolytic action. In the case of IKKα/β-NEMO complex, it specifically targets the regulatory subunit NEMO via its ubiquitin binding in ABIN (A20 binding and inhibitor of NF-κB) and NEMO (UBAN) motif and inhibits its K63-linked ubiquitination [193]. As expected, USP18 deficient mice are more prone to developing endotoxemia or sepsis upon LPS exposure [191]. Furthermore, human macrophages in which UPS18 is silenced with siRNA secrete higher levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) upon treatment with LPS [193]. USP18 also controls activation of inflammatory pathway in microglial cells of the brain [194]. Thus, USP18 acts as negative regulator of TLR4 signaling in both mice and humans, and may provide a new therapeutic approach for controlling exaggerated inflammation observed in sepsis patients.
Deubiquitinating enzyme A (DUBA) or OTUD5 is an OUT (ovarian tumor) domain family member cysteine protease. It suppresses type 1 IFN synthesis downstream to TLR signaling by removing K63-ubiquitin chains from TRAF3 to prevent the TBK1 recruitment and other downstream proteins necessary for NF-κB and IRF activation (Table 2) [195], [196], [197]. The interaction between DUBA and TRAF3 occurs during TLR3 activation [197]. The catalytic residues of DUBA comprise D221, C224, and H334, and C224S mutation abrogates its potential to deubiquitinate TRAF3 [197]. Thus, DUBA mediated inhibition of TRF3 inhibition blocks TLR-dependent type 1 IFN production, but does not inhibit NF-κB activation [86]. IL-1R-induced signaling down-regulates expression of DUBA, enhances K63-ubiquitination of TRAF-3, and increases production of anti-inflammatory cytokines (IL-10 and IFN-I) upon TLR9 stimulation [198]. OTUB1 and OTUB2 are members of the OTUB (Otubain) family of deubiquitinating cysteine protease family. They also negatively regulate the TLR signaling via deubiquitinating TRAF3 and TRAF6 and inhibiting induction of type I IFN [199].
5.4. Nuclear receptors/proteins negatively regulating TLR signaling
Nur77 (NR4A1, TR3 or NGFIB) is a key member of the Nuclear receptor 4A (NR4A) receptor subfamily. This subfamily of nuclear receptors plays crucial roles in various biological processes, including early embryogenesis regulation, thymocytes negative selection, gene expression in hypothalamic-pituitary adrenal axis, chronic inflammatory response, and vascular smooth muscle cell proliferation [200], [201], [202], [203]. They are orphan receptors (no known endogenous ligand) and lack a conventional DNA binding domain. Nur77 KO mice are more prone to develop sepsis upon LPS treatment [204]. Nur77 expression upregulates upon exposure to LPS and binds to TRAF6 within one hour of sepsis induction in vivo, and prevents auto-ubiquitination of TRAF6 [204]. The nuclear receptor also directly represses IRF4 and induces expansion of CD8+T cells [205]. Furthermore, Nur77 inhibits norepinephrine (NE) production by macrophages via trans-repressing NE promoter and attenuates neuro-inflammation [206]. Thus, Nur77 inhibits NF-κB signaling downstream to TLR4 activation directly by binding to TRAF6 and trans-repression of different gene promoters and may be important in preventing of the CNS inflammation in sepsis.
Nuclear receptor 4A2 or NR4A2 (Nurr1, HZF-3, RNR1, NOT or DHR38) is the second member of the NR4A subfamily and has similar structural motifs as shown by NR4A1 and NR4A3 [201]. It inhibits macrophage-mediated pro-inflammatory immune response [207]. This anti-inflammatory action of NR4A2 involves NF-κB signaling inhibition downstream to LPS-TLR4 signaling pathway (Table 2) [201]. The negative feedback mechanism involves GSK3β-mediated phosphorylation of p65 (RelA) subunit of NF-κB at serine 468. The phosphorylated p65 recruits Nurr1, which is induced and sumoylated in response to the inflammatory stimuli (IL-1β). The sumoylated Nurr1 recruits the Co-repressor for element-1-silencing transcription factor or CoREST receptor complex containing LSD1 (lysine specific demethylase-1), the histone methyltransferase G9a, histone demethylase, and histone deacetylases (HDACs). The complex clears NF-κB-p65 from promoters of inflammatory genes and brings back their transcription to the basal level. The mechanism operates well in microglia (brain macrophages) and astrocytes, and is important for protecting dopaminergic neurons from death [208]. Nurr1 also binds to the Ras guanyl-releasing protein 1 (RasGRP1) intron to regulate its expression [209]. RasGRP1 regulates the Ras-Raf-MEK-ERK signaling cascade downstream to TLR4 signaling [209]. The binding of prostaglandin E1 (PGE1) and PGA2 to the Nurr1 or NR4A2 enhances its anti-inflammatory action [210]. The knock down (KD) of the nuclear receptor from macrophages and microglia makes mice susceptible to atherosclerosis and Parkinson’s disease (PD). Thus, NR4A2 acts as negative regulator of TLR4 signaling by trans-repression of p65-stimulated genes, maintains homeostasis, and has a potential to protect individuals from inflammatory diseases, including sepsis.
Small heterodimer partner (SHP) is also known as NR0B2 and is considered as an atypical orphan receptor. It also lacks a conventional DNA-binding domain and has no known endogenous ligand except some recently identified adamantyl-substituted retinoid-related molecules [211]. SHP is highly expressed in spleen, liver, and BMDMs [212]. SHP-/- mice are more prone to develop LPS-induced endotoxemia and septic shock due to overproduction of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) as compared to WT mice [212]. LPS exposure to macrophages increases SHP expression through adenosine monophosphate (AMP)-activated protein kinase (AMPK). The LPS-induced SHP prevents Lys63-linked polyubiquitination of TRAF6, subsequent NF-κB activation, and production of pro-inflammatory cytokines (Table 2) [212], [213]. SHP also downregulates the mitochondrial anion carrier protein, UCP (uncoupling protein)-2 to lower the ROS production. It is noteworthy that ROS causes assembly of the inflammasome NLRP-3/ASC complex (which is pre-primed by LPS exposure), activation of caspase-1 (CASP-1), and maturation/release of IL-1β and IL-18 [214]. SHP also directly inhibits assembly of NLRP3 by inhibiting ASC recruitment to assembled NLRP3, a process required for recruitment and activation of CASP-1. In addition to TLR4, SHP also inhibits the activation of NF-κB via other TLRs, including TLR1, TLR2, TLR6, and TLR3 in both MyD88-dependent and -independent manner via binding with and inhibiting p65 and TRIF, respectively [212]. Drugs that increase expression of SHP (macrophage stimulating protein/MSP and fenofibrates) could reduce cytokine storm and tissue damage in sepsis.
Mitogen- and stress-activated kinases (MSK) 1 and 2 are nuclear proteins which share homology with the 90 kDa ribosomal S6 kinase (p90rsk) family. They are activated by ERK1/2 and p38MAPK in response to TLR (except TLR3) stimulation in immune cells, including macrophages and DC [215]. They usually have 2 kinase domains, namely, an N-terminal kinase domain (NTKD) in the AGC kinase (cAMP-dependent, cGMP-dependent and protein kinase C) family and a C-terminal kinase domain (CTKD) from the calmodulin kinase family [215], [216]. Their substrates include histone H3 (main substrate), cAMP response element binding protein (CREB), and activating transcription factor (ATF)-1 [215]. However, various other molecules, including NF-κB, High mobility group protein (HMG)-14, Retinoic acid receptor (RAR)-related orphan receptor (ROR)-α, Lysine demethylase-3A (KDM3A), Tripartite motif protein (Trim)-7, and Trim-28 also serve as substrate for MSKs [217], [218], [219], [220], [221]. MSK1/2 KO mice are hypersensitive to LPS. Upon exposure to LPS, these mice produce higher levels of pro-inflammatory cytokines (TNF-α, IL-6, and IL-12) with decreased production of anti-inflammatory cytokine IL-10, and develop sepsis [222], [223]. MSK1 and 2 phosphorylate histone H3 and CREB that negatively regulate TLR signaling (Table 2), inducing several anti-inflammatory genes including IL-10, IL-1Ra (IL-1Receptor antagonist), TTP (Tristetraprolin; the mRNA binding protein that causes TNF-α mRNA degradation), DUSP1 (Dual specificity protein phosphatase that inactivates p38 and JNK), and Prostaglandin synthetase (Ptgs)-2 that produces PGE2 [222], [223]. It is noteworthy that PGE2 inhibits TLR-induced production of pro-inflammatory cytokines from macrophages via binding to Nurr1 and activating its anti-inflammatory function.
As a homeostatic mechanism, MSK1 and 2 induced IL-10 inhibits TLR-induced cyclooxygenase (COX)-2 induction by degrading its mRNA and inhibits the prostaglandins (PGs) production, both in vitro and in vivo [224]. MSK1 and 2 KO mice exhibit increased PGE2 levels upon intraperitoneal (ip) injection of LPS or during sepsis [224]. Thus, MSKs play a crucial role in the negative regulation of TLR signaling, and maintaining homeostasis in macrophages and DC. Interestingly, MSKs may induce pro-inflammatory mediators like IL-8 in other cell types such as human fibroblast-like synoviocytes, neutrophils, keratinocytes, and smooth muscle cells due to different signaling circuits [215], [225].
TANK (TRAF-associated NF-κB activator) is a TRAF-binding protein and out of 7 identified TRAFs, it binds to TRAF1, 2, 3, 5, and 6 [226], [227], [228]. TANK is known for exerting its positive effect on TLR-induced NF-κB activation by acting as an adaptor to bridge TRAF3 and TBK1-IKK-i complex, and plays a crucial role in type I IFN production in viral infections, and in vitro TLR stimulation [229], [230]. However, TANK-/- mice have shown that TANK is not involved in TLR-induced type 1 IFN production, instead it acts as a negative regulator of TLR and B cell receptor (BCR) signaling [229]. TLR (TLR2 and TLR4, but not TLR3)-mediated ubiquitination of TRAF6 gets elevated in TANK-/- macrophages resulting in enhanced NF-κB activation and higher production of pro-inflammatory cytokines (IL-6 and TNF-α) [229]. Thus binding of TANK to TRAF6 inhibits its ubiquitination at K-63 chain and downstream signaling, activating NF-κB and AP-1 (Table 2). TANK also suppresses constitutive TLR signaling by gut’s commensal microbiota [229]. In addition to TRAF6 inhibition, TANK also inhibits canonical IKKs activated downstream to TLR signaling [231]. For example, macrophages isolated from TANK-/ - mice exhibit IKKε inhibition upon TLR stimulation, decreased activation of TBK1, and interaction between IKK-related kinases and canonical IKKs, which prevents the IKK-related kinases from negatively regulating the canonical IKKs. Thus, TANK exerts a negative impact on TLR signaling by preventing ubiquitination of TRAF6 and secondly by facilitating a cross-talk within the IκB kinase family members. Thus, it would be interesting to observe the impact of TANK on cytokine storm and sepsis.
PDLIM-2 is a PDZ (Postsynaptic density 95, discs large and zonula occludens-1) and LIM (Lin-11, Isl1 and Mec-3) domain containing protein. It is also known as Mystique or SLIM in mice [232], [233]. Mystique is the newest member of the alkaline phosphatase (ALP) subfamily of PDZ-LIM domain proteins [234], [235]. PDZ (also known as DHR or GLGF) domains are comprised of 80–100 amino acid (AA) domains mediating highly specific protein–protein interactions with a crucial role in the assembly of protein complexes [236]. While, LIM domains are cysteine rich zinc finger motifs, which act as protein binding interfaces in various transcription factors, protein kinases and scaffolding proteins, including zyxin and paxillin [237]. LIM domain containing proteins play important roles in cell fate decision during development, cytoskeletal arrangement, and in oncogenesis [237]. Mystique functions as an ubiquitin E3 ligase and targets STAT proteins for proteasome-mediated degradation. However, a report has shown PDLIM2 negatively impacts TLR (TLR3, TLR4 and TLR9)-mediated activation of NF-κB and inhibits the production of pro-inflammatory cytokines [238]. It acts as a nuclear E3 ubiquitin ligase and polyubiquitinates and transfers p65 subunit of NF-κB to discrete sub-nuclear domains called promyelocytic leukemia protein (PML) nuclear bodies (or PML oncogenic domains) for proteasomal degradation (Table 2) [238].
On LPS exposure, PDLIM2-/- mice exhibit uncontrolled NF-κB-mediated pro-inflammatory innate immune response, and produce increased levels of pro-inflammatory IL-6 and IL-12p40 cytokines [238]. Macrophages, DCs, and T cells highly express PDLIM2 [233]. The PDLIM2-induced p65 degradation is mediated by the activation of chaperone protein [heat shock protein 70 (Hsp70)], in LPS stimulated DCs [239]. The LPS-mediated TLR4 stimulation in DCs translocates Hsp70 to the nucleus, where it binds to the PDLIM2 and the proteasome-associated protein BAG (BCL2-associated athanogene)-1 causing translocation of the polyubiquinated NF-κB-PDLIM2 complex to the proteasome for degradation [239]. Their findings are further supported by the evidence that DCs deficient in either Hsp70 or BAG-1 release higher amount of pro-inflammatory cytokines in response to the exaggerated NF-κB activation due to high availability of active p65 upon TLR4 stimulation [239]. Consistent with these results, heat killed Propionebacteruim acnes elicit a profound pro-inflammatory immune response (higher levels of IL-1 and IL-12) in Hsp70-/- mice, showing their higher tendency to develop sepsis [239]. In human THP-1 cells, PDLIM2 is expressed in the nucleus, whereas upon their differentiation into macrophages, it is phosphorylated and sequestered in the cytoplasm. Contrary to the nuclear PDLIM2, its cytoplasmic counterpart profoundly activates NF-κB, increases TNF-α release, and increases adhesion in macrophages [240]. Thus, cytosolic and nuclear PDLIM2 behave differently with respect to their impact on TLR stimulation.
Makorin ring finger protein 2 (MKRN2) is also known as RNF62 or HSPC070. It has a tendency to bind to PDLIM2 and negatively regulates TLR signaling via inhibiting the NF-κB activation [241], [242]. MKRN2 contains four zinc finger domains and one RING finger domain [242]. RING finger domains are made up of eight conserved cysteine (Cys) or histidine (His) residues and are found in several hundreds of proteins acting as ubiquitin E3 ligases [243]. Consistent with its structural features, MKRN2 directly interacts with PDLIM2 via LIM domain of the PDLIM2, and promotes polyubiquitination and proteasomal degradation of p65 (Table 2) [242]. The two proteins act synergistically to polyubiquitinate the nuclear factor. MRKN2-/- DCs respond to LPS stimulation with higher NF-κB levels and produce higher levels of pro-inflammatory cytokines [242].
PDLIM1 is also known as CLP36 or Elfin. It is another LIM and PDZ domain-containing protein belonging to the APL sub-family. Like PDLIM2 and MRKN2, it also exerts its negative regulatory impact on TLR signaling and is present in the cytosol in most immune cell types, where it associates with α-actinin-1 and −4 [244], [245]. Interestingly, the inhibition of TLR signaling mediated by PDLIM1 occurs in the cytoplasm and involves sequestration of p65 subunit of NF-κB in the cytosol (Table 2). It binds and suppresses nuclear translocation of p65 in an α-actinin-4-dependent but IκBα-independent manner [244]. DCs and B cells from mice lacking PDLIM1 also show profound activation of pro-inflammatory immune response induced by TLR signaling due to over-activation of NF-κB [244].
TRIM30α is a member of the tripartite-motif (TRIM) protein family whose members play important roles in the regulation of cell development, differentiation and proliferation etc. [246], [247]. However, TRIM30α also serves as an important negative regulator of TLR signaling. It does so via interacting with the TAK1-TAB2-TAB3 complex formed upon TLR4 stimulation with LPS [248]. This interaction inhibits autoubiquitination of TRAF6 by degrading TAB2 and TAB3, and inhibits NF-κB activation, and IL-6 and TNF-α production (Table 2) [248]. Mice lacking TRIM30α are more prone to develop endotoxic shock and sepsis due to increased circulating levels of pro-inflammatory cytokine [248]. Thus, TRIM30α is an important regulator of TLR signaling and associated immune response.
TRIM8 is also a member of the TRIM family having a RING domain, a characteristic of ubiquitin E3 ligases, which plays a crucial role in posttranslational modifications of proteins and regulation of innate and adaptive immunity [246], [247]. This protein was previously shown to regulate TNF/IL-1β triggered inflammatory pathway via catalyzing K63-linked polyubiquitination of TAK1 and preventing phosphorylation of TAK1 [249]. However, TRIM8 also negatively regulates TLR3 and TLR4 signaling pathways via polyubiquitination of TRIF and inhibiting TRIF-TBK1 interaction (Table 2) [250]. LPS administration to TRIM8-/- mice causes an early induction of endotoxic shock and higher mortality as compared to WT animals [250]. In addition, TRIM8 KO mice infected with Salmonella typhimurium show greater losses in body weight and early development of sepsis [250]. Deficiency of TRIM8 induces a higher serum concentration of pro-inflammatory mediators (IL-6, TNF-α, RANTES, and IFN-β) upon exposure to TLR4 and TLR3 ligands than WT animals [250]. Thus, TRIM8 forms another member of TRIM family proteins that negatively regulates TLR signaling and sepsis immunopathogenesis.
Triad3A is a RING finger type E3 ubiquitin ligase that binds the cytoplasmic tail of several TLRs (TLR3, TLR4, TLR5, and TLR9 except TLR2) [251]. Triad3A is a protein comprising of 866 AA residues having a TRIAD domain with two RING motifs and an ‘in-between-RING’ motif at its C terminus [251]. It inhibits TLR signaling via degrading some TLR (TLR4 and TLR9 but not TLR2) proteins due to its E3 ubiquitin-protein ligase activity (Table 2) [251]. Triad3A decreases production of pro-inflammatory mediators responsible for the development of sepsis in response to LPS and CpG oligonucleotides, whereas its deletion from the cells causes a profound activation of TLR4 upon treatment with LPS [251]. In addition to inhibiting TLR4 and TLR9 via their direct degradation through ubiquitin–proteasome system, Triad3A also interacts with and promotes down-regulation of TRIF and TRIF-related adaptor molecule (TIRAP) [252]. Thus, this molecule acts as an important internal negative regulator of TLR signaling due to its E3 ubiquitin ligase activity. A recent study has shown that the monoclonal antibody (mAb) against transmembrane-TNF-α (tmTNF-α) protects against sepsis and septic shock involves the LPS-induced TLR4 internalization and its degradation to the TLR4 by recruiting Triad3A [253]. The mAb against tmTNF-α also protects against septic shock via increasing the A20 level and its anti-TLR signaling function. The treatment with adenovirus expressing Triad3A also protects against cardiac hypertrophy and improves cardiac function via degrading TLR4 and TLR9 through their ubiquitination that suppresses pro-inflammatory NF-κB activation [254]. Further studies are required in direction to design therapeutics activating Triad3A and A20 like endogenous pro-inflammatory TLR signaling inhibitors.
6. SUMOylation in TLR signaling negative regulation
SUMOylation is a post-translational modification pathway that is mechanistically analogous to the ubiquitylation pathway, but involves distinct enzymes [255]. Vertebrates have three SUMO (Small ubiquitin-like modifier) proteins: SUMO-1, −2, and −3. SUMO-1 is the founding member of the family and is also known as sentrin, PIC-1 or GAP-modifying protein 1 (GMP1) [256], [257], [258]. All SUMO proteins are initially synthesized as inactive precursors, and require maturation via proteolytic cleavage and removal of their carboxy (–coo) terminal glycine residues [255]. SUMOylation involves formation of an isopeptide bond between –coo terminus of SUMO and ε-amino group of a lysine residue of a target protein [255]. All SUMoylation reactions are catalyzed by a group of ubiquitin-like protein-processing enzymes that include E1 activating enzyme, an E2 conjugating enzyme and an E3 protein ligase [259], [260], [261]. The SUMOylation process is reversible and SUMO deconjugation (desSUMOylation) involves two ubiquitin-like protein-specific proteases in yeast (Ulp1 and Ulp2) and six sentrin-specific proteases (SENPs) in humans (SENP1-3 and SENP5-7) [261]. The SUMOylation of a protein alters its three-dimensional conformation, stability, ability to interact with other proteins and sub-cellular localization [261].
Several studies have shown that SUMOylation, like ubiquitination, also alters TLR signaling. For example, SUMOylation of IκBα component of NF-κB via SUMO1 inhibits NF-κB activation via inhibiting its phosphorylation [262]. SUMO2 and SUMO3 bind to Lysine residue 277 of NEMO and prevent its deubiquitination by CYLD (a deubiquitinase) and thus, causing stronger activation of IKK upon stimulation of cells with TLR4, TLR3, and TLR7-specific agonists [263]. The SUMO deconjugating enzyme SENP6 has the potential to inhibit this process of SUMO2 and SUMO3-mediated SUMOylation of NEMO. The SENP6-/- mice show an increased NF-κB activation and a profound production of pro-inflammatory mediators upon TLR stimulation. Thus, SENP6 is another host-derived negative regulator of TLR signaling with a potential to decrease sepsis-associated mortality. Myeloid cells (macrophages and DCs) deficient in SUMOylation due to absence of UBC9 (Ube2i)/SUMO E2 conjugating enzyme show a higher activation of NF-κB accompanied by exaggerated secretion of pro-inflammatory cytokines and type 1 IFNs [264]. The UBC9-/- mice with deficient/defective SUMOylation are more susceptible to the LPS-induced endotoxic shock and mortality. However, they exhibit more resistance to Chikungunya infection [264]. Another recent study has indicated that DC-specific UBC9-deficient (UBC9ΔDC) mice subjected to sepsis have higher mortality than WT mice over the time course of seven days [265]. These UBCΔDC mice also have higher circulating pro-inflammatory cytokines (IL-1β and IL-18) and their DCs also secrete their higher amount. As mentioned above, SUMOylation of Nurr1 recruits the repressor complex CoREST to GSK-3β-phosphorylated p65 in the nucleus. The complex clears NF-κB bound to the promoters of pro-inflammatory genes and down-regulates their expression to basal levels [207], [208], [266]. Thus, SUMOylation has emerged as a master regulator of NF-κB signaling downstream to TLR stimulation and pharmacologic targeting of this pathway may prove helpful in designing future therapeutic approaches for controlling bacterial sepsis and certain viral infections. For example, different pharmacological modulators (N106, Topotecan, spectomycin B1 (an antibiotic against Gram-positive bacteria), ginkgolic acid, tannic acid (potent anti-inflammatory agents), tenatoprazole, L-ascorbic acid, ebselen, and disulfiram etc.) of SUMOylation have been developed and tested for different diseases, including heart failure, ischemic diseases, neurological disorders, and cancers (glioblastoma, breast cancer) in animal models that can be studied in context to sepsis and cytokine storm for their anti-TLR signaling activity [267], [268], [269], [270], [271] .
7. NOD (Nucleotide binding oligomerization domain)-like receptor (NLR) family members as negative regulators of TLR signaling
NLRX1 (NLR family member X1) is a member of the NLR family that localizes to mitochondria, contains an N-terminal X domain, a NOD domain, and a C-terminal leucine-rich repeat domain (LRR) domain [272]. It regulates mitochondrial ROS (mROS) generation in mitochondria. It negatively regulates TLR4-mediated NF-κB activation upon LPS stimulation [272]. The stimulation of the TLR with LPS ubiquitinates NLRX1 causing its detachment from TRAF6. The free NLRX1 binds to the IKK complex inhibiting IKKα and IKKβ phosphorylation, and NF-κB activation (Table 2) [272]. The finding is further strengthened by the observation that NLRX1-/- mice are more prone to develop endotoxemia and sepsis after their exposure to LPS, and succumb to septic shock due to the increased production of IL-6 and TNF-α [272]. Thus, NLRX1-mediated inhibition of TLR-induced NF-κB activation is mediated by its interaction with TRAF6 and IKK complex. Interestingly, NLRX1 also sequesters STING (stimulator of interferon genes) and inhibit its interaction with TBK-1, required to produce IFN-I in response to viral DNA [273].
NLRC3 (NLR containing CARD domain-3) is also known as CLR16.2 or NOD3. It is a poorly characterized member of the NLR family, and acts as a cytosolic regulator of innate immunity. The negative regulation of TLR signaling and inhibition of NF-κB by NLRC3 involves its interaction with TRAF6 to attenuate its Lys63 (K63)-linked ubiquitination (Table 2) [274]. The LPS-mediated stimulation of macrophages in vivo decreases the NLRC3 mRNA levels and restores them once the inflammatory response subsided [274]. Macrophages isolated from NLRC3-/- mice, upon stimulation with the LPS, generate higher levels of pro-inflammatory cytokines (IL-6, IL-1β, IL-12, and TNF-α) due to increased K63 ubiquitination of TRAF6 and consequently enhanced NF-κB activation [274]. Furthermore, NLRC3-/- mice, upon treatment with even low dose of LPS, develop lethal endotoxemia [274]. Thus, NLRC3 has a protective role in sepsis via negatively regulating TLR signaling mediated by its inhibitory action of MyD88 and TRAF6. Upon stimulation with LPS, NLRC3 associates with PI3-K, blocks activation of AKT and mTOR (mammalian target of rapamycin, a serine/threonine kinase, is the catalytic subunit for both the mTORC1 and mTORC2 complexes) [275].
The TLR4-induced activation of AKT in macrophages is very important for the initiation of oxidative phosphorylation (OXPHOS) and induction of the mitochondrial transcription factor A [276]. Alginate-derived guluronate oligosaccharide (GOS) readily activates macrophages via binding to TLR4 and mediated TLR4/Akt/NF-κB, TLR4/Akt/mTOR, and MAPK signaling pathways [277]. An increased activation of the mTOR signaling pathway has been observed in NLRC3-/- BMDMs upon stimulation with LPS [275]. This activation can be responsible for the altered cytokine release as inhibiting mTOR signaling by rapamycin (selectively inhibits the complex comprising of mTOR, mLST8, and Raptor (mTORC1) via associating with FK506-binding protein 12 (FKBP12), but rapamycin does not inhibit the complex comprising of mTOR, mLST8, and mTORC2) promotes synthesis and release of pro-inflammatory cytokines (IL-12p40, IL-12p70 and IL-23 heterodimer, TNF-α, IL-6) via the transcription factor NF-κB, but suppresses the IL-10 release via STAT3 [278]. The tuberous sclerosis complex protein 2 (TSC2, a key negative regulator of mTOR) knockout murine embryonic fibroblasts (MEFs) do not produce IL-12p40, and IL-6 and TNF-α production also decrease, instead produce higher levels of IL-10 due to increased STAT3 activity [278]. Thus TSC2 negatively regulates mTOR activation-mediated pro-inflammatory cytokine release. IL-10 does not affect the IL-12p40 production in response to the rapamycin-mediated mTOR inhibition. Rapamycin-mediated IL-12p40 and TNF-α increase occurs in response to the overactivation of JNK-1 and NF-κB in response to the LPS. Thus, NLRC3 has a potential to regulate the TLR4 signaling through several mechanisms, including inhibition of MyD88-TRAF6 interaction, via regulating PI3K and Akt signaling pathway, and mTOR signaling. Because of these pathways, mice lacking NLRC3 are more prone to develop sepsis upon LPS exposure.
NLRC5 (NLR family, CARD domain containing 5) is the largest NLR family member, consisting of 1866 AA with C-terminal 27 LRRs [279]. It plays an essential role in the expression of basal levels of MHC class I [280]. Cellular expression of NLRC5 is induced by TLR stimulation with their specific ligands. The induced NLRC5 shuttles between cytosol and nucleus in a CrmA-dependent manner [281]. Inhibition of NLRC5 in murine macrophages generates exaggerated level of pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β) upon stimulation with LPS [281]. The NLRC5-mediated inhibition of TLR signaling is mediated by inhibition of NF-κB and type 1 IFN signaling pathway through its interaction with, and blocking phosphorylation of, IKK complexes (Table 2) [282]. NLRC5 deficiency in mice is associated with enhanced IL-6 and IFN-β production by embryonic fibroblasts (MEFs), peritoneal macrophages, and BMDMs upon LPS stimulation [283]. These mice also show higher levels of IL-6 in serum upon their exposure to LPS [283]. However, some contradictory reports have also been published [284], [285]. Meng et al (2015) have provided more insight in aspect of NLRC5 and showed that NLRC5 undergoes robust and rapid ubiquitination by TRAF2/6 upon LPS exposure, causing dissociation of NLRC5 from IκB kinase complex [286]. The ubiquitin specific protease 14 (USP14) removes the polyubiquitin chains from NLRC5 for enhancing its negative impact on NF-κB signaling [286]. Investigation by these authors has also indicated that NLRC5-mediated negative impact on TLR signaling is cell type specific and depends upon the cell’s repertoire of deubiquitinases. For example, BMDCs do not exhibit the NLRC5 mediated negative impact on TLR signaling due to their reduced expression of NLRC5 and USP14 [286]. Whereas, BMDMs express higher levels of NLRC5 and USP14, and peritoneal macrophages express even their higher levels. Therefore these cell types are differentially sensitive to NLRC5-mediated regulation of TLR signaling [286]. Thus a feedforward loop of NLRC5 mediated (de)ubiquitination keeps IKKα and IKKβ mediated NF-κB activation in check depending on the cell type and level of NLRC5 and USP14 expression upon exposure to TLR ligands [287].
8. Inhibition of TLR signaling by soluble decoy TLRs
ST2 (Serum stimulation 2 factor) is also known as the Interleukin 1 receptor-like 1 receptor (IL1RL1). Its other names include T1, Fit-1 (Fos-induced transcript 1) and DER4 [135], [288]. Tominaga et al in 1989 described ST2 for the first time in 1989, and it was cloned as a delayed early response gene induced in Th2 cells by cell proliferation inducing stimuli [289], [290], [291], [292]. It serves as a part of IL-33 receptor and plays a crucial role in inflammation in allergic asthma [293]. At least 3 (ST2L, sST2, and ST2V) protein variants of ST2 are known [294]. ST2L is the longest protein variant of ST2, which is present as a type I transmembrane protein having three extracellular immunoglobulin (Ig)-like domains and an intracellular TIR domain [135], [295]. This variant is expressed in Th2 cells and mast cells, and heterodimerises with the co-receptor IL1RAP (IL1R accessory protein) to constitute the cellular receptor for the IL-33 cytokine [293]. Whereas sST2 (the soluble form) is similar to the extracellular region of ST2L and has 9 extra AAs at its C terminus [294]. It has only an N-terminal extracellular Ig-like domain, but lacks both the transmembrane (TM) and the cytoplasmic TIR domains [288]. On the other hand, sST2 acts as a decoy receptor for ST2L, the function of ST2V, which also lacks the immunoglobulin (Ig) domain, remains unknown [288], [289], [296].
There is another form, ST2LV, found in birds. It lacks trans-membrane domain and its function is also not known (2 6 2). ST2L in humans negatively regulates TLR4, TLR2, and TLR9 signaling via binding to and sequestering MyD88 and MAL through its BB-loop region located in its TIR domain albeit without impacting TRIF or IRAK activity [297], [298], [299], [300]. However, ST2L deficient mice are not susceptible to LPS-induced endotoxic shock as compared to WT mice and did not develop endotoxin tolerance both under in vitro and in vivo conditions [297]. This phenotype of ST2-deficient mice can be explained by the fact that ST2L, under normal conditions, is present inside the cytosol and is translocated onto the cell membrane at least after 4 h post stimulation with LPS [135]. This 4 h delay in the expression of ST2L on the cell surface provides a time frame for the generation of endotoxic or septic shock, but provides enough time for the development of endotoxin/LPS tolerance in the animal [135]. Hence, ST2L provides an effective negative-feedback mechanism in selective TLRs (TLR2, TLR4 and TLR9 but not TLR3)-mediated signaling including contribution to endotoxin tolerance and inhibition of TH1 responses [135].
sST2 also acts as a negative regulator of TLR-induced pro-inflammatory immune response. The levels of sST2 increase under inflammatory conditions (upon stimulation with LPS), and thereafter it binds to macrophages through ST2 receptors, which also increase on macrophages upon exposure to LPS [301], [302]. sST2-IgG fusion protein inhibits the generation of pro-inflammatory cytokines (IL-6, IL-12, and TNF-α) from LPS-stimulated macrophages [303]. It also decreases mRNA expression for TLR4 and TLR1, the exact mechanism of which remains to be explored [303]. Mice treated with anti-sST2 antibody exacerbate LPS-induced septic shock and mortality [303]. Additionally, recent studies have also shown elevated plasma levels of sST2 in human sepsis patients as early as day 0, and these levels correlate with severity of sepsis and associated mortality [304], [305].
SIGIRR (Single immunoglobulin IL-1R-related receptor) is also known as TIR (Toll IL-1 receptor)-8. It is a member of the TLR/IL-1R superfamily and is the only TIR-domain–containing receptor with one immunoglobulin (Ig) domain in its extracellular region [306], [307]. The sigirr gene was first identified in 1998 and is localized on human chromosome 11p15.5 [308]. It is highly expressed in epithelial cells of gastrointestinal tract (GIT), liver, lungs, spleen, kidneys (tubular cells, on the luminal brush border and basolateral cell membranes), and other lymphoid organs [308], [309], [310]. At the cellular level, SIGIRR is expressed on NK, B, and Treg cells, monocytes, and immature myeloid DC, and its level of expression keeps on decreasing as the cells mature [309], [311], [312]. SIGIRR/TIR8 plays a crucial role as a negative regulator of TLR signaling and IL-1R family via directly inhibiting NF-κB and JNK activation (Table 2) [306], [309], [313], [314], [315], [316]. For example, SIGIRR-mediated inhibition of TLR4 signaling pathway involves only its TIR domain to inhibit the recruitment of IRAK and TRAF6 towards MyD88 [314]. Thus, SIGIRR plays a crucial role in TLR signaling, and SIGIRR-/- mice are more prone to develop skin, lung, and kidney infections along with their enhanced susceptibility to develop sepsis depending on the strain of laboratory mice [317], [318]. For example, SIGIRR-deficient BALB/c mice are more prone to develop sepsis, but SIGIRR-deficient C57BL/6 × 129/Sv mice exhibit normal systemic inflammatory immune response upon challenge with LPS [306], [311]. However, monocytes isolated from human septic patients exhibit higher levels of SIGIRR as compared to monocytes isolated from healthy volunteers [313]. The results may explain a ‘reprogramming’ of these cells in sepsis patients. The increased expression of SIGIRR may represent a compensatory mechanism of the patients for downregulating their inflammation. SIGIRR serves as a subunit of IL-37 receptor in human. IL-37 inhibits TLR and members of the IL-1R family-induced signaling and could serve as an important therapeutic tool for chronic inflammatory conditions in humans [319].
TLR10 is a member of the TLR family expressed by humans for which no specific ligand has been identified and hence remains an orphan receptor [320], [321]. The human TLR10 gene was first cloned in 2001 and was found to be strongly expressed in various lymphoid tissues including spleen, lymph nodes, tonsils and thymus [322], [323]. It is located on chromosome 4 within the TLR2 gene cluster, along with the genes encoding TLR1, TLR2, and TLR6 [323], [324]. In mice, it is present as a pseudogene, therefore mouse models remain non-informative regarding its biological and immunological functions in the host [320]. However, a complete TLR10 gene sequence has been detected in the rat genome indicating that this species may have functional TLR10 [324].
The expression of TLR10 is highly restricted; it is expressed in a highly N-glycosylated form in certain human B and monocytic cell lines, pDC isolated from human tonsils, Langerhans cells (LCs), peripheral blood B cells, and monocytes [320], [324]. Phylogenetically, TLR10 is closely related to TLR1 and TLR6, and forms a heterodimer with TLR1 [321]. The TLR1-TLR10 heterodimer localizes in the phagosomes and recognizes the same PAMPs that are specific for TLR2 [321]. On its own, TLR10 is not known to recognize any PAMP. Even after ligation with specific PAMPs, TLR10-TLR2 heterodimer does not activate the typical TLR signaling pathway highlighting an inhibitory role of TLR10 in the heterodimer-induced signaling [321]. Thus, human TLR10 exhibits a tendency to cooperate with TLR2 in the recognition of different bacterial and fungal PAMPs and modifies TLR-mediated signaling [321].
The genetic variation within the TLR10/1/6 gene locus is a major common genetic factor that explains inter-individual variation in TLR1/2-mediated cytokine responses in humans and most highly-associated SNPs fall within the TLR10 gene region [325]. Thus, variation in the expression of TLR10 gene determines an individual’s response to TLR2 specific PAMPs and has implications for the development of sepsis and inflammatory diseases. This notion is further supported by a study indicating that humans with loss-of-function SNPs in the TLR10 gene locus exhibit an increased TLR2-mediated cytokine production upon exposure to Gram-positive bacteria or their PAMPs [326]. For example, PBMCs isolated from individuals harboring N241H (rs11096957) and M326T (rs11466653) SNPs (responsible for degrading TLR10) upon stimulation with Pam3Cys (a TLR2 ligand) or live Borrelia burgdorferi (a spirochete that is the causative agent of Lyme Disease) show a higher production of IL-1β, IL-6, IL-8, and TNF-α due to a lack of functional TLR10 [326].
The anti-inflammatory effect of TLR10 has further been confirmed in mice expressing human TLR10 as a transgene. The mice exhibited decreased innate immune response with lower plasma IL-6, CXCL1, and IL-8 levels in response to challenge with TLR2 ligands [326]. In addition to its inhibitory action on TLR2 signaling, TLR10 also exerts its negative impact on LPS-induced TLR4 signaling via inhibiting MyD88- and TRIF-inducing IFN-β–mediated signaling (Table 2). More specifically, it inhibits degradation of IkBα and phosphorylation of MAPK. For these inhibitory actions on TLR4, a homodimeric form of TLR10 is required [322]. The transgenic mice expressing human TLR10, upon exposure to LPS, also secrete lower levels of pro-inflammatory mediators of endotoxemia (IL-6, TNF-a, and type 1 IFNs) [322]. Thus TLR10 also has a potential to decrease the pro-inflammatory immune response observed during sepsis via negatively inhibiting the TLR4 signaling. TLR10 negatively impacts activation and differentiation of monocytes via inhibiting TLR- as well as CD40-activated signaling pathways [327]. Thus, TLR10 negatively regulates TLR1, TLR2, and TLR4 signaling that are major TLRs playing crucial roles in the pathogenesis of bacterial sepsis and its outcomes. Future studies will be helpful in this direction to design better therapeutic agents activating TLR10 to reduce sepsis-associated multi organ damage and morbidity.
9. Miscellaneous inhibitors of TLR signaling that are not direct players in the TLR-activated signaling cascade
Activating transcription factor-3 (ATF3) belongs to the activating transcription factor/cAMP response element (ATF/CREB) family of bZip (basic Leucine Zipper) transcription factors that bind to the consensus cAMP response element (CRE) sequences (Table 2) [328]. Stimulation of macrophages with LPS or Bacillus Calmette Guerin (BCG) induces ATF3 synthesis [329], [330]. This increase in ATF3 in stimulated macrophages initiates the induction of negative feedback loop to modulate the TLR4 signaling, and inhibits the synthesis of IL-6 and IL-12 cytokines [331]. Furthermore, ATF3 not only inhibits TLR4 signaling, but also other TLRs, including TLR2/6, TLR3, TLR5, TLR7, and TLR9 in both macrophages and DCs [332]. Macrophages and DCs deficient in ATF3 secrete higher levels of IL-6 and IL-12 upon stimulation with different TLR agonists [332]. ATF3-/- mice also show inhibitory action of ATF3 on TLR signaling and the release of pro-inflammatory cytokines (IL-6 and IL-12). Furthermore, ATF3-/- mice also show increased mortality and decreased bacterial clearance following Streptococcus pneumoniae-induced pneumonia [333]. The urinary ATF3 levels can be used as a biomarker for sepsis-associated AKI [334]. Thus, ATF3 is a crucial player in the pathogenesis of sepsis due to its negative impact on multiple TLRs and could serve as a biomarker for sepsis-associated kidney injury.
The therapeutics activating ATF3 or treatment with Adenovirus-associated ATF3 should be explored during bacterial sepsis or sepsis-associated with severe CVOID-19. The adeno-associated virus-mediated ATF3 gene transfer in ATF3 KO mice protects them from sepsis-induced mortality and decreases the concentration of pro-inflammatory mediators, including IL-6, TNF-α, NO, MCP-1, and HMGB1 in the lung tissues or serum [335]. Hence, ATF3 has a potential to inhibit sepsis and severe COVID-19-induced cytokine storm. Further studies are needed in the direction. However, ATF3-based therapeutics should be indicated in during the early stages of sepsis with increased pro-inflammatory immune response as during SAIS it may cause further immunosuppression that may increase the predisposition of the host to secondary infections due to the increased suppression of IL-6 [336]. Hence, ATF3-based therapeutics should be contradicted in patients with SAIS.
Interleukin 37 (IL-37) is also known as IL-1 family member 7 (IL-1F7). The cytokine exists in its five splice variants (a-d), which were discovered and given different names in the past [337], [338]. Of these splice variants, isoform b (or IL-37b) is the most prevalent and widely studied. IL-37 is a member of the IL-18 subfamily and IL-1 cytokine family, and was identified in 2000 by three independent research groups using in silico technology [339], [340]. IL-37 induction occurs via TLR4 and TLR2 activation in response to their corresponding ligands (LPS or Pam3CSK4) [341]. The gene is constitutively expressed at mRNA level, but is rapidly degraded due to the presence of an mRNA instability motif in its exon 5. Upon cell activation by TLR or IL-1RF members, IL-37 mRNA is stabilized and translated [342]. For this reason, inflammatory stimuli induce an increase in IL-37 levels in tissues and plasma. For example, sepsis patients have elevated serum IL-37 level that well correlates with IL-1β levels [343]. No IL-37 homologous gene has so far been detected in mice. IL-37 binds IL-18Rα (which is also used by IL-18) and recruits SIGIRR for transducing its inhibitory signal [344]. Although mice do not produce IL-37, human IL-37 functions as a potent anti-inflammatory cytokine. On one hand SIGIRR KO mice are hyper-responsive to LPS and other TLR agonists, on the other hand, IL-37 transgenic mice are relatively resistant to TLR agonists [337], [338]. Inhibition of IL-37 activity by siRNA significantly increase the production of several pro-inflammatory mediators (IL-1β, IL-1α, GM-CSF, C-CSF, IL-6, and TNF-α) from human PBMCs when stimulated with either LPS or Pam3CSK4 [341]. The inhibition also increasingly activates the TLR2-TLR6 heterodimer signaling upon its stimulation with MALP-2 (Mycoplasma-derived lipoprotein-2) [341]. The RAW 264.7 macrophage cell line stimulated with different TLR4 agonists also shows similar results [341].
The inhibitory action of IL-37 on the release of pro-inflammatory cytokine occurs due to its interaction with Smad3 (Mothers against decapentaplegic homolog 3), an important mediator for TGF-β-induced signaling [341]. Furthermore, it also inhibits mTOR and GSK-3α/β, and activates Mer-PTEN-DOK and AKT pathways. The group has further shown the protective effects of IL-37 in mice against LPS induced endotoxemia due to decreased production of pro-inflammatory cytokines, decreased activation of DCs via LPS and modulation of other pathophysiological markers of sepsis [341]. IL-37-mediated inhibitory effect on LPS or Pam3CSK4 by TLR4 and TLR2 activation, respectively, is dependent on the inhibition of MyD88 adaptor activity without affecting the TRIF-dependent TLR3 activation (Table 2) [345]. IL-37 also inhibits inflammasome assembly, caspase-1 activation, and maturation and release of cytokines such as IL-1β and IL-18 [346]. In addition, IL-37 released as consequence of TLR signaling forms a tripartite complex with IL18Rα and IL-1R8 (SIGIRR) that has a potent inhibitory action through many MyD88-independent mechanisms [344], [347], [348]. For example, IL-37 and IL-1R8 (SIGIRR) signaling has a boosting effect on TAM family of receptor kinases that are well known inhibitors of TLR signaling via inducing the activation of SOCS1 and SOCS3 [118]. IL-37 via SIGIRR also increases the expression of adaptor p62 (DOK) and inhibits the activation of Erk through TLR4-LPS signaling [112]. IL-37 via SIGIRR or IL-1R8 also inhibits TAK1 a central mediator of various inflammatory pathways (that is IL-1R1, IL-18R, TNF and antigen receptors on T and B cells) along with all TLR-signaling pathways [344], [349]. The increased IL-37 levels in the serum of sepsis patients have been reported and correlate with IL-1β levels [343]. Thus, IL-37 is another host-derived cytokine with a great potential to inhibit TLR2 and TLR4 signaling involved in the immunopathogenesis of bacterial sepsis. This TLR inhibitory action of IL-37 during sepsis can be further studied to design a new therapeutic approach for sepsis. Thus, IL-37 is another host-derived molecule that could use multiple mechanisms to inhibit TLR-induced cytokine storm could be target to design a new therapeutic approach for sepsis.
10. Future perspective and conclusion
TLRs are one of the major players in the recognition of microbial PAMPs and the induction of a pro-inflammatory innate immune response to protect the host from the invading pathogen. However, exaggerated and continuous activation of the TLR signaling pathway in response to continuous exposure to PAMPs (for example, LPS released from bacteria) and DAMPs released from dying host cells and damaged tissues results in the generation of cytokine storm, which proves detrimental to the host via causing development of multi-organ dysfunction syndrome (MODS) or more severe multi-organ failure, and ultimate death of the patient. The TLR-activated signaling pathways not only induce pro-inflammatory mediators but are also programmed to activate several negative feedback mechanisms that counter the actions of the pro-inflammatory mediators, downregulate their production, and exert anti-inflammatory, and tissue repairing effects. Their main aim is to eliminate the pathogen, conclude the TLR-initiated inflammatory response, restore immune homeostasis and repair damaged tissues.
Still the systemic inflammatory response causing organ damage and high mortality among sepsis patients remains high and uncontrollable. We do not know how the TLR-induced negative regulatory mechanisms fail to fulfil their job of inhibiting exaggerated activation of TLRs and associated cytokine storm. Many studies have shown that knocking out the gene responsible for these negative regulatory molecules in mice make them more prone to develop LPS-induced endotoxemia and associated endotoxic shock, and increased mortality as compared to WT mice [95], [98]. By and large, we have remained unsuccessful to translate these findings into clinical settings. For example, the most recent TLR-based therapeutic agent known as Eritoran (a synthetic MD2-TLR4 antagonist) or E5564 has shown effective negative regulation of TLR4 stimulation and sepsis in animal models [350], [351] but proved nonsignificant in patients of sepsis and did not lead to decreased 28-day mortality in comparison to placebo [352], [353]. The failure may be due to different reasons, including the involvement of more than one TLR (TLR4) in sepsis patients and patients in the phase III trial were not enrolled on the basis of presence of LPS in the circulation. Also, in clinical trial for sepsis in future a homogeneous sepsis population with same source of sepsis (community acquired pneumonia-induced sepsis or peritonitis-induced sepsis) along with other clinical trial-based criteria should be included to escape the failure of clinical trial. This will help to get better clinical trials-based results for therapeutics targeting immune system and TLR signaling-based therapeutics. This failure of Eritoran to manage sepsis has suggested us the requirement for the complete rethink in evaluating the novel agents/molecules for sepsis management. However, Eritoran has successfully targeted the cytokine storm generated during acute influenza virus infection in experimental studies [354]. The protective action of Eritoran also involves CD14 and TLR2. The Eritoran directly binds to the CD14 and prohibits the ligand binding to MD-2 (myeloid differentiation factor-2) or lymphocyte antigen 96 [354] . Hence, further studies of Eritoran are required to observe its impact on cytokine storm in severe COVID-19 also. Thus, failure of Eritoran in the management of sepsis in clinical settings opened our Pandora box with the hope to use Eritoran and these host-derived negative regulators of TLRs differently to design future therapeutics to target inflammatory diseases, including cytokine storm associated with recent pandemic called SARS-CoV2 infection-induced severe COVID-19 and bacterial sepsis.
For example, research in this direction has established the new role of Wnt-β catenin signaling pathway as a negative regulator of LPS-induced TLR4 activation and NF-κB mediated generation of pro-inflammatory immune response without any direct interaction between NF-κB and β-catenin [355]. Also, further study has indicated Wnt3A (Wnt family member 3A) or dishevelled 3 (Dvl3) activates during TRL4 stimulation and inhibits the exaggerated production of pro-inflammatory cytokines (TNF-α, IL-6, and IL-12) and neutrophil infiltration via regulating GSK-β-catenin and NF-κB signaling [356]. In addition, S100 alarmins, comprising of S100 calcium-binding protein A8 (S100A8) [also called myeloid related protein-8 (Mrp-8)] and S100 calcium-binding protein A9 (S100A9) [myeloid related protein-14 (Mrp-14)] are endogenous TLR4 ligands and typically form a heterodimer complex called S100A8/A9 (or Calprotectin) upon their release from myeloid cells [357]. They prove detrimental to adult animals suffering from sepsis [357]. These S100-alarmins induce stress tolerance in phagocytes under sterile inflammatory conditions [358]. Ulas et al (2017) have shown that high levels of these S100 alarmins at birth exert their protective effect on neonatal sepsis via inducing MyD88-dependent hyporesponsiveness to LPS, but preserve the antimicrobial action of innate immune system [359]. Thus, host-derived TLR regulatory mechanisms which are protective to newborn but detrimental to later in life in adults or vice versa needs to be carefully studied at all levels, and different age groups of people as sepsis affects everyone without making a discrimination on the basis of age, gender and race.
The genetic variants of A20, a negative regulator of TLR signaling, has been studied in relation to other inflammatory disease, including inflammatory bowel disease (IBD), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), diabetes, and atherosclerosis [360], [361]. However, study of incidence of sepsis in these group of people exhibiting genetic variations in the A20 gene may provide a direct evidence to target sepsis via A20-based therapeutic approach in humans. In this direction, a study has shown that a specific group of people in China (Chinese Hans) exhibiting rs5743867 in TOLLIP gene are resistant to develop sepsis [137], and TOLLIP based therapeutic approach has a great potential in sepsis management. Other than genetics, post-translational modifications (ubiquitination, phosphorylation, SUMOylation etc.) of TLR-signaling components can also be used as novel therapeutic approaches to target sepsis and associated cytokine storm. Future studies in the direction will open the Pandora Box where not the evil but the novel therapeutics will come out to take care of cytokine storm during sepsis and severe COVID-19.
In conclusion, TLR-signaling mediated induction of cytokine storm plays a crucial role in bacterial sepsis and acute viral infection (severe COVID-19)-associated cytokine storm responsible for multi organ failure and death among patients. Thus understanding and studying endogenous negative regulators of TLR signaling may provide novel therapeutic targets against cytokine storm generated during different diseases, including sepsis and sever COVID-19. Endogenous negative regulators of TLR signaling have potential for gene-based therapeutics or gene therapy for cytokine storm during sepsis and severe COVID-19. However, these endogenous negative regulators of TLR signaling should be used cautiously depending on the pathogen-type inducing the cytokine storm and sepsis, age, immune status, comorbidities, and sex of the patients during experimental and clinical setting to escape from failure. Further studies will prove helpful in the direction.
Declaration of Competing Interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.intimp.2020.107087.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References:
- 1.Fleischmann C., Scherag A., Adhikari N.K., Hartog C.S., Tsaganos T., Schlattmann P., Angus D.C., Reinhart K. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations, Am J Respir Crit Care Med. 2016;193(3):259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 2.Rudd K.E., Johnson S.C., Agesa K.M., Shackelford K.A., Tsoi D., Kievlan D.R., Colombara D.V., Ikuta K.S., Kissoon N., Finfer S., Fleischmann-Struzek C., Machado F.R., Reinhart K.K., Rowan K., Seymour C.W., Watson R.S., West T.E., Marinho F., Hay S.I., Lozano R., Lopez A.D., Angus D.C., Murray C.J.L., Naghavi M. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi: 10.1016/S0140-6736(19)32989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finfer S., Machado F.R. The Global Epidemiology of Sepsis. Does It Matter That We Know So Little? Am J Respir Crit Care Med. 2016;193(3):228–230. doi: 10.1164/rccm.201510-1976ED. [DOI] [PubMed] [Google Scholar]
- 4.Rosenthal V.D., Maki D.G., Mehta Y., Leblebicioglu H., Memish Z.A., Al-Mousa H.H., Balkhy H., Hu B., Alvarez-Moreno C., Medeiros E.A., Apisarnthanarak A., Raka L., Cuellar L.E., Ahmed A., Navoa-Ng J.A., El-Kholy A.A., Kanj S.S., Bat-Erdene I., Duszynska W., Van Truong N., Pazmino L.N., See-Lum L.C., Fernandez-Hidalgo R., Di-Silvestre G., Zand F., Hlinkova S., Belskiy V., Al-Rahma H., Luque-Torres M.T., Bayraktar N., Mitrev Z., Gurskis V., Fisher D., Abu-Khader I.B., Berechid K., Rodriguez-Sanchez A., Horhat F.G., Requejo-Pino O., Hadjieva N., Ben-Jaballah N., Garcia-Mayorca E., Kushner-Davalos L., Pasic S., Pedrozo-Ortiz L.E., Apostolopoulou E., Mejia N., Gamar-Elanbya M.O., Jayatilleke K., de Lourdes-Duenas M., Aguirre-Avalos G. International Nosocomial Infection Control Consortium (INICC) report, data summary of 43 countries for 2007–2012. Device-associated module, Am J Infect Control. 2014;42(9):942–956. doi: 10.1016/j.ajic.2014.05.029. [DOI] [PubMed] [Google Scholar]
- 5.Silva J.M., dos Santos S.D.S. Sepsis in AIDS patients: clinical, etiological and inflammatory characteristics. Journal of the International AIDS Society. 2013;16(1):17344. doi: 10.7448/IAS.16.1.17344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danai P.A., Moss M., Mannino D.M., Martin G.S. The epidemiology of sepsis in patients with malignancy. Chest. 2006;129(6):1432–1440. doi: 10.1378/chest.129.6.1432. [DOI] [PubMed] [Google Scholar]
- 7.Gingo M.R., Morris A. HIV-infection and severe sepsis: a bitter pill to swallow. Critical care medicine. 2015;43(8):1779–1781. doi: 10.1097/CCM.0000000000001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angus D.C., Linde-Zwirble W.T., Lidicker J., Clermont G., Carcillo J., Pinsky M.R. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Jawad I., Lukšić I., Rafnsson S.B. Assessing available information on the burden of sepsis: global estimates of incidence, prevalence and mortality. Journal of Global Health. 2012;2(1) doi: 10.7189/jogh.02.010404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar V. Emerging Human Coronavirus Infections (SARS, MERS, and COVID-19): Where They Are Leading Us. International Reviews of Immunology. 2020:1–49. doi: 10.1080/08830185.2020.1800688. [DOI] [PubMed] [Google Scholar]
- 11.Kumar V. Understanding the complexities of SARS-CoV2 infection and its immunology: A road to immune-based therapeutics. Int Immunopharmacol. 2020;88 doi: 10.1016/j.intimp.2020.106980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayr F.B., Yende S., Linde-Zwirble W.T., Peck-Palmer O.M., Barnato A.E., Weissfeld L.A., Angus D.C. Infection rate and acute organ dysfunction risk as explanations for racial differences in severe sepsis. Jama. 2010;303(24):2495–2503. doi: 10.1001/jama.2010.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.C. Healthcare, P. Utilization, HCUP Facts and Figures, HCUP Facts and Figures, 2006: Statistics on Hospital-Based Care in the United States, Agency for Healthcare Research and Quality (US), Rockville (MD), 2008. [PubMed]
- 14.C.M. Torio, B.J. Moore, National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013: Statistical Brief #204, Healthcare Cost and Utilization Project (HCUP) Statistical Briefs, Agency for Healthcare Research and Quality (US), Rockville (MD), 2006. [PubMed]
- 15.Reinhart K., Daniels R., Kissoon N., Machado F.R., Schachter R.D., Finfer S. Recognizing Sepsis as a Global Health Priority - A WHO Resolution. N Engl J Med. 2017 doi: 10.1056/NEJMp1707170. [DOI] [PubMed] [Google Scholar]
- 16.Chousterman B.G., Swirski F.K., Weber G.F. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–528. doi: 10.1007/s00281-017-0639-8. [DOI] [PubMed] [Google Scholar]
- 17.N.R. London, W. Zhu, F.A. Bozza, M.C. Smith, D.M. Greif, L.K. Sorensen, L. Chen, Y. Kaminoh, A.C. Chan, S.F. Passi, Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza, Science translational medicine 2(23) (2010) 23ra19-23ra19. [DOI] [PMC free article] [PubMed]
- 18.Harrison C. Calming the cytokine storm. Nature Reviews Drug Discovery. 2010;9(5):360–361. doi: 10.1038/nrd3162. [DOI] [PubMed] [Google Scholar]
- 19.Ragab D., Salah Eldin H., Taeimah M., Khattab R., Salem R. The COVID-19 Cytokine Storm; What We Know So Far. Frontiers in Immunology. 2020;11(1446) doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinha P., Matthay M.A., Calfee C.S. Is a “Cytokine Storm” Relevant to COVID-19?, JAMA. Internal Medicine. 2020 doi: 10.1001/jamainternmed.2020.3313. [DOI] [PubMed] [Google Scholar]
- 21.Coperchini F., Chiovato L., Croce L., Magri F., Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020;53:25–32. doi: 10.1016/j.cytogfr.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara J.L., Abhyankar S., Gilliland D.G. Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1. Transplant Proc. 1993;25(1 Pt 2):1216–1217. [PubMed] [Google Scholar]
- 23.Yuen K., Wong S. Human infection by avian influenza A H5N1. Hong Kong Medical Journal. 2005 [PubMed] [Google Scholar]
- 24.Tisoncik J.R., Korth M.J., Simmons C.P., Farrar J., Martin T.R., Katze M.G. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montecino-Rodriguez E., Berent-Maoz B., Dorshkind K. Causes, consequences, and reversal of immune system aging. The Journal of Clinical Investigation. 2013;123(3):958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singer M., Deutschman C.S., Seymour C. The third international consensus definitions for sepsis and septic shock (sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V. Innate lymphoid cells: new paradigm in immunology of inflammation. Immunol Lett. 2014;157(1–2):23–37. doi: 10.1016/j.imlet.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Kumar V., Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10(11):1325–1334. doi: 10.1016/j.intimp.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Silva M.T. When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol. 2010;87(1):93–106. doi: 10.1189/jlb.0809549. [DOI] [PubMed] [Google Scholar]
- 30.Galli S.J., Borregaard N., Wynn T.A. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12(11):1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolaczkowska E., Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 32.de Oliveira S., Rosowski E.E., Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16(6):378–391. doi: 10.1038/nri.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan B.P., Harris C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14(12):857–877. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasupuleti M., Schmidtchen A., Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol. 2012;32(2):143–171. doi: 10.3109/07388551.2011.594423. [DOI] [PubMed] [Google Scholar]
- 35.Wiesner J., Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. 2010;1(5):440–464. doi: 10.4161/viru.1.5.12983. [DOI] [PubMed] [Google Scholar]
- 36.Wagner E., Frank M.M. Therapeutic potential of complement modulation. Nat Rev Drug Discov. 2010;9(1):43–56. doi: 10.1038/nrd3011. [DOI] [PubMed] [Google Scholar]
- 37.Kumar V., Sharma A. Innate immunity in sepsis pathogenesis and its modulation: new immunomodulatory targets revealed. J Chemother. 2008;20(6):672–683. doi: 10.1179/joc.2008.20.6.672. [DOI] [PubMed] [Google Scholar]
- 38.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2011;11(4):275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- 39.Areschoug T., Gordon S. Pattern recognition receptors and their role in innate immunity: focus on microbial protein ligands. Contrib Microbiol. 2008;15:45–60. doi: 10.1159/000135685. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 41.Liu J., Cao X. Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol. 2016;13(6):711–721. doi: 10.1038/cmi.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. Journal of Neuroimmunology. 2019;332:16–30. doi: 10.1016/j.jneuroim.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 43.K. V, Toll-like receptors in immunity and inflammatory diseases: Past, present, and future, Int Immunopharmacol 59 (2018) 391-412. [DOI] [PMC free article] [PubMed]
- 44.Delano M.J., Ward P.A. The immune system's role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. doi: 10.1111/imr.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao C., Yu M., Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death & Disease. 2019;10(10):782. doi: 10.1038/s41419-019-2015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uronen-Hansson H., Allen J., Osman M., Squires G., Klein N., Callard R.E. Toll-like receptor 2 (TLR2) and TLR4 are present inside human dendritic cells, associated with microtubules and the Golgi apparatus but are not detectable on the cell surface: integrity of microtubules is required for interleukin-12 production in response to internalized bacteria. Immunology. 2004;111(2):173–178. doi: 10.1111/j.0019-2805.2003.01803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng M., Scott M.J., Loughran P., Gibson G., Sodhi C., Watkins S., Hackam D., Billiar T.R. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol. 2013;190(10):5152–5160. doi: 10.4049/jimmunol.1300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.R. Salomão, P.S. Martins, M.K.C. Brunialti, M.d.L. Fernandes, L.S.W. Martos, M.E. Mendes, N.E. Gomes, O. Rigato, TLR SIGNALING PATHWAY IN PATIENTS WITH SEPSIS, Shock 30(7) (2008) 73-77. [DOI] [PubMed]
- 49.Li W., Deng M., Loughran P.A., Yang M., Lin M., Yang C., Gao W., Jin S., Li S., Cai J., Lu B., Billiar T.R., Scott M.J. LPS Induces Active HMGB1 Release From Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front Immunol. 2020;11:229. doi: 10.3389/fimmu.2020.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng M., Tang Y., Li W., Wang X., Zhang R., Zhang X., Zhao X., Liu J., Tang C., Liu Z., Huang Y., Peng H., Xiao L., Tang D., Scott M.J., Wang Q., Liu J., Xiao X., Watkins S., Li J., Yang H., Wang H., Chen F., Tracey K.J., Billiar T.R., Lu B. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity. 2018;49(4):740–753.e7. doi: 10.1016/j.immuni.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andersson U., Ottestad W., Tracey K.J. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol Med. 2020;26(1):42. doi: 10.1186/s10020-020-00172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.T.A.M. Claushuis, A.I.P. Van Der Veen, J. Horn, M.J. Schultz, R.H. Houtkooper, C. Van ’T Veer, T. Van Der Poll, Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis, Platelets 30(3) (2019) 296-304. [DOI] [PubMed]
- 53.Liu B., Iwanowycz S., Fan H. Dendritic cell-intrinsic TLR signaling regulates polymicrobial sepsis. The Journal of Immunology 198(1 Supplement) 2017;131(6-131):6. [Google Scholar]
- 54.Yang Y., Liu B., Dai J., Srivastava P.K., Zammit D.J., Lefrançois L., Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26(2):215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song L., Kim D.S., Gou W., Wang J., Wang P., Wei Z., Liu B., Li Z., Gou K., Wang H. GRP94 regulates M1 macrophage polarization and insulin resistance. Am J Physiol Endocrinol Metab. 2020;318(6):E1004–E1013. doi: 10.1152/ajpendo.00542.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cosin-Roger J., Spalinger M.R., Ruiz P.A., Stanzel C., Terhalle A., Wolfram L., Melhem H., Atrott K., Lang S., Frey-Wagner I., Fried M., Scharl M., Hausmann M., Rogler G. Gp96 deficiency affects TLR4 functionality and impairs ERK and p38 phosphorylation. PLOS ONE. 2018;13(2) doi: 10.1371/journal.pone.0193003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang M., Zou L., Feng Y., Chen Y.-J., Zhou Q., Ichinose F., Chao W. Toll-like Receptor 4 Is Essential to Preserving Cardiac Function and Survival in Low-grade Polymicrobial Sepsis. Anesthesiology: The Journal of the American Society of Anesthesiologists. 2014;121(6):1270–1280. doi: 10.1097/ALN.0000000000000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bakopoulos A., Kapelouzou A., Tsilimigras D.I., Katsimpoulas M., Schizas D., Aravanis C., Balafas E., Mavroidis M., Pavlakis K., Machairas A., Liakakos T. Expression of Toll-like receptors (TLRs) in the lungs of an experimental sepsis mouse model. PLOS ONE. 2017;12(11) doi: 10.1371/journal.pone.0188050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abreu M.T. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10(2):131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 60.Cario E., Podolsky D.K. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu S., Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci. 2015;72(17):3343–3353. doi: 10.1007/s00018-015-1931-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamonic G., Pasternak J.A., Wilson H.L. Recognizing conserved non-canonical localization patterns of toll-like receptors in tissues and across species. Cell Tissue Res. 2018;372(1):1–11. doi: 10.1007/s00441-017-2767-9. [DOI] [PubMed] [Google Scholar]
- 63.Hug H., Mohajeri M.H., La Fata G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients. 2018;10(2):203. doi: 10.3390/nu10020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Redondo A.C.C., Ceccon M.E.J.R., Silveira-Lessa A.L., Quinello C., Palmeira P., Carvalho W.B., Carneiro-Sampaio M. TLR-2 and TLR-4 expression in monocytes of newborns with late-onset sepsis. Jornal de Pediatria. 2014;90(5):472–478. doi: 10.1016/j.jped.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 65.A.A. FQ Younis, NH Zaki, TLR2 AND TLR4 AS A BIOMARKER IN ADULT AND CHILDREN IRAQI BACTERIAL SEPSIS SYNDROME PATIENTS, Pak. J. Biotechnol. 15(3) (2018) 837-842.
- 66.R.H. Elsherif, H.A.F. Algebaly, D.K. Ismail, B. Meligy, M. Magdy, Aziz, D.M. Ghaith, E. Salah, Toll-like receptors 2 and 9 gene polymorphisms in severe sepsis and septic shock : a single center study in the pediatric intensive care unit, 2019.
- 67.Atalan N., Karagedik H., Acar L., Isbir S., Yilmaz S.G., Ergen A., Isbir T. Analysis of Toll-like Receptor 9 Gene Polymorphisms in Sepsis. Vivo. 2016;30(5):639–643. [PubMed] [Google Scholar]
- 68.Atalan N., Acar L., Yapici N., Kudsioglu T., Ergen A., Yilmaz S.G., Isbir T. The Relationship Between Sepsis-induced Immunosuppression and Serum Toll-like Receptor 9 Level. Vivo. 2018;32(6):1653–1658. doi: 10.21873/invivo.11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Frontiers in Immunology. 2020;11(1722) doi: 10.3389/fimmu.2020.01722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krivan S., Kapelouzou A., Vagios S., Tsilimigras D.I., Katsimpoulas M., Moris D., Aravanis C.V., Demesticha T.D., Schizas D., Mavroidis M., Pavlakis K., Machairas A., Misiakos E., Liakakos T. Increased expression of Toll-like receptors 2, 3, 4 and 7 mRNA in the kidney and intestine of a septic mouse model. Scientific Reports. 2019;9(1):4010. doi: 10.1038/s41598-019-40537-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Q., Wu C., Liu Z., Zhang H., Du Y., Liu Y., Song K., Shi Q., Li R. Increased TLR4 Expression Aggravates Sepsis by Promoting IFN-<i>γ</i> Expression in CD38<sup>−/−</sup> Mice. Journal of Immunology Research. 2019;2019:3737890. doi: 10.1155/2019/3737890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Castoldi A., Braga T.T., Correa-Costa M., Aguiar C.F., Bassi Ê.J., Correa-Silva R., Elias R.M., Salvador F., Moraes-Vieira P.M., Cenedeze M.A., Reis M.A., Hiyane M.I., Pacheco-Silva Á., Gonçalves G.M., Câmara N.O.S. TLR2, TLR4 and the MYD88 Signaling Pathway Are Crucial for Neutrophil Migration in Acute Kidney Injury Induced by Sepsis. PLOS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sônego F., Castanheira F.V.S., Czaikoski P.G., Kanashiro A., Souto F.O., França R.O., Nascimento D.C., Freitas A., Spiller F., Cunha L.D., Zamboni D.S., Alves-Filho J.C., Cunha F.Q. MyD88-, but not Nod1- and/or Nod2-deficient mice, show increased susceptibility to polymicrobial sepsis due to impaired local inflammatory response. PloS one. 2014;9(8) doi: 10.1371/journal.pone.0103734. e103734-e103734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plant L., Wan H., Jonsson A.-B. MyD88-dependent signaling affects the development of meningococcal sepsis by nonlipooligosaccharide ligands. Infect Immun. 2006;74(6):3538–3546. doi: 10.1128/IAI.00128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feuillet V., Medjane S., Mondor I., Demaria O., Pagni P.P., Galán J.E., Flavell R.A., Alexopoulou L. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci U S A. 2006;103(33):12487–12492. doi: 10.1073/pnas.0605200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akira S., Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 77.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 78.Zhang D., Zhang G., Hayden M.S., Greenblatt M.B., Bussey C., Flavell R.A., Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303(5663):1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 79.Hatai H., Lepelley A., Zeng W., Hayden M.S., Ghosh S. Toll-Like Receptor 11 (TLR11) Interacts with Flagellin and Profilin through Disparate Mechanisms. PLOS ONE. 2016;11(2) doi: 10.1371/journal.pone.0148987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hussain S., Johnson C.G., Sciurba J., Meng X., Stober V.P., Liu C., Cyphert-Daly J.M., Bulek K., Qian W., Solis A., Sakamachi Y., Trempus C.S., Aloor J.J., Gowdy K.M., Foster W.M., Hollingsworth J.W., Tighe R.M., Li X., Fessler M.B., Garantziotis S. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. eLife. 2020;9 doi: 10.7554/eLife.50458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hawn T.R., Verbon A., Lettinga K.D., Zhao L.P., Li S.S., Laws R.J., Skerrett S.J., Beutler B., Schroeder L., Nachman A., Ozinsky A., Smith K.D., Aderem A. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires' disease. J Exp Med. 2003;198(10):1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar V. Innate Lymphoid Cells: Immunoregulatory Cells of Mucosal Inflammation. European Journal of Inflammation. 2014;12(1):11–20. [Google Scholar]
- 83.El-Zayat S.R., Sibaii H., Mannaa F.A. Toll-like receptors activation, signaling, and targeting: an overview. Bulletin of the National Research Centre. 2019;43(1):187. [Google Scholar]
- 84.Liew F.Y., Xu D., Brint E.K., O'Neill L.A. Negative regulation of toll-like receptor-mediated immune responses. Nature reviews. Immunology. 2005;5(6):446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 85.Palsson-McDermott E.M., Doyle S.L., McGettrick A.F., Hardy M., Husebye H., Banahan K., Gong M., Golenbock D., Espevik T., O'Neill L.A. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol. 2009;10(6):579–586. doi: 10.1038/ni.1727. [DOI] [PubMed] [Google Scholar]
- 86.Kondo T., Kawai T., Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33(9):449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 87.Zhou M., Cheng S., Yu J., Lu Q. Interleukin-8 for Diagnosis of Neonatal Sepsis: A Meta-Analysis. PLOS ONE. 2015;10(5) doi: 10.1371/journal.pone.0127170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kraft R., Herndon D.N., Finnerty C.C., Cox R.A., Song J., Jeschke M.G. Predictive Value of IL-8 for Sepsis and Severe Infections After Burn Injury: A Clinical Study. Shock. 2015;43(3):222–227. doi: 10.1097/SHK.0000000000000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carty M., Goodbody R., Schroder M., Stack J., Moynagh P.N., Bowie A.G. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7(10):1074–1081. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 90.Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 91.Peng J., Yuan Q., Lin B., Panneerselvam P., Wang X., Luan X.L., Lim S.K., Leung B.P., Ho B., Ding J.L. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur J Immunol. 2010;40(6):1738–1747. doi: 10.1002/eji.200940034. [DOI] [PubMed] [Google Scholar]
- 92.Gong Y., Zou L., Cen D., Chao W., Chen D. Reduced Expression of SARM in Mouse Spleen during Polymicrobial Sepsis. Inflammation. 2016;39(6):1930–1938. doi: 10.1007/s10753-016-0428-x. [DOI] [PubMed] [Google Scholar]
- 93.Carty M., Kearney J., Shanahan K.A., Hams E., Sugisawa R., Connolly D., Doran C.G., Muñoz-Wolf N., Gürtler C., Fitzgerald K.A., Lavelle E.C., Fallon P.G., Bowie A.G. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity. 2019;50(6):1412–1424.e6. doi: 10.1016/j.immuni.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 94.Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S., Honda K., Ohba Y., Mak T.W., Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434(7030):243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 95.Negishi H., Ohba Y., Yanai H., Takaoka A., Honma K., Yui K., Matsuyama T., Taniguchi T., Honda K. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102(44):15989–15994. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li M., Yu H., Wang Y., Qin L., Sun W. Role of IRF4 in the Protection of Metformin-Mediated Sepsis Myocarditis. Dose Response. 2019;17(1) doi: 10.1177/1559325819827436. 1559325819827436-1559325819827436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang M., Sun W., Du J., Gou Y., Liu L., Wang R., Xu X. Protective Effect of Metformin on Sepsis Myocarditis in Zebrafish. Dose-Response. 2020;18(3) doi: 10.1177/1559325820938543. 1559325820938543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun H., Gong S., Carmody R.J., Hilliard A., Li L., Sun J., Kong L., Xu L., Hilliard B., Hu S., Shen H., Yang X., Chen Y.H. TIPE2, a novel negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133(3):415–426. doi: 10.1016/j.cell.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oho M., Nakano R., Nakayama R., Sakurai W., Miyamoto A., Masuhiro Y., Hanazawa S. TIPE2 (Tumor Necrosis Factor alpha-induced Protein 8-like 2) Is a Novel Negative Regulator of TAK1 Signal. J Biol Chem. 2016;291(43):22650–22660. doi: 10.1074/jbc.M116.733451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Q., Ma L., Liu T., Ge C., Zhou Q., Wei C., Shi W. TIPE2 Suppresses Pseudomonas aeruginosa Keratitis by Inhibiting NF-κB Signaling and the Infiltration of Inflammatory Cells. J Infect Dis. 2019;220(6):1008–1018. doi: 10.1093/infdis/jiz246. [DOI] [PubMed] [Google Scholar]
- 101.Zhang H., Zhu T., Liu W., Qu X., Chen Y., Ren P., Wang Z., Wei X., Zhang Y., Yi F. TIPE2 acts as a negative regulator linking NOD2 and inflammatory responses in myocardial ischemia/reperfusion injury. J Mol Med (Berl) 2015;93(9):1033–1043. doi: 10.1007/s00109-015-1288-9. [DOI] [PubMed] [Google Scholar]
- 102.Luan Y.Y., Yao Y.M., Zhang L., Dong N., Zhang Q.H., Yu Y., Sheng Z.Y. Expression of tumor necrosis factor-α induced protein 8 like-2 contributes to the immunosuppressive property of CD4(+)CD25(+) regulatory T cells in mice. Mol Immunol. 2011;49(1–2):219–226. doi: 10.1016/j.molimm.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 103.Liu M.W., Liu R., Wu H.Y., Zhang W., Xia J., Dong M.N., Yu W., Wang Q., Xie F.M., Wang R., Huang Y.Q., Qian C.Y. Protective effect of Xuebijing injection on D-galactosamine- and lipopolysaccharide-induced acute liver injury in rats through the regulation of p38 MAPK, MMP-9 and HO-1 expression by increasing TIPE2 expression. Int J Mol Med. 2016;38(5):1419–1432. doi: 10.3892/ijmm.2016.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu M.W., Wang Y.H., Qian C.Y., Li H. Xuebijing exerts protective effects on lung permeability leakage and lung injury by upregulating Toll-interacting protein expression in rats with sepsis. Int J Mol Med. 2014;34(6):1492–1504. doi: 10.3892/ijmm.2014.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou J., Chen P., Li Z., Zuo Q. Gene delivery of TIPE2 attenuates collagen-induced arthritis by modulating inflammation. Int Immunopharmacol. 2020;79 doi: 10.1016/j.intimp.2019.106044. [DOI] [PubMed] [Google Scholar]
- 106.Hamerman J.A., Tchao N.K., Lowell C.A., Lanier L.L. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6(6):579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomasello E., Vivier E. KARAP/DAP12/TYROBP: three names and a multiplicity of biological functions. Eur J Immunol. 2005;35(6):1670–1677. doi: 10.1002/eji.200425932. [DOI] [PubMed] [Google Scholar]
- 108.Peng Q., Long C.L., Malhotra S., Humphrey M.B. A physical interaction between the adaptor proteins DOK3 and DAP12 is required to inhibit lipopolysaccharide signaling in macrophages. Sci Signal. 2013;6(289):ra72. doi: 10.1126/scisignal.2003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Turnbull I.R., Colonna M. Activating and inhibitory functions of DAP12. Nat Rev Immunol. 2007;7(2):155–161. doi: 10.1038/nri2014. [DOI] [PubMed] [Google Scholar]
- 110.Turnbull I.R., McDunn J.E., Takai T., Townsend R.R., Cobb J.P., Colonna M. DAP12 (KARAP) amplifies inflammation and increases mortality from endotoxemia and septic peritonitis. J Exp Med. 2005;202(3):363–369. doi: 10.1084/jem.20050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bouchon A., Facchetti F., Weigand M.A., Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410(6832):1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 112.Shinohara H., Inoue A., Toyama-Sorimachi N., Nagai Y., Yasuda T., Suzuki H., Horai R., Iwakura Y., Yamamoto T., Karasuyama H., Miyake K., Yamanashi Y. Dok-1 and Dok-2 are negative regulators of lipopolysaccharide-induced signaling. J Exp Med. 2005;201(3):333–339. doi: 10.1084/jem.20041817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mashima R., Hishida Y., Tezuka T., Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol Rev. 2009;232(1):273–285. doi: 10.1111/j.1600-065X.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- 114.Suzu S., Tanaka-Douzono M., Nomaguchi K., Yamada M., Hayasawa H., Kimura F., Motoyoshi K. p56(dok-2) as a cytokine-inducible inhibitor of cell proliferation and signal transduction. Embo j. 2000;19(19):5114–5122. doi: 10.1093/emboj/19.19.5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11) doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Linger R.M., Keating A.K., Earp H.S., Graham D.K. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rothlin C.V., Carrera-Silva E.A., Bosurgi L., Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355–391. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rothlin C.V., Ghosh S., Zuniga E.I., Oldstone M.B., Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 119.Yoshimura A., Naka T., Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 120.van der Meer J.H., van der Poll T. C. van 't Veer, TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123(16):2460–2469. doi: 10.1182/blood-2013-09-528752. [DOI] [PubMed] [Google Scholar]
- 121.Sharif M.N., Sosic D., Rothlin C.V., Kelly E., Lemke G., Olson E.N., Ivashkiv L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203(8):1891–1901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O'Neill L.A. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29(1):12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 123.Guignant C., Venet F., Planel S., Demaret J., Gouel-Cheron A., Nougier C., Friggeri A., Allaouchiche B., Lepape A., Monneret G. Increased MerTK expression in circulating innate immune cells of patients with septic shock. Intensive Care Med. 2013;39(9):1556–1564. doi: 10.1007/s00134-013-3006-9. [DOI] [PubMed] [Google Scholar]
- 124.Girardis M., Cossarizza A. A Janus role for MerTK in the outcome of septic shock. Intensive Care Medicine. 2013;39(12):2217–2219. doi: 10.1007/s00134-013-3106-6. [DOI] [PubMed] [Google Scholar]
- 125.Tibrewal N., Wu Y., D'Mello V., Akakura R., George T.C., Varnum B., Birge R.B. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283(6):3618–3627. doi: 10.1074/jbc.M706906200. [DOI] [PubMed] [Google Scholar]
- 126.Scott R.S., McMahon E.J., Pop S.M., Reap E.A., Caricchio R., Cohen P.L., Earp H.S., Matsushima G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 127.Kobayashi K., Hernandez L.D., Galan J.E., Janeway C.A., Jr., Medzhitov R., Flavell R.A. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110(2):191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 128.Nakayama K., Okugawa S., Yanagimoto S., Kitazawa T., Tsukada K., Kawada M., Kimura S., Hirai K., Takagaki Y., Ota Y. Involvement of IRAK-M in peptidoglycan-induced tolerance in macrophages. J Biol Chem. 2004;279(8):6629–6634. doi: 10.1074/jbc.M308620200. [DOI] [PubMed] [Google Scholar]
- 129.Deng J.C., Cheng G., Newstead M.W., Zeng X., Kobayashi K., Flavell R.A., Standiford T.J. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. The Journal of Clinical Investigation. 2006;116(9):2532–2542. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Steiger S., Kumar S.V., Honarpisheh M., Lorenz G., Günthner R., Romoli S., Gröbmayr R., Susanti H.E., Potempa J., Koziel J., Lech M. Immunomodulatory Molecule IRAK-M Balances Macrophage Polarization and Determines Macrophage Responses during Renal Fibrosis. J Immunol. 2017;199(4):1440–1452. doi: 10.4049/jimmunol.1601982. [DOI] [PubMed] [Google Scholar]
- 131.Xia Q., Zhou Y., Wang X., Fu S. Interleukin-1 receptor-associated kinase 3 downregulation in peripheral blood mononuclear cells attenuates immunosuppression in sepsis. Exp Ther Med. 2018;15(2):1586–1593. doi: 10.3892/etm.2017.5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dong G.-H., Gong J.-P., Li J.-Z., Luo Y.-H., Li Z.-D., Li P.-Z., He K. Association Between Gene Polymorphisms of IRAK-M and the Susceptibility of Sepsis. Inflammation. 2013;36(5):1087–1093. doi: 10.1007/s10753-013-9641-z. [DOI] [PubMed] [Google Scholar]
- 133.Burns K., Clatworthy J., Martin L., Martinon F., Plumpton C., Maschera B., Lewis A., Ray K., Tschopp J., Volpe F. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2(6):346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 134.Zhang G., Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277(9):7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 135.Liew F.Y., Xu D., Brint E.K., O’Neill L.A. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5 doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 136.Li T., Hu J., Li L. Characterization of Tollip protein upon Lipopolysaccharide challenge. Mol Immunol. 2004;41(1):85–92. doi: 10.1016/j.molimm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 137.Song Z., Yin J., Yao C., Sun Z., Shao M., Zhang Y., Tao Z., Huang P., Tong C. Variants in the Toll-interacting protein gene are associated with susceptibility to sepsis in the Chinese Han population. Critical Care. 2011;15(1) doi: 10.1186/cc9413. R12-R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Diao N., Zhang Y., Chen K., Yuan R., Lee C., Geng S., Kowalski E., Guo W., Xiong H., Li M., Li L. Deficiency in Toll-interacting protein (Tollip) skews inflamed yet incompetent innate leukocytes in vivo during DSS-induced septic colitis. Scientific Reports. 2016;6:34672. doi: 10.1038/srep34672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maitra U., Gan L., Chang S., Li L. Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating CCAAT/enhancer-binding protein delta. J Immunol. 2011;186(7):4467–4473. doi: 10.4049/jimmunol.1003300. [DOI] [PubMed] [Google Scholar]
- 140.Kowalski E.J.A., Li L. Toll-Interacting Protein in Resolving and Non-Resolving Inflammation. Front Immunol. 2017;8:511. doi: 10.3389/fimmu.2017.00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen K., Yuan R., Zhang Y., Geng S., Li L. Tollip Deficiency Alters Atherosclerosis and Steatosis by Disrupting Lipophagy. J Am Heart Assoc. 2017;6(4) doi: 10.1161/JAHA.116.004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abram C.L., Lowell C.A. Shp1 function in myeloid cells. J Leukoc Biol. 2017;102(3):657–675. doi: 10.1189/jlb.2MR0317-105R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.An H., Hou J., Zhou J., Zhao W., Xu H., Zheng Y., Yu Y., Liu S., Cao X. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9(5):542–550. doi: 10.1038/ni.1604. [DOI] [PubMed] [Google Scholar]
- 144.Abu-Dayyeh I., Shio M.T., Sato S., Akira S., Cousineau B., Olivier M. Leishmania-Induced IRAK-1 Inactivation Is Mediated by SHP-1 Interacting with an Evolutionarily Conserved KTIM Motif. PLOS Neglected Tropical Diseases. 2008;2(12) doi: 10.1371/journal.pntd.0000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rego D., Kumar A., Nilchi L., Wright K., Huang S., Kozlowski M. IL-6 production is positively regulated by two distinct Src homology domain 2-containing tyrosine phosphatase-1 (SHP-1)-dependent CCAAT/enhancer-binding protein β and NF-κB pathways and an SHP-1-independent NF-κB pathway in lipopolysaccharide-stimulated bone marrow-derived macrophages. J Immunol. 2011;186(9):5443–5456. doi: 10.4049/jimmunol.1003551. [DOI] [PubMed] [Google Scholar]
- 146.Zhou D., Collins C.A., Wu P., Brown E.J. Protein tyrosine phosphatase SHP-1 positively regulates TLR-induced IL-12p40 production in macrophages through inhibition of phosphatidylinositol 3-kinase. J Leukoc Biol. 2010;87(5):845–855. doi: 10.1189/jlb.0409289. [DOI] [PubMed] [Google Scholar]
- 147.Lin L., Jian J., Song C.Y., Chen F., Ding K., Xie W.F., Hu P.F. SHP-1 ameliorates nonalcoholic steatohepatitis by inhibiting proinflammatory cytokine production. FEBS Lett. 2020 doi: 10.1002/1873-3468.13879. [DOI] [PubMed] [Google Scholar]
- 148.Hao D., Wang Y., Li L., Qian G., Liu J., Li M., Zhang Y., Zhou R., Yan D. SHP-1 suppresses the antiviral innate immune response by targeting TRAF3. Faseb j. 2020 doi: 10.1096/fj.202000600RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhou Y., He C., Yan D., Liu F., Liu H., Chen J., Cao T., Zuo M., Wang P., Ge Y., Lu H., Tong Q., Qin C., Deng Y., Ge B. The kinase CK1ɛ controls the antiviral immune response by phosphorylating the signaling adaptor TRAF3. Nat Immunol. 2016;17(4):397–405. doi: 10.1038/ni.3395. [DOI] [PubMed] [Google Scholar]
- 150.An H., Zhao W., Hou J., Zhang Y., Xie Y., Zheng Y., Xu H., Qian C., Zhou J., Yu Y., Liu S., Feng G., Cao X. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity. 2006;25(6):919–928. doi: 10.1016/j.immuni.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 151.Park J.H., Ko R., Lee S.Y. Reciprocal regulation of TLR2-mediated IFN-β production by SHP2 and Gsk3β. Scientific Reports. 2017;7(1):6807. doi: 10.1038/s41598-017-07316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rusnak F., Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80(4):1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 153.Vandewalle A., Tourneur E., Bens M., Chassin C., Werts C. Calcineurin/NFAT signaling and innate host defence: a role for NOD1-mediated phagocytic functions. Cell Commun Signal. 2014;12:8. doi: 10.1186/1478-811X-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kang Y.J., Kusler B., Otsuka M., Hughes M., Suzuki N., Suzuki S., Yeh W.C., Akira S., Han J., Jones P.P. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. 2007;179(7):4598–4607. doi: 10.4049/jimmunol.179.7.4598. [DOI] [PubMed] [Google Scholar]
- 155.Ranjan R., Deng J., Chung S., Lee Y.G., Park G.Y., Xiao L., Joo M., Christman J.W., Karpurapu M. The transcription factor nuclear factor of activated T cells c3 modulates the function of macrophages in sepsis. J Innate Immun. 2014;6(6):754–764. doi: 10.1159/000362647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jennings C., Kusler B., Jones P.P. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immun. 2009;15(2):109–120. doi: 10.1177/1753425908100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bendickova K., Tidu F., Fric J. Calcineurin-NFAT signalling in myeloid leucocytes: new prospects and pitfalls in immunosuppressive therapy. EMBO Mol Med. 2017;9(8):990–999. doi: 10.15252/emmm.201707698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M., Moghal N., Lubkin M., Kim Y.B., Sharpe A.H., Stricker-Krongrad A., Shulman G.I., Neel B.G., Kahn B.B. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20(15):5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu H., An H., Hou J., Han C., Wang P., Yu Y., Cao X. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol Immunol. 2008;45(13):3545–3552. doi: 10.1016/j.molimm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 160.Traves P.G., Pardo V., Pimentel-Santillana M., Gonzalez-Rodriguez A., Mojena M., Rico D., Montenegro Y., Cales C., Martin-Sanz P., Valverde A.M., Bosca L. Pivotal role of protein tyrosine phosphatase 1B (PTP1B) in the macrophage response to pro-inflammatory and anti-inflammatory challenge. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yue L., Xie Z., Li H., Pang Z., Junkins R.D., Tremblay M.L., Chen X., Lin T.J. Protein Tyrosine Phosphatase-1B Negatively Impacts Host Defense against Pseudomonas aeruginosa Infection. Am J Pathol. 2016;186(5):1234–1244. doi: 10.1016/j.ajpath.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 162.Ma A., Malynn B.A. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012;12(11):774–785. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Catrysse L., Vereecke L., Beyaert R., van Loo G. A20 in inflammation and autoimmunity. Trends Immunol. 2014;35(1):22–31. doi: 10.1016/j.it.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 164.Heyninck K., De Valck D., Vanden Berghe W., Van Criekinge W., Contreras R., Fiers W., Haegeman G., Beyaert R. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. The Journal of cell biology. 1999;145(7):1471–1482. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee E.G., Boone D.L., Chai S., Libby S.L., Chien M., Lodolce J.P., Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Heyninck K., Beyaert R. The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-kappaB activation at the level of TRAF6. FEBS Lett. 1999;442(2–3):147–150. doi: 10.1016/s0014-5793(98)01645-7. [DOI] [PubMed] [Google Scholar]
- 167.Boone D.L., Turer E.E., Lee E.G., Ahmad R.C., Wheeler M.T., Tsui C., Hurley P., Chien M., Chai S., Hitotsumatsu O., McNally E., Pickart C., Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5(10):1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 168.Shembade N., Ma A., Harhaj E.W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327(5969):1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shembade N., Harhaj E. A20 inhibition of NFkappaB and inflammation: targeting E2:E3 ubiquitin enzyme complexes. Cell Cycle. 2010;9(13):2481–2482. doi: 10.4161/cc.9.13.12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hutti J.E., Turk B.E., Asara J.M., Ma A., Cantley L.C., Abbott D.W. IkappaB kinase beta phosphorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-kappaB pathway. Mol Cell Biol. 2007;27(21):7451–7461. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.A.J. Hoogendijk, M.I. Garcia-Laorden, L.A. van Vught, M.A. Wiewel, H. Belkasim-Bohoudi, J. Duitman, J. Horn, M.J. Schultz, B.P. Scicluna, C. van ‘t Veer, A.F. de Vos, T. van der Poll, Sepsis Patients Display a Reduced Capacity to Activate Nuclear Factor-κB in Multiple Cell Types*, Critical care medicine 45(5) (2017) e524-e531. [DOI] [PubMed]
- 172.Tiruppathi C., Soni D., Wang D.-M., Xue J., Singh V., Thippegowda P.B., Cheppudira B.P., Mishra R.K., Debroy A., Qian Z., Bachmaier K., Zhao Y.-Y., Christman J.W., Vogel S.M., Ma A., Malik A.B. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nature immunology. 2014;15(3):239–247. doi: 10.1038/ni.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Carrión A.M., Link W.A., Ledo F., Mellström B., Naranjo J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398(6722):80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- 174.Yoshida H., Jono H., Kai H., Li J.D. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem. 2005;280(49):41111–41121. doi: 10.1074/jbc.M509526200. [DOI] [PubMed] [Google Scholar]
- 175.Xu L.G., Li L.Y., Shu H.B. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. 2004;279(17):17278–17282. doi: 10.1074/jbc.C400063200. [DOI] [PubMed] [Google Scholar]
- 176.Kovalenko A., Chable-Bessia C., Cantarella G., Israel A., Wallach D., Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424(6950):801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 177.Trompouki E., Hatzivassiliou E., Tsichritzis T., Farmer H., Ashworth A., Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424(6950):793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 178.Sun S.C. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17(1):25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lim J.H., Ha U.H., Woo C.H., Xu H., Li J.D. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. 2008;10(11):2247–2256. doi: 10.1111/j.1462-5822.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- 180.Jono H., Lim J.H., Chen L.F., Xu H., Trompouki E., Pan Z.K., Mosialos G., Li J.D. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. 2004;279(35):36171–36174. doi: 10.1074/jbc.M406638200. [DOI] [PubMed] [Google Scholar]
- 181.Lee B.-C., Miyata M., Lim J.H., Li J.-D. Deubiquitinase CYLD acts as a negative regulator for bacterium NTHi-induced inflammation by suppressing K63-linked ubiquitination of MyD88. Proceedings of the National Academy of Sciences. 2016;113(2):E165–E171. doi: 10.1073/pnas.1518615113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shuto T., Xu H., Wang B., Han J., Kai H., Gu X.X., Murphy T.F., Lim D.J., Li J.D. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci U S A. 2001;98(15):8774–8779. doi: 10.1073/pnas.151236098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schworer S.A., Smirnova I., II, Kurbatova U., Bagina M., Churova T., Fowler A.L., Roy A., Degterev A. Poltorak. Toll-like receptor-mediated down-regulation of the deubiquitinase cylindromatosis (CYLD) protects macrophages from necroptosis in wild-derived mice. J Biol Chem. 2014;289(20):14422–14433. doi: 10.1074/jbc.M114.547547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Patel S., Webster J.D., Varfolomeev E., Kwon Y.C., Cheng J.H., Zhang J., Dugger D.L., Wickliffe K.E., Maltzman A., Sujatha-Bhaskar S., Bir Kohli P., Ramaswamy S., Deshmukh G., Liederer B.M., Fong R., Hamilton G., Lupardus P., Caplazi P., Lee W.P., van Lookeren Campagne M., Johnson A., McKenzie B.S., Junttila M.R., Newton K., Vucic D. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death & Differentiation. 2020;27(1):161–175. doi: 10.1038/s41418-019-0347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou F., Zhang X., van Dam H., Ten Dijke P., Huang H., Zhang L. Ubiquitin-specific protease 4 mitigates Toll-like/interleukin-1 receptor signaling and regulates innate immune activation. J Biol Chem. 2012;287(14):11002–11010. doi: 10.1074/jbc.M111.328187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.V. Francois-Newton, G. Magno de Freitas Almeida, B. Payelle-Brogard, D. Monneron, L. Pichard-Garcia, J. Piehler, S. Pellegrini, G. Uze, USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response, PLoS One 6(7) (2011) e22200. [DOI] [PMC free article] [PubMed]
- 187.Liu L.Q., Ilaria R., Jr., Kingsley P.D., Iwama A., van Etten R.A., Palis J., Zhang D.E. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol. 1999;19(4):3029–3038. doi: 10.1128/mcb.19.4.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Malakhov M.P., Malakhova O.A., Kim K.I., Ritchie K.J., Zhang D.E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277(12):9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 189.Honke N., Shaabani N., Cadeddu G., Sorg U.R., Zhang D.E., Trilling M., Klingel K., Sauter M., Kandolf R., Gailus N., van Rooijen N., Burkart C., Baldus S.E., Grusdat M., Lohning M., Hengel H., Pfeffer K., Tanaka M., Haussinger D., Recher M., Lang P.A., Lang K.S. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. 2011;13(1):51–57. doi: 10.1038/ni.2169. [DOI] [PubMed] [Google Scholar]
- 190.Honke N., Shaabani N., Zhang D.E., Iliakis G., Xu H.C., Haussinger D., Recher M., Lohning M., Lang P.A., Lang K.S. Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog. 2013;9(10) doi: 10.1371/journal.ppat.1003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Honke N., Shaabani N., Zhang D.-E., Hardt C., Lang K.S. Multiple functions of USP18. Cell Death Dis. 2016;7 doi: 10.1038/cddis.2016.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Burkart C., Fan J.B., Zhang D.E. Two independent mechanisms promote expression of an N-terminal truncated USP18 isoform with higher DeISGylation activity in the nucleus. J Biol Chem. 2012;287(7):4883–4893. doi: 10.1074/jbc.M111.255570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang Z., Xian H., Hu J., Tian S., Qin Y., Wang R.F., Cui J. USP18 negatively regulates NF-kappaB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci Rep. 2015;5:12738. doi: 10.1038/srep12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Goldmann T., Zeller N., Raasch J., Kierdorf K., Frenzel K., Ketscher L., Basters A., Staszewski O., Brendecke S.M., Spiess A., Tay T.L., Kreutz C., Timmer J., Mancini G.M., Blank T., Fritz G., Biber K., Lang R., Malo D., Merkler D., Heikenwalder M., Knobeloch K.P., Prinz M. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. The EMBO journal. 2015;34(12):1612–1629. doi: 10.15252/embj.201490791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bibeau-Poirier A., Servant M.J. Roles of ubiquitination in pattern-recognition receptors and type I interferon receptor signaling. Cytokine. 2008;43(3):359–367. doi: 10.1016/j.cyto.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 196.Hacker H., Tseng P.H., Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11(7):457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 197.Kayagaki N., Phung Q., Chan S., Chaudhari R., Quan C., O'Rourke K.M., Eby M., Pietras E., Cheng G., Bazan J.F., Zhang Z., Arnott D., Dixit V.M. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318(5856):1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 198.González-Navajas J.M., Law J., Nguyen K.P., Bhargava M., Corr M.P., Varki N., Eckmann L., Hoffman H.M., Lee J., Raz E. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9–driven antiinflammatory cytokine production. The Journal of Experimental Medicine. 2010;207(13):2799–2807. doi: 10.1084/jem.20101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li S., Zheng H., Mao A.P., Zhong B., Li Y., Liu Y., Gao Y., Ran Y., Tien P., Shu H.B. Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. The Journal of biological chemistry. 2010;285(7):4291–4297. doi: 10.1074/jbc.M109.074971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hsu H.C., Zhou T., Mountz J.D. Nur77 family of nuclear hormone receptors. Curr Drug Targets Inflamm Allergy. 2004;3(4):413–423. doi: 10.2174/1568010042634523. [DOI] [PubMed] [Google Scholar]
- 201.Safe S., Jin U.H., Morpurgo B., Abudayyeh A., Singh M., Tjalkens R.B. Nuclear receptor 4A (NR4A) family - orphans no more. J Steroid Biochem Mol Biol. 2016;157:48–60. doi: 10.1016/j.jsbmb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rodriguez-Calvo R., Tajes M., Vazquez-Carrera M. The NR4A subfamily of nuclear receptors: potential new therapeutic targets for the treatment of inflammatory diseases. Expert Opin Ther Targets. 2017;21(3):291–304. doi: 10.1080/14728222.2017.1279146. [DOI] [PubMed] [Google Scholar]
- 203.McMorrow J.P., Murphy E.P. Inflammation: a role for NR4A orphan nuclear receptors? Biochemical Society transactions. 2011;39(2):688–693. doi: 10.1042/BST0390688. [DOI] [PubMed] [Google Scholar]
- 204.Li X.M., Zhang S., He X.S., Guo P.D., Lu X.X., Wang J.R., Li J.M., Wu H. Nur77-mediated TRAF6 signalling protects against LPS-induced sepsis in mice. J Inflamm (Lond) 2016;13:4. doi: 10.1186/s12950-016-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nowyhed H.N., Huynh T.R., Thomas G.D., Blatchley A., Hedrick C.C. Cutting Edge: The Orphan Nuclear Receptor Nr4a1 Regulates CD8+ T Cell Expansion and Effector Function through Direct Repression of Irf4. J Immunol. 2015;195(8):3515–3519. doi: 10.4049/jimmunol.1403027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shaked I., Hanna R.N., Shaked H., Chodaczek G., Nowyhed H.N., Tweet G., Tacke R., Basat A.B., Mikulski Z., Togher S., Miller J., Blatchley A., Salek-Ardakani S., Darvas M., Kaikkonen M.U., Thomas G.D., Lai-Wing-Sun S., Rezk A., Bar-Or A., Glass C.K., Bandukwala H., Hedrick C.C. Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat Immunol. 2015;16(12):1228–1234. doi: 10.1038/ni.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Glass C.K., Saijo K., Winner B., Marchetto M.C., Gage F.H. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Saijo K., Winner B., Carson C.T., Collier J.G., Boyer L., Rosenfeld M.G., Gage F.H., Glass C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137(1):47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Oh M., Kim S.Y., Gil J.E., Byun J.S., Cha D.W., Ku B., Lee W., Kim W.K., Oh K.J., Lee E.W., Bae K.H., Lee S.C., Han B.S. Nurr1 performs its anti-inflammatory function by regulating RasGRP1 expression in neuro-inflammation. Sci Rep. 2020;10(1):10755. doi: 10.1038/s41598-020-67549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rajan S., Jang Y., Kim C.H., Kim W., Toh H.T., Jeon J., Song B., Serra A., Lescar J., Yoo J.Y., Beldar S., Ye H., Kang C., Liu X.W., Feitosa M., Kim Y., Hwang D., Goh G., Lim K.L., Park H.M., Lee C.H., Oh S.F., Petsko G.A., Yoon H.S., Kim K.S. PGE1 and PGA1 bind to Nurr1 and activate its transcriptional function. Nat Chem Biol. 2020;16(8):876–886. doi: 10.1038/s41589-020-0553-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Farhana L., Dawson M.I., Leid M., Wang L., Moore D.D., Liu G., Xia Z., Fontana J.A. Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor. Cancer Res. 2007;67(1):318–325. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yuk J.M., Shin D.M., Lee H.M., Kim J.J., Kim S.W., Jin H.S., Yang C.S., Park K.A., Chanda D., Kim D.K., Huang S.M., Lee S.K., Lee C.H., Kim J.M., Song C.H., Lee S.Y., Hur G.M., Moore D.D., Choi H.S., Jo E.K. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol. 2011;12(8):742–751. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- 213.Yuk J.-M., Jin H.S., Jo E.-K. Small Heterodimer Partner and Innate Immune Regulation. Endocrinology and Metabolism. 2016;31(1):17–24. doi: 10.3803/EnM.2016.31.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.He Y., Hara H., Núñez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends in Biochemical Sciences. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Reyskens K.M.S.E., Arthur J.S.C. Emerging Roles of the Mitogen and Stress Activated Kinases MSK1 and MSK2. Frontiers in Cell and Developmental Biology. 2016;4(56) doi: 10.3389/fcell.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Caenepeel S., Charydczak G., Sudarsanam S., Hunter T., Manning G. The mouse kinome: discovery and comparative genomics of all mouse protein kinases. Proc Natl Acad Sci U S A. 2004;101(32):11707–11712. doi: 10.1073/pnas.0306880101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Soloaga A., Thomson S., Wiggin G.R., Rampersaud N., Dyson M.H., Hazzalin C.A., Mahadevan L.C., Arthur J.S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. Embo j. 2003;22(11):2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) Embo j. 2003;22(6):1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bruck N., Vitoux D., Ferry C., Duong V., Bauer A., de The H., Rochette-Egly C. A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARalpha to target promoters. Embo j. 2009;28(1):34–47. doi: 10.1038/emboj.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cheng M.B., Zhang Y., Cao C.Y., Zhang W.L., Zhang Y., Shen Y.F. Specific phosphorylation of histone demethylase KDM3A determines target gene expression in response to heat shock. PLoS Biol. 2014;12(12) doi: 10.1371/journal.pbio.1002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Reyskens K.M.S.E., Arthur J.S.C. Emerging Roles of the Mitogen and Stress Activated Kinases MSK1 and MSK2. Frontiers in Cell and Developmental Biology. 2016;4:56. doi: 10.3389/fcell.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ananieva O., Darragh J., Johansen C., Carr J.M., McIlrath J., Park J.M., Wingate A., Monk C.E., Toth R., Santos S.G., Iversen L., Arthur J.S. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol. 2008;9(9):1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]
- 223.Kim C., Sano Y., Todorova K., Carlson B.A., Arpa L., Celada A., Lawrence T., Otsu K., Brissette J.L., Arthur J.S., Park J.M. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9(9):1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.MacKenzie K.F., Van Den Bosch M.W., Naqvi S., Elcombe S.E., McGuire V.A., Reith A.D., Blackshear P.J., Dean J.L., Arthur J.S. MSK1 and MSK2 inhibit lipopolysaccharide-induced prostaglandin production via an interleukin-10 feedback loop. Mol Cell Biol. 2013;33(7):1456–1467. doi: 10.1128/MCB.01690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhao C., Hui W., Fernandes M.J., Poubelle P.E., Bourgoin S.G. Lysophosphatidic acid-induced IL-8 secretion involves MSK1 and MSK2 mediated activation of CREB1 in human fibroblast-like synoviocytes. Biochem Pharmacol. 2014;90(1):62–72. doi: 10.1016/j.bcp.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 226.Cheng G., Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-kappaB activation. Genes Dev. 1996;10(8):963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- 227.Rothe M., Xiong J., Shu H.B., Williamson K., Goddard A., Goeddel D.V. I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc Natl Acad Sci U S A. 1996;93(16):8241–8246. doi: 10.1073/pnas.93.16.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chin A.I., Shu J., Shan Shi C., Yao Z., Kehrl J.H., Cheng G. TANK potentiates tumor necrosis factor receptor-associated factor-mediated c-Jun N-terminal kinase/stress-activated protein kinase activation through the germinal center kinase pathway. Mol Cell Biol. 1999;19(10):6665–6672. doi: 10.1128/mcb.19.10.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kawagoe T., Takeuchi O., Takabatake Y., Kato H., Isaka Y., Tsujimura T., Akira S. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol. 2009;10(9):965–972. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Guo B., Cheng G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem. 2007;282(16):11817–11826. doi: 10.1074/jbc.M700017200. [DOI] [PubMed] [Google Scholar]
- 231.Clark K., Takeuchi O., Akira S., Cohen P. The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2011;108(41):17093–17098. doi: 10.1073/pnas.1114194108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Loughran G., Healy N.C., Kiely P.A., Huigsloot M., Kedersha N.L., O'Connor R. Mystique Is a New Insulin-like Growth Factor-I-regulated PDZ-LIM Domain Protein That Promotes Cell Attachment and Migration and Suppresses Anchorage-independent Growth. Molecular Biology of the Cell. 2005;16(4):1811–1822. doi: 10.1091/mbc.E04-12-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Tanaka T., Soriano M.A., Grusby M.J. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22(6):729–736. doi: 10.1016/j.immuni.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 234.Pomies P., Macalma T., Beckerle M.C. Purification and characterization of an alpha-actinin-binding PDZ-LIM protein that is up-regulated during muscle differentiation. J Biol Chem. 1999;274(41):29242–29250. doi: 10.1074/jbc.274.41.29242. [DOI] [PubMed] [Google Scholar]
- 235.Bashirova A.A., Markelov M.L., Shlykova T.V., Levshenkova E.V., Alibaeva R.A., Frolova E.I. The human RIL gene: mapping to human chromosome 5q31.1, genomic organization and alternative transcripts. Gene. 1998;210(2):239–245. doi: 10.1016/s0378-1119(98)00080-8. [DOI] [PubMed] [Google Scholar]
- 236.Ponting C.P., Phillips C., Davies K.E., Blake D.J. PDZ domains: targeting signalling molecules to sub-membranous sites. Bioessays. 1997;19(6):469–479. doi: 10.1002/bies.950190606. [DOI] [PubMed] [Google Scholar]
- 237.Bach I. The LIM domain: regulation by association. Mech Dev. 2000;91(1–2):5–17. doi: 10.1016/s0925-4773(99)00314-7. [DOI] [PubMed] [Google Scholar]
- 238.Tanaka T., Grusby M.J., Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8(6):584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 239.Tanaka T., Shibazaki A., Ono R., Kaisho T. HSP70 mediates degradation of the p65 subunit of nuclear factor kappaB to inhibit inflammatory signaling. Sci Signal. 2014;7(356):ra119. doi: 10.1126/scisignal.2005533. [DOI] [PubMed] [Google Scholar]
- 240.Healy N.C., O'Connor R. Sequestration of PDLIM2 in the cytoplasm of monocytic/macrophage cells is associated with adhesion and increased nuclear activity of NF-kappaB. J Leukoc Biol. 2009;85(3):481–490. doi: 10.1189/jlb.0408238. [DOI] [PubMed] [Google Scholar]
- 241.Yang P.H., Cheung W.K., Peng Y., He M.L., Wu G.Q., Xie D., Jiang B.H., Huang Q.H., Chen Z., Lin M.C., Kung H.F. Makorin-2 is a neurogenesis inhibitor downstream of phosphatidylinositol 3-kinase/Akt (PI3K/Akt) signal. J Biol Chem. 2008;283(13):8486–8495. doi: 10.1074/jbc.M704768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Shin C., Ito Y., Ichikawa S., Tokunaga M., Sakata-Sogawa K., Tanaka T. MKRN2 is a novel ubiquitin E3 ligase for the p65 subunit of NF-kappaB and negatively regulates inflammatory responses. Sci Rep. 2017;7:46097. doi: 10.1038/srep46097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Deshaies R.J., Joazeiro C.A. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 244.Ono R., Kaisho T., Tanaka T. PDLIM1 inhibits NF-κB-mediated inflammatory signaling by sequestering the p65 subunit of NF-κB in the cytoplasm. Scientific Reports. 2015;5:18327. doi: 10.1038/srep18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Vallenius T., Luukko K., Makela T.P. CLP-36 PDZ-LIM protein associates with nonmuscle alpha-actinin-1 and alpha-actinin-4. J Biol Chem. 2000;275(15):11100–11105. doi: 10.1074/jbc.275.15.11100. [DOI] [PubMed] [Google Scholar]
- 246.Nisole S., Stoye J.P., Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3(10):799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- 247.Ozato K., Shin D.-M., Chang T.-H., Morse H.C. TRIM family proteins and their emerging roles in innate immunity. Nature reviews. Immunology. 2008;8(11):849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Shi M., Deng W., Bi E., Mao K., Ji Y., Lin G., Wu X., Tao Z., Li Z., Cai X., Sun S., Xiang C., Sun B. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat Immunol. 2008;9(4):369–377. doi: 10.1038/ni1577. [DOI] [PubMed] [Google Scholar]
- 249.Li Q., Yan J., Mao A.P., Li C., Ran Y., Shu H.B., Wang Y.Y. Tripartite motif 8 (TRIM8) modulates TNFalpha- and IL-1beta-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc Natl Acad Sci U S A. 2011;108(48):19341–19346. doi: 10.1073/pnas.1110946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ye W., Hu M.M., Lei C.Q., Zhou Q., Lin H., Sun M.S., Shu H.B. TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF-TBK1 Interaction. J Immunol. 2017 doi: 10.4049/jimmunol.1601647. [DOI] [PubMed] [Google Scholar]
- 251.Chuang T.H., Ulevitch R.J. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5(5):495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 252.Nakhaei P., Mesplede T., Solis M., Sun Q., Zhao T., Yang L., Chuang T.H., Ware C.F., Lin R., Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5(11) doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Li C., Gu H., Yu M., Yang P., Zhang M., Ba H., Yin Y., Wang J., Yin B., Zhou X., Li Z. Inhibition of transmembrane TNF-α shedding by a specific antibody protects against septic shock. Cell Death & Disease. 2019;10(8):586. doi: 10.1038/s41419-019-1808-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lu X., He Y., Tang C., Wang X., Que L., Zhu G., Liu L., Ha T., Chen Q., Li C., Xu Y., Li J., Li Y. Triad3A attenuates pathological cardiac hypertrophy involving the augmentation of ubiquitination-mediated degradation of TLR4 and TLR9. Basic Research in Cardiology. 2020;115(2):19. doi: 10.1007/s00395-020-0779-1. [DOI] [PubMed] [Google Scholar]
- 255.Muller S., Hoege C., Pyrowolakis G., Jentsch S. SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell Biol. 2001;2(3):202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 256.Kamitani T., Kito K., Nguyen H.P., Fukuda-Kamitani T., Yeh E.T. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. 1998;273(18):11349–11353. doi: 10.1074/jbc.273.18.11349. [DOI] [PubMed] [Google Scholar]
- 257.Boddy M.N., Howe K., Etkin L.D., Solomon E., Freemont P.S. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene. 1996;13(5):971–982. [PubMed] [Google Scholar]
- 258.Okura T., Gong L., Kamitani T., Wada T., Okura I., Wei C.F., Chang H.M., Yeh E.T. Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J Immunol. 1996;157(10):4277–4281. [PubMed] [Google Scholar]
- 259.Kerscher O., Felberbaum R., Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 260.Capili A.D., Lima C.D. Taking it step by step: mechanistic insights from structural studies of ubiquitin/ubiquitin-like protein modification pathways. Curr Opin Struct Biol. 2007;17(6):726–735. doi: 10.1016/j.sbi.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gareau J.R., Lima C.D. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nature reviews. Molecular cell biology. 2010;11(12):861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Desterro J.M., Rodriguez M.S., Hay R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2(2):233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 263.Liu X., Chen W., Wang Q., Li L., Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9(6) doi: 10.1371/journal.ppat.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Decque A., Joffre O., Magalhaes J.G., Cossec J.-C., Blecher-Gonen R., Lapaquette P., Silvin A., Manel N., Joubert P.-E., Seeler J.-S., Albert M.L., Amit I., Amigorena S., Dejean A. Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat Immunol. 2016;17(2):140–149. doi: 10.1038/ni.3342. [DOI] [PubMed] [Google Scholar]
- 265.Wang G., Yang F., Gao T., Wu D., Huang J., Li J. Effect of deletion of SUMOylation on dendritic cell function in septic mice and its role in sepsis. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2020;45(3):314–321. doi: 10.11817/j.issn.1672-7347.2020.190684. [DOI] [PubMed] [Google Scholar]
- 266.Glass C.K., Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10(5):365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 267.Kho C., Lee A., Jeong D., Oh J.G., Gorski P.A., Fish K., Sanchez R., DeVita R.J., Christensen G., Dahl R., Hajjar R.J. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun. 2015;6:7229. doi: 10.1038/ncomms8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bernstock J.D., Ye D., Gessler F.A., Lee Y.-J., Peruzzotti-Jametti L., Baumgarten P., Johnson K.R., Maric D., Yang W., Kögel D., Pluchino S., Hallenbeck J.M. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Scientific Reports. 2017;7(1):7425. doi: 10.1038/s41598-017-07631-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Yang Y., Xia Z., Wang X., Zhao X., Sheng Z., Ye Y., He G., Zhou L., Zhu H., Xu N., Liang S. Small molecular inhibitors targeting protein SUMOylation as novel anticancer compounds. Molecular Pharmacology. 2018 doi: 10.1124/mol.118.112300. mol.118.112300. [DOI] [PubMed] [Google Scholar]
- 270.Bernstock J.D., Ye D., Smith J.A., Lee Y.J., Gessler F.A., Yasgar A., Kouznetsova J., Jadhav A., Wang Z., Pluchino S., Zheng W., Simeonov A., Hallenbeck J.M., Yang W. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO conjugation via the inhibition of SUMO-specific protease (SENP)2. Faseb j. 2018;32(3):1677–1691. doi: 10.1096/fj.201700711R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bernstock J.D., Ye D.G., Lee Y.-J., Gessler F., Friedman G.K., Zheng W., Hallenbeck J.M. Drugging SUMOylation for neuroprotection and oncotherapy. Neural Regen Res. 2018;13(3):415–416. doi: 10.4103/1673-5374.228718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Xia X., Cui J., Wang H.Y., Zhu L., Matsueda S., Wang Q., Yang X., Hong J., Songyang Z., Chen Z.J., Wang R.F. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity. 2011;34(6):843–853. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Guo H., Konig R., Deng M., Riess M., Mo J., Zhang L., Petrucelli A., Yoh S.M., Barefoot B., Samo M., Sempowski G.D., Zhang A., Colberg-Poley A.M., Feng H., Lemon S.M., Liu Y., Zhang Y., Wen H., Zhang Z., Damania B., Tsao L.C., Wang Q., Su L., Duncan J.A., Chanda S.K., Ting J.P. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe. 2016;19(4):515–528. doi: 10.1016/j.chom.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Schneider M., Zimmermann A.G., Roberts R.A., Zhang L., Swanson K.V., Wen H., Davis B.K., Allen I.C., Holl E.K., Ye Z., Rahman A.H., Conti B.J., Eitas T.K., Koller B.H., Ting J.P.Y. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-[kappa]B. Nat Immunol. 2012;13(9):823–831. doi: 10.1038/ni.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Karki R., Man S.M., Malireddi R.K.S., Kesavardhana S., Zhu Q., Burton A.R., Sharma B.R., Qi X., Pelletier S., Vogel P., Rosenstiel P., Kanneganti T.-D. NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature. 2016;540(7634):583–587. doi: 10.1038/nature20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bauerfeld C.P., Rastogi R., Pirockinaite G., Lee I., Huttemann M., Monks B., Birnbaum M.J., Franchi L., Nunez G., Samavati L. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol. 2012;188(6):2847–2857. doi: 10.4049/jimmunol.1102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.W. Fang, D. Bi, R. Zheng, N. Cai, H. Xu, R. Zhou, J. Lu, M. Wan, X. Xu, Identification and activation of TLR4-mediated signalling pathways by alginate-derived guluronate oligosaccharide in RAW264.7 macrophages, Scientific Reports 7(1) (2017) 1663. [DOI] [PMC free article] [PubMed]
- 278.Weichhart T., Costantino G., Poglitsch M., Rosner M., Zeyda M., Stuhlmeier K.M., Kolbe T., Stulnig T.M., Horl W.H., Hengstschlager M., Muller M., Saemann M.D. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29(4):565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 279.Zhao Y., Shao F. NLRC5: a NOD-like receptor protein with many faces in immune regulation. Cell Research. 2012;22(7):1099–1101. doi: 10.1038/cr.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Meissner T.B., Li A., Kobayashi K.S. NLRC5: a newly discovered MHC class I transactivator (CITA) Microbes and Infection / Institut Pasteur. 2012;14(6):477–484. doi: 10.1016/j.micinf.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Benko S., Magalhaes J.G., Philpott D.J., Girardin S.E. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185(3):1681–1691. doi: 10.4049/jimmunol.0903900. [DOI] [PubMed] [Google Scholar]
- 282.Cui J., Zhu L., Xia X., Wang H.Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z.J., Wang R.F. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141(3):483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tong Y., Cui J., Li Q., Zou J., Wang H.Y., Wang R.F. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012;22(5):822–835. doi: 10.1038/cr.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Yao Y., Wang Y., Chen F., Huang Y., Zhu S., Leng Q., Wang H., Shi Y., Qian Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012;22(5):836–847. doi: 10.1038/cr.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kumar H., Pandey S., Zou J., Kumagai Y., Takahashi K., Akira S., Kawai T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol. 2011;186(2):994–1000. doi: 10.4049/jimmunol.1002094. [DOI] [PubMed] [Google Scholar]
- 286.Meng Q., Cai C., Sun T., Wang Q., Xie W., Wang R., Cui J. Reversible ubiquitination shapes NLRC5 function and modulates NF-kappaB activation switch. J Cell Biol. 2015;211(5):1025–1040. doi: 10.1083/jcb.201505091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Hu Y. A feedforward loop of NLRC5 (de)ubiquitination keeps IKK–NF-κB in check. The Journal of Cell Biology. 2015 doi: 10.1083/jcb.201511039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Rebl A., Rebl H., Köbis J.M., Goldammer T., Seyfert H.-M. ST2 from rainbow trout quenches TLR signalling, localises at the nuclear membrane and allows the nuclear translocation of MYD88. Developmental & Comparative Immunology. 2017;67:139–152. doi: 10.1016/j.dci.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 289.Tominaga A., Mita S., Kikuchi Y., Hitoshi Y., Takatsu K., Nishikawa S., Ogawa M. Establishment of IL-5-dependent early B cell lines by long-term bone marrow cultures. Growth Factors. 1989;1(2):135–146. doi: 10.3109/08977198909029123. [DOI] [PubMed] [Google Scholar]
- 290.Kakkar R., Lee R.T. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nature reviews. Drug discovery. 2008;7(10):827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.K. Bulek, S. Swaidani, J. Qin, Y. Lu, M.F. Gulen, T. Herjan, B. Min, R.A. Kastelein, M. Aronica, M. Kosz-Vnenchak, X. Li, The essential role of SIGIRR/TIR8 in regulation of Th2 immune response, Journal of immunology (Baltimore, Md. : 1950) 182(5) (2009) 2601-2609. [DOI] [PMC free article] [PubMed]
- 292.Li X., Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med (Berl) 2005;83(4):258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 293.Schmitz J., Owyang A., Oldham E., Song Y., Murphy E., McClanahan T.K., Zurawski G., Moshrefi M., Qin J., Li X., Gorman D.M., Bazan J.F., Kastelein R.A. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 294.Bergers G., Reikerstorfer A., Braselmann S., Graninger P., Busslinger M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. Embo j. 1994;13(5):1176–1188. doi: 10.1002/j.1460-2075.1994.tb06367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Yanagisawa K., Takagi T., Tsukamoto T., Tetsuka T., Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett. 1993;318(1):83–87. doi: 10.1016/0014-5793(93)81333-u. [DOI] [PubMed] [Google Scholar]
- 296.Iwahana H., Hayakawa M., Kuroiwa K., Tago K., Yanagisawa K., Noji S., Tominaga S. Molecular cloning of the chicken ST2 gene and a novel variant form of the ST2 gene product, ST2LV. Biochim Biophys Acta. 2004;1681(1):1–14. doi: 10.1016/j.bbaexp.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 297.Brint E.K., Xu D., Liu H., Dunne A., McKenzie A.N.J., O'Neill L.A.J., Liew F.Y. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5(4):373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 298.Brint E.K., Fitzgerald K.A., Smith P., Coyle A.J., Gutierrez-Ramos J.C., Fallon P.G., O'Neill L.A. Characterization of signaling pathways activated by the interleukin 1 (IL-1) receptor homologue T1/ST2. A role for Jun N-terminal kinase in IL-4 induction. J Biol Chem. 2002;277(51):49205–49211. doi: 10.1074/jbc.M209685200. [DOI] [PubMed] [Google Scholar]
- 299.Liu J., Buckley J.M., Redmond H.P., Wang J.H. ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J Immunol. 2010;184(10):5802–5808. doi: 10.4049/jimmunol.0904127. [DOI] [PubMed] [Google Scholar]
- 300.Basith S., Manavalan B., Govindaraj R.G., Choi S. In Silico Approach to Inhibition of Signaling Pathways of Toll-Like Receptors 2 and 4 by ST2L. PLOS ONE. 2011;6(8) doi: 10.1371/journal.pone.0023989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Saccani S., Polentarutti N., Penton-Rol G., Sims J.E., Mantovani A. Divergent effects of LPS on expression of IL-1 receptor family members in mononuclear phagocytes in vitro and in vivo. Cytokine. 1998;10(10):773–780. doi: 10.1006/cyto.1998.0359. [DOI] [PubMed] [Google Scholar]
- 302.Kumar S., Tzimas M.N., Griswold D.E., Young P.R. Expression of ST2, an interleukin-1 receptor homologue, is induced by proinflammatory stimuli. Biochem Biophys Res Commun. 1997;235(3):474–478. doi: 10.1006/bbrc.1997.6810. [DOI] [PubMed] [Google Scholar]
- 303.Sweet M.J., Leung B.P., Kang D., Sogaard M., Schulz K., Trajkovic V., Campbell C.C., Xu D., Liew F.Y. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J Immunol. 2001;166(11):6633–6639. doi: 10.4049/jimmunol.166.11.6633. [DOI] [PubMed] [Google Scholar]
- 304.Hoogerwerf J.J., Tanck M.W.T., van Zoelen M.A.D., Wittebole X., Laterre P.-F., van der Poll T. Soluble ST2 plasma concentrations predict mortality in severe sepsis. Intensive Care Medicine. 2010;36(4):630–637. doi: 10.1007/s00134-010-1773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Parenica J., Malaska J., Jarkovsky J., Lipkova J., Dastych M., Helanova K., Litzman J., Tomandl J., Littnerova S., Sevcikova J., Gal R., Sevcik P., Spinar J., Goldbergova M.P. Soluble ST2 levels in patients with cardiogenic and septic shock are not predictors of mortality. Experimental & Clinical Cardiology. 2012;17(4):205–209. [PMC free article] [PubMed] [Google Scholar]
- 306.Wald D., Qin J., Zhao Z., Qian Y., Naramura M., Tian L., Towne J., Sims J.E., Stark G.R., Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4(9):920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 307.Li D., Zhang X., Chen B. SIGIRR participates in negative regulation of LPS response and tolerance in human bladder epithelial cells. BMC Immunol. 2015;16:73. doi: 10.1186/s12865-015-0137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Thomassen E., Renshaw B.R., Sims J.E. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine. 1999;11(6):389–399. doi: 10.1006/cyto.1998.0452. [DOI] [PubMed] [Google Scholar]
- 309.Lech M., Garlanda C., Mantovani A., Kirschning C.J., Schlondorff D., Anders H.J. Different roles of TiR8/Sigirr on toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int. 2007;72(2):182–192. doi: 10.1038/sj.ki.5002293. [DOI] [PubMed] [Google Scholar]
- 310.Xiao H., Gulen M.F., Qin J., Yao J., Bulek K., Kish D., Altuntas C.Z., Wald D., Ma C., Zhou H., Tuohy V.K., Fairchild R.L., de la Motte C., Cua D., Vallance B.A., Li X. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26(4):461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 311.Garlanda C., Riva F., Polentarutti N., Buracchi C., Sironi M., De Bortoli M., Muzio M., Bergottini R., Scanziani E., Vecchi A., Hirsch E., Mantovani A. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci U S A. 2004;101(10):3522–3526. doi: 10.1073/pnas.0308680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Polentarutti N., Rol G.P., Muzio M., Bosisio D., Camnasio M., Riva F., Zoja C., Benigni A., Tomasoni S., Vecchi A., Garlanda C., Mantovani A. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur Cytokine Netw. 2003;14(4):211–218. [PubMed] [Google Scholar]
- 313.Adib-Conquy M., Adrie C., Fitting C., Gattolliat O., Beyaert R., Cavaillon J.M. Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med. 2006;34(9):2377–2385. doi: 10.1097/01.CCM.0000233875.93866.88. [DOI] [PubMed] [Google Scholar]
- 314.Qin J., Qian Y., Yao J., Grace C., Li X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem. 2005;280(26):25233–25241. doi: 10.1074/jbc.M501363200. [DOI] [PubMed] [Google Scholar]
- 315.Garlanda C., Anders H.J., Mantovani A. TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 2009;30(9):439–446. doi: 10.1016/j.it.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 316.Zhang C., Wu X., Zhao Y., Deng Z., Qian G. SIGIRR inhibits toll-like receptor 4, 5, 9-mediated immune responses in human airway epithelial cells. Mol Biol Rep. 2011;38(1):601–609. doi: 10.1007/s11033-010-0146-7. [DOI] [PubMed] [Google Scholar]
- 317.Huang X., Hazlett L.D., Du W., Barrett R.P. SIGIRR promotes resistance against Pseudomonas aeruginosa keratitis by down-regulating type-1 immunity and IL-1R1 and TLR4 signaling. J Immunol. 2006;177(1):548–556. doi: 10.4049/jimmunol.177.1.548. [DOI] [PubMed] [Google Scholar]
- 318.Garlanda C., Di Liberto D., Vecchi A., La Manna M.P., Buracchi C., Caccamo N., Salerno A., Dieli F., Mantovani A. Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol. 2007;179(5):3119–3125. doi: 10.4049/jimmunol.179.5.3119. [DOI] [PubMed] [Google Scholar]
- 319.Abulkhir A., Samarani S., Amre D., Duval M., Haddad E., Sinnett D., Leclerc J.-M., Diorio C., Ahmad A. A protective role of IL-37 in cancer: a new hope for cancer patients. Journal of leukocyte biology. 2016 doi: 10.1189/jlb.5RU0816-341R. [DOI] [PubMed] [Google Scholar]
- 320.Lee S.M., Kok K.H., Jaume M., Cheung T.K., Yip T.F., Lai J.C., Guan Y., Webster R.G., Jin D.Y., Peiris J.S. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U S A. 2014;111(10):3793–3798. doi: 10.1073/pnas.1324266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Guan Y., Ranoa D.R.E., Jiang S., Mutha S.K., Li X., Baudry J., Tapping R.I. Human TLRs 10 and 1 Share Common Mechanisms of Innate Immune Sensing but Not Signaling. The Journal of Immunology. 2010;184(9):5094–5103. doi: 10.4049/jimmunol.0901888. [DOI] [PubMed] [Google Scholar]
- 322.Jiang S., Li X., Hess N.J., Guan Y., Tapping R.I. TLR10 Is a Negative Regulator of Both MyD88-Dependent and -Independent TLR Signaling. J Immunol. 2016;196(9):3834–3841. doi: 10.4049/jimmunol.1502599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Chuang T., Ulevitch R.J. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta. 2001;1518(1–2):157–161. doi: 10.1016/s0167-4781(00)00289-x. [DOI] [PubMed] [Google Scholar]
- 324.Hasan U., Chaffois C., Gaillard C., Saulnier V., Merck E., Tancredi S., Guiet C., Briere F., Vlach J., Lebecque S., Trinchieri G., Bates E.E. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174(5):2942–2950. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- 325.Mikacenic C., Reiner A.P., Holden T.D., Nickerson D.A., Wurfel M.M. Variation in the TLR10/TLR1/TLR6 Locus is the Major Genetic Determinant of Inter-Individual Difference in TLR1/2-Mediated Responses. Genes and immunity. 2013;14(1):52–57. doi: 10.1038/gene.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Oosting M., Cheng S.-C., Bolscher J.M., Vestering-Stenger R., Plantinga T.S., Verschueren I.C., Arts P., Garritsen A., van Eenennaam H., Sturm P., Kullberg B.-J., Hoischen A., Adema G.J., van der Meer J.W.M., Netea M.G., Joosten L.A.B. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proceedings of the National Academy of Sciences. 2014;111(42):E4478–E4484. doi: 10.1073/pnas.1410293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Hess N.J., Felicelli C., Grage J., Tapping R.I. TLR10 suppresses the activation and differentiation of monocytes with effects on DC-mediated adaptive immune responses. Journal of Leukocyte Biology. 2017 doi: 10.1189/jlb.3A1116-492R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hai T., Hartman M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273(1):1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- 329.Drysdale B.E., Howard D.L., Johnson R.J. Identification of a lipopolysaccharide inducible transcription factor in murine macrophages. Mol Immunol. 1996;33(11–12):989–998. doi: 10.1016/s0161-5890(96)00043-0. [DOI] [PubMed] [Google Scholar]
- 330.Farber J.M. A collection of mRNA species that are inducible in the RAW 264.7 mouse macrophage cell line by gamma interferon and other agents. Mol Cell Biol. 1992;12(4):1535–1545. doi: 10.1128/mcb.12.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Gilchrist M., Thorsson V., Li B., Rust A.G., Korb M., Roach J.C., Kennedy K., Hai T., Bolouri H., Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441(7090):173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 332.Whitmore M.M., Iparraguirre A., Kubelka L., Weninger W., Hai T., Williams B.R. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179(6):3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 333.Lee S., Kim G.-L., Kim N.Y., Kim S.-J., Ghosh P., Rhee D.-K. ATF3 Stimulates IL-17A by Regulating Intracellular Ca2+/ROS-Dependent IL-1β Activation During Streptococcus pneumoniae Infection. Frontiers in Immunology. 2018;9(1954) doi: 10.3389/fimmu.2018.01954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Panich T., Chancharoenthana W., Somparn P., Issara-Amphorn J., Hirankarn N., Leelahavanichkul A. Urinary exosomal activating transcriptional factor 3 as the early diagnostic biomarker for sepsis-induced acute kidney injury. BMC Nephrol. 2017;18(1):10. doi: 10.1186/s12882-016-0415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Lai P.-F., Cheng C.-F., Lin H., Tseng T.-L., Chen H.-H., Chen S.-H. ATF3 Protects against LPS-Induced Inflammation in Mice via Inhibiting HMGB1 Expression. Evid Based Complement Alternat Med. 2013;2013 doi: 10.1155/2013/716481. 716481-716481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Hoetzenecker W., Echtenacher B., Guenova E., Hoetzenecker K., Woelbing F., Brück J., Teske A., Valtcheva N., Fuchs K., Kneilling M., Park J.-H., Kim K.-H., Kim K.-W., Hoffmann P., Krenn C., Hai T., Ghoreschi K., Biedermann T., Röcken M. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nature Medicine. 2012;18(1):128–134. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhuang X., Wu B., Li J., Shi H., Jin B., Luo X. The emerging role of interleukin-37 in cardiovascular diseases. Immun Inflamm Dis. 2017 doi: 10.1002/iid3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Quirk S., Agrawal D.K. Immunobiology of Interleukin-37: Mechanism of Action and Clinical Perspectives. Expert review of clinical immunology. 2014;10(12):1703–1709. doi: 10.1586/1744666X.2014.971014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Dinarello C.A., Bufler P. Interleukin-37. Semin Immunol. 2013;25(6):466–468. doi: 10.1016/j.smim.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 340.Chen H.M., Fujita M. IL-37: a new player in immune tolerance. Cytokine. 2015;72(1):113–114. doi: 10.1016/j.cyto.2014.11.025. [DOI] [PubMed] [Google Scholar]
- 341.Nold M.F., Nold-Petry C.A., Zepp J.A., Palmer B.E., Bufler P., Dinarello C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11(11):1014–1022. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Bufler P., Gamboni-Robertson F., Azam T., Kim S.H., Dinarello C.A. Interleukin-1 homologues IL-1F7b and IL-18 contain functional mRNA instability elements within the coding region responsive to lipopolysaccharide. Biochem J. 2004;381(Pt 2):503–510. doi: 10.1042/BJ20040217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Wang Y.C., Weng G.P., Liu J.P., Li L., Cheng Q.H. Elevated serum IL-37 concentrations in patients with sepsis. Medicine (Baltimore) 2019;98(10) doi: 10.1097/MD.0000000000014756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Nold-Petry C.A., Lo C.Y., Rudloff I., Elgass K.D., Li S., Gantier M.P., Lotz-Havla A.S., Gersting S.W., Cho S.X., Lao J.C., Ellisdon A.M., Rotter B., Azam T., Mangan N.E., Rossello F.J., Whisstock J.C., Bufler P., Garlanda C., Mantovani A., Dinarello C.A., Nold M.F. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol. 2015;16(4):354–365. doi: 10.1038/ni.3103. [DOI] [PubMed] [Google Scholar]
- 345.Zhan Q., Zeng Q., Song R., Zhai Y., Xu D., Fullerton D.A., Dinarello C.A., Meng X. IL-37 Suppresses MyD88-mediated Inflammatory Responses in Human Aortic Valve Interstitial Cells. Molecular Medicine. 2017;23:83–91. doi: 10.2119/molmed.2017.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Moretti S., Bozza S., Oikonomou V., Renga G., Casagrande A., Iannitti R.G., Puccetti M., Garlanda C., Kim S., Li S., van de Veerdonk F.L., Dinarello C.A., Romani L. IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog. 2014;10(11) doi: 10.1371/journal.ppat.1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Dinarello C.A., Nold-Petry C., Nold M., Fujita M., Li S., Kim S., Bufler P. Suppression of innate inflammation and immunity by interleukin-37. Eur J Immunol. 2016;46(5):1067–1081. doi: 10.1002/eji.201545828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Li S., Neff C.P., Barber K., Hong J., Luo Y., Azam T., Palmer B.E., Fujita M., Garlanda C., Mantovani A., Kim S., Dinarello C.A. Extracellular forms of IL-37 inhibit innate inflammation in vitro and in vivo but require the IL-1 family decoy receptor IL-1R8. Proc Natl Acad Sci U S A. 2015;112(8):2497–2502. doi: 10.1073/pnas.1424626112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33(10):522–530. doi: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 350.Mullarkey M., Rose J.R., Bristol J., Kawata T., Kimura A., Kobayashi S., Przetak M., Chow J., Gusovsky F., Christ W.J., Rossignol D.P. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304(3):1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- 351.Solomon S.B., Cui X., Gerstenberger E., Danner R.L., Fitz Y., Banks S.M., Natanson C., Eichacker P.Q. Effective dosing of lipid A analogue E5564 in rats depends on the timing of treatment and the route of Escherichia coli infection. J Infect Dis. 2006;193(5):634–644. doi: 10.1086/500147. [DOI] [PubMed] [Google Scholar]
- 352.Opal S.M., Laterre P.F., Francois B., LaRosa S.P., Angus D.C., Mira J.P., Wittebole X., Dugernier T., Perrotin D., Tidswell M., Jauregui L., Krell K., Pachl J., Takahashi T., Peckelsen C., Cordasco E., Chang C.S., Oeyen S., Aikawa N., Maruyama T., Schein R., Kalil A.C., Van Nuffelen M., Lynn M., Rossignol D.P., Gogate J., Roberts M.B., Wheeler J.L., Vincent J.L. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. Jama. 2013;309(11):1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 353.Barochia A., Solomon S., Cui X., Natanson C., Eichacker P.Q. Eritoran tetrasodium (E5564) Treatment for Sepsis: Review of Preclinical and Clinical Studies. Expert opinion on drug metabolism & toxicology. 2011;7(4):479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Shirey K.A., Lai W., Scott A.J., Lipsky M., Mistry P., Pletneva L.M., Karp C.L., McAlees J., Gioannini T.L., Weiss J., Chen W.H., Ernst R.K., Rossignol D.P., Gusovsky F., Blanco J.C.G., Vogel S.N. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 2013;497(7450):498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ke B., Shen X.D., Kamo N., Ji H., Yue S., Gao F., Busuttil R.W., Kupiec-Weglinski J.W. beta-catenin regulates innate and adaptive immunity in mouse liver ischemia-reperfusion injury. Hepatology. 2013;57(3):1203–1214. doi: 10.1002/hep.26100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Yang D., Li S., Duan X., Ren J., Liang S., Yakoumatos L., Kang Y., Uriarte S.M., Shang J., Li W., Wang H. TLR4 induced Wnt3a-Dvl3 restrains the intensity of inflammation and protects against endotoxin-driven organ failure through GSK3β/β-catenin signaling. Mol Immunol. 2020;118:153–164. doi: 10.1016/j.molimm.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M.A., Nacken W., Foell D., van der Poll T., Sorg C., Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 358.Austermann J., Friesenhagen J., Fassl S.K., Petersen B., Ortkras T., Burgmann J., Barczyk-Kahlert K., Faist E., Zedler S., Pirr S., Rohde C., Muller-Tidow C., von Kockritz-Blickwede M., von Kaisenberg C.S., Flohe S.B., Ulas T., Schultze J.L., Roth J., Vogl T., Viemann D. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep. 2014;9(6):2112–2123. doi: 10.1016/j.celrep.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 359.Ulas T., Pirr S., Fehlhaber B., Bickes M.S., Loof T.G., Vogl T., Mellinger L., Heinemann A.S., Burgmann J., Schoning J., Schreek S., Pfeifer S., Reuner F., Vollger L., Stanulla M., von Kockritz-Blickwede M., Glander S., Barczyk-Kahlert K., von Kaisenberg C.S., Friesenhagen J., Fischer-Riepe L., Zenker S., Schultze J.L., Roth J., Viemann D. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol. 2017;18(6):622–632. doi: 10.1038/ni.3745. [DOI] [PubMed] [Google Scholar]
- 360.Vereecke L., Beyaert R., van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30(8):383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 361.Vereecke L., Beyaert R., van Loo G. Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011;39(4):1086–1091. doi: 10.1042/BST0391086. [DOI] [PubMed] [Google Scholar]
- 362.Fleer A., Krediet T.G. Innate immunity: toll-like receptors and some more. A brief history, basic organization and relevance for the human newborn, Neonatology. 2007;92(3):145–157. doi: 10.1159/000102054. [DOI] [PubMed] [Google Scholar]
- 363.Nguyen B.N., Chávez-Arroyo A., Cheng M.I., Krasilnikov M., Louie A., Portnoy D.A. TLR2 and endosomal TLR-mediated secretion of IL-10 and immune suppression in response to phagosome-confined Listeria monocytogenes. PLOS Pathogens. 2020;16(7) doi: 10.1371/journal.ppat.1008622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Patidar A., Selvaraj S., Sarode A., Chauhan P., Chattopadhyay D., Saha B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine. 2018;104:114–123. doi: 10.1016/j.cyto.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 365.Blasius A.L., Beutler B. Intracellular Toll-like Receptors. Immunity. 2010;32(3):305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 366.Tartey S., Takeuchi O. Pathogen recognition and Toll-like receptor targeted therapeutics in innate immune cells. International Reviews of Immunology. 2017;36(2):57–73. doi: 10.1080/08830185.2016.1261318. [DOI] [PubMed] [Google Scholar]
- 367.Das N., Dewan V., Grace P.M., Gunn R.J., Tamura R., Tzarum N., Watkins L.R., Wilson I.A., Yin H. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Rep. 2016;17(4):1128–1140. doi: 10.1016/j.celrep.2016.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.De Leo M.G., Staiano L., Vicinanza M., Luciani A., Carissimo A., Mutarelli M., Di Campli A., Polishchuk E., Di Tullio G., Morra V., Levtchenko E., Oltrabella F., Starborg T., Santoro M., Di Bernardo D., Devuyst O., Lowe M., Medina D.L., Ballabio A., De Matteis M.A. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat Cell Biol. 2016;18(8):839–850. doi: 10.1038/ncb3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Tian J., Avalos A.M., Mao S.Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L.L., Audoly L., La Rosa G., Bierhaus A., Naworth P., Marshak-Rothstein A., Crow M.K., Fitzgerald K.A., Latz E., Kiener P.A., Coyle A.J. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 370.Henrick B.M., Yao X.D., Zahoor M.A., Abimiku A., Osawe S., Rosenthal K.L. TLR10 Senses HIV-1 Proteins and Significantly Enhances HIV-1 Infection. Front Immunol. 2019;10:482. doi: 10.3389/fimmu.2019.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Fore F., Indriputri C., Mamutse J., Nugraha J. TLR10 and Its Unique Anti-Inflammatory Properties and Potential Use as a Target in Therapeutics. Immune Netw. 2020;20(3) doi: 10.4110/in.2020.20.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Raetz M., Kibardin A., Sturge C.R., Pifer R., Li H., Burstein E., Ozato K., Larin S., Yarovinsky F. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J Immunol. 2013;191(9):4818–4827. doi: 10.4049/jimmunol.1301301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Koblansky A.A., Jankovic D., Oh H., Hieny S., Sungnak W., Mathur R., Hayden M.S., Akira S., Sher A., Ghosh S. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity. 2013;38(1):119–130. doi: 10.1016/j.immuni.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Oldenburg M., Kruger A., Ferstl R., Kaufmann A., Nees G., Sigmund A., Bathke B., Lauterbach H., Suter M., Dreher S., Koedel U., Akira S., Kawai T., Buer J., Wagner H., Bauer S., Hochrein H., Kirschning C.J. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science. 2012;337(6098):1111–1115. doi: 10.1126/science.1220363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.