

Abstract
Background:
Chronic pulmonary graft-versus-host disease (cpGVHD) after hematopoietic cell transplant (HCT) manifests as progressive airway and parenchymal lung fibrosis. Based on our prior data, mice that undergo allogeneic HCT with Tbet-knockout donors (AlloTbet−/−) have increased lung Th17 cells and IL-17A and develop fibrosis resembling human cpGVHD. The role of IL-17A in post-transplant pulmonary fibrosis remains incompletely understood. We hypothesized that IL-17A is necessary for development of murine cpGVHD in this model.
Methods:
AlloTbet−/− mice received weekly intraperitoneal anti-IL-17A or IgG (200μg/mouse) starting 2 weeks post-HCT and were sacrificed after week 5. Histologic airway and parenchymal fibrosis was semi-quantitatively graded in a blinded fashion. Lung cells and proteins were measured by flow cytometry, ELISA, and multi-cytokine assays.
Results:
Anti-IL-17A modestly decreased airway and parenchymal lung fibrosis, along with a striking reduction in pulmonary neutrophilia, IL-6, MIP-1α, MIP-1β, CXCL1, and CXCL5 in AlloTbet−/− mice. Additionally, anti-IL-17A decreased CCL2, inflammatory monocytes and macrophages, and Th17 cells.
Conclusions:
In the setting of murine AlloHCT with Tbet−/− donors, IL-17A blockade decreases fibrotic features of cpGVHD. This may be mediated by the observed reduction in neutrophils and/or specific lung monocyte and macrophage populations, or alternatively via a direct effect on fibroblasts. Collectively, our results further suggest that anti-IL-17A strategies could prove useful in preventing alloimmune-driven fibrotic lung diseases.
INTRODUCTION
Chronic airway fibrosis, usually manifest as obliterative bronchiolitis (OB), and lung parenchymal fibrosis represent a major complication and cause of mortality in hematopoietic cell transplant (HCT) patients.1,2 This fibrotic process remains a fatal and untreatable condition, and its pathogenesis is poorly understood. Due to similar fibrotic pathology development in allogeneic HCT as well as in recipients of lung transplants, alloimmune mechanisms are thought to significantly contribute to the post-transplant pulmonary fibrosis, through chronic graft-versus-host disease (GVHD) in HCT recipients and chronic allograft rejection in lung transplant recipients. However, classic anti-T cell and standard immunosuppressive therapies appear to neither treat nor completely prevent OB.3 Improved understanding of mechanisms driving chronic pulmonary GVHD (cpGVHD) is urgently needed.
Recent data from our laboratory and others have indicated that T helper 17 (Th17) cells, known to mediate autoimmunity and fibrosis and to produce IL-17A,4 play an important role in chronic GVHD. Th17 and IL-17A have been associated with chronic GVHD of the skin5,6 and liver.7 Furthermore, inhibition of Th17 differentiation resulted in reduced cpGVHD pathology in an animal model, although the specific role of IL-17A was not assessed.5 Studies of chronic lung allograft rejection and fibrosis have also pointed to a role for IL-17A.8–15 While these data suggest that the Th17 pathway is important in development of alloimmune-mediated lung fibrosis, the precise downstream effects of IL-17A in cpGVHD remain unclear and require further elucidation.
Our laboratory has previously evaluated the role of T cell polarization in a model of cpGVHD after major-MHC-mismatched HCT. In this model, donor cells deficient in the T-bet transcription factor do not generate a Th1 phenotype,16 which results in the development of airway fibrosis in the recipient mouse, consistent with cpGVHD.17 While there is no airway obliteration, there is increased lung collagen content and clear airway-centric peribronchiolar fibrosis confirmed with Masson-Trichrome staining. Airway fibrosis is associated with decreased levels of IFN-γ and increased IL-17A and Th17 T cells. IL-4 and IL-13 are also elevated in HCT mice transplanted with T-bet−/− donors compared to WT donors, but are equally elevated in allogeneic and syngeneic HCT settings.17
The overall objective of this study was to further delineate the role of IL-17A in the pathogenesis of cpGVHD in our major-MHC-mismatched allogeneic HCT mouse model.17 Our hypothesis was that IL-17A is necessary for the development of airway fibrosis in allogeneic HCT mice transplanted with T-bet-deficient donors. We specifically aimed to determine whether IL-17A neutralization reduces fibrosis as well as inflammation in this model.
MATERIALS AND METHODS
Animals:
All experiments were approved by the Institutional Animal Care and Use Committees at Duke University (protocol A017-12-01) and followed the National Institutes of Health recommendations cited in the Guide for the Care and Use of Laboratory Animals. Male 8–10 week old C57Bl/6J (H2b), B10.BR-H2kH2-T18a/SgSnJ (B10.BR) (H2k), and B6.129-Tbx21tm2Srnr/J (Tbet−/− on C57BL/6J background)16 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were housed in individually-ventilated cages in a pathogen-free facility at Duke University on endotoxin-free bedding (Alpha Dri bedding, Shepherd Specialty Papers Inc., Kalamazoo, MI) and were fed irradiated food (PicoLab Mouse Diet 20 5058, Purina Mills, Richmond, IN).
Murine hematopoietic cell transplant (HCT):
Recipient B10.BR mice underwent HCT with 4×106 bone marrow cells and 1×106 splenocytes from donor C57BL/6 or Tbet−/− mice.18 Briefly, donor C57BL/6 or Tbet−/− mice were euthanized using CO2 and bone marrow was isolated from their tibia and femurs and splenocytes were isolated from their spleens via homogenization and filtration. All donor cells were washed in media, filtered through 70 um filters (BD, Franklin Lakes, NJ), counted on a hemocytometer, and resuspended at an appropriate concentration in media containing 10% FBS (Hyclone, Logan, UT), 1% L-Glutamine (Sigma-Aldrich, St. Louis, MO) and 1% Penicillin/Streptomycin (Sigma-Aldrich, St. Louis, MO). Recipient B10.BR mice were lethally irradiated using a Cesium irradiator (8.5 Gy) and, within 4 hours, injected intravenously with 4×106 donor bone marrow cells and 1×106 donor splenocytes in a total volume of 0.5 mL. After HCT, all animals were maintained with Septra-containing water (Sulfamethoxazole/Trimethoprim at 1.2/0.24 mg/mL). Recipient engraftment was evaluated 3 to 4 weeks following transplantation using peripheral blood flow cytometry. Peripheral blood was obtained using maxillary sinus puncture. Cells were stained with FITC-conjugated anti-mouse H2Kk, PE-conjugated anti-mouse H2Db, and PerCP-Cy5.5-conjugated anti-mouse CD45. (eBioscience, San Diego, CA). Flow cytometry was performed as described below. Only animals with greater than 95% donor-derived peripheral blood cells were used for further experiments.
Anti-IL-17A antibody treatment:
Rat anti-mouse IL-17A (Clone #50104, catalog #MAB421) or IgG2A isotype control (Clone #54447, catalog #MAB006) antibody (R&D Systems, Inc., Minneapolis, MN) was administered weekly via intra-peritoneal injections (200μg/mouse, resuspended in 500μl sterile phosphate buffered saline [PBS]) starting 2 weeks post-HCT.19–21 Based on pilot studies (data not shown), earlier and higher doses of anti-IL-17A were avoided due to concerns over potential impairment in hematopoietic cell engraftment.
Necropsy, bronchoalveolar lavage (BAL) and lung tissue collection:
Mice were euthanized 5 weeks after HCT using CO2 and their lungs were surgically exposed. The trachea was cannulated and lungs were lavaged with 5 aliquots of 800 μL of 0.9% saline (VWR International, West Chester, PA). BAL fluid supernatant from the first 2 lavage aliquots was stored at −80°C until further use. BAL cells underwent red blood cell lysis and were subsequently counted using a hemocytometer. Lungs were perfused with 0.9% saline. The right lung was snap-frozen in liquid nitrogen and stored at −80°C for subsequent collagen (hydroxyproline) analysis. The left lung was gravity-inflated with 10% formalin (VWR International, West Chester, PA), fixed in 10% formalin solution for 24 hours and then transferred into 70% EtOH. The entire left lung was subsequently embedded in paraffin, three to four 5 μm sagittal sections were placed on each slide and stained with hematoxylin and eosin (H&E) and Masson-Trichrome stains. Histologic analysis for each sample was based on three sections spanning the entire left lung.
Histologic analysis:
For each left lung sample, all visible airways were evaluated on Masson-Trichrome-stained slides. Each airway was classified as fibrotic or not-fibrotic and the percentage of fibrotic airways was then calculated. The percentage of the area of each lung section affected by parenchymal fibrosis was estimated. Grading was performed by an observer blinded to group assignment.
Cytokine analysis:
CXCL5 and IL-17A were measured in the BAL supernatant using specific ELISA kits (R&D systems, Minneapolis, MN). For additional cytokine analysis in the BAL supernatant, a commercially-available mouse 23-plex was chosen for detection of common T cell cytokines, neutrophil chemoattractants, and monocyte/macrophage-related cytokines, including: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17A, Eotaxin, G-CSF, GM-CSF, IFN- γ, CXCL1 (KC), CCL2, CCL3 (MIP-1α), CCL4 (MIP-1 β), CCL5 (RANTES), and TNF- α (Bio-Rad Laboratories, Hercules, CA). Results for 23-plex cytokines that were detected in less than 50% of the samples are not shown: IL-1β, IL-2, IL-3, IL-12(p70), Eotaxin.
Quantification of fibrosis and extracellular matrix production:
Total lung collagen was quantified in the right lung using the hydroxyproline assay as previously described.22 Levels of Hyaluronan were measured in the BAL supernatant using a specific ELISA (R&D systems).
BAL cell flow cytometry:
BAL cells were blocked using a flow cytometry buffer containing phosphate-buffered sodium solution (Sigma-Aldrich, St. Louis, MO) with 3% FBS (Hyclone, Logan, UT), 0.05% sodium azide (VWR International, West Chester, PA), and 10 mM EDTA (Sigma-Aldrich, St. Louis, MO) with the addition of 5% normal mouse serum, 5% normal rat serum, and 1% Fc receptor block (Affinity purified anti-mouse CD16/32) (eBioscience).
For myeloid cell staining, cells were incubated with conjugated antibodies for 30 minutes. For intracellular cytokine staining, cells were stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin (Sigma-Aldrich) and incubated with GolgiStop (BD Biosciences) before blocking, staining with surface antibodies for 30 min, and subsequently fixing, permeabilizing, and staining with intracellular antibodies. The myeloid cell-staining panel included FITC-conjugated anti-mouse-MHCII, PE-conjugated anti-mouse-Ly6G, APC-Cy7-conjugated anti-mouse-Gr1(Ly6G/Ly6C) (BD Biosciences, San Jose, CA), APC-conjugated anti-mouse-CD11c (eBioscience, San Diego, CA), PE-Cy7-conjugated anti-mouse-CD11b, and PerCP-Cy5.5-conjugated anti-mouse-CD45 (Biolegend). The T cell staining panel included FITC-conjugated anti-mouse-CD3, PerCP-Cy5.5-conjugated anti-mouse-CD4 (Biolegend), and APC-Cy7-conjugated anti-mouse-CD8 (BioLegend, San Diego, CA). Intracellular staining was performed with PE-conjugated anti-mouse-IL-17A (clone eBio17B7) and APC-conjugated anti-mouse-IFN-γ (eBioscience). Cell fluorescence was measured using a BD FACS Canto II flow cytometer with BD FACSDiva software (BD, Franklin Lakes, NJ). Flow cytometry analysis was performed using FlowJo software (Tree Star Inc., Ashland, OR). A singlet gate was used to exclude cell aggregates, followed by an all-cell gate to exclude small debris and dead cells, followed by a CD45+ cell gate to define all white blood cells. Myeloid cell gating was done based on previously published studies23,24 defining inflammatory macrophages as CD11c+MHCII+CD11b+Gr1+ cells and inflammatory monocytes CD11c−MHCII−Ly6G−CD11b+Gr1+ cells. Cell percentages are expressed as percentage of all cells and converted to absolute numbers by multiplying by live-cell counts.
Statistical analysis:
Data are expressed as mean ± SEM. All data were analyzed using a one-way ANOVA with a post-test comparison of select groups with Bonferroni correction. For the baseline model data (Figures 1 and Supplemental Figure 1), the post-test was applied to compare AlloTbet−/− to AlloWT and AlloTbet−/− to SynTbet−/− groups. For all other figures, the post-test was used to compare AlloWT to AlloTbet−/− and AlloTbet−/− to AlloTbet−/−αIL17A. Values of p<0.05 are reported to be significant. All statistical analyses were performed using Prism 5 (GraphPad Software Inc., La Jolla, CA).
Figure 1. Allogeneic HCT with Tbet−/− cells leads to airway and parenchymal lung fibrosis in the setting of IL-17A+ T cell accumulation:
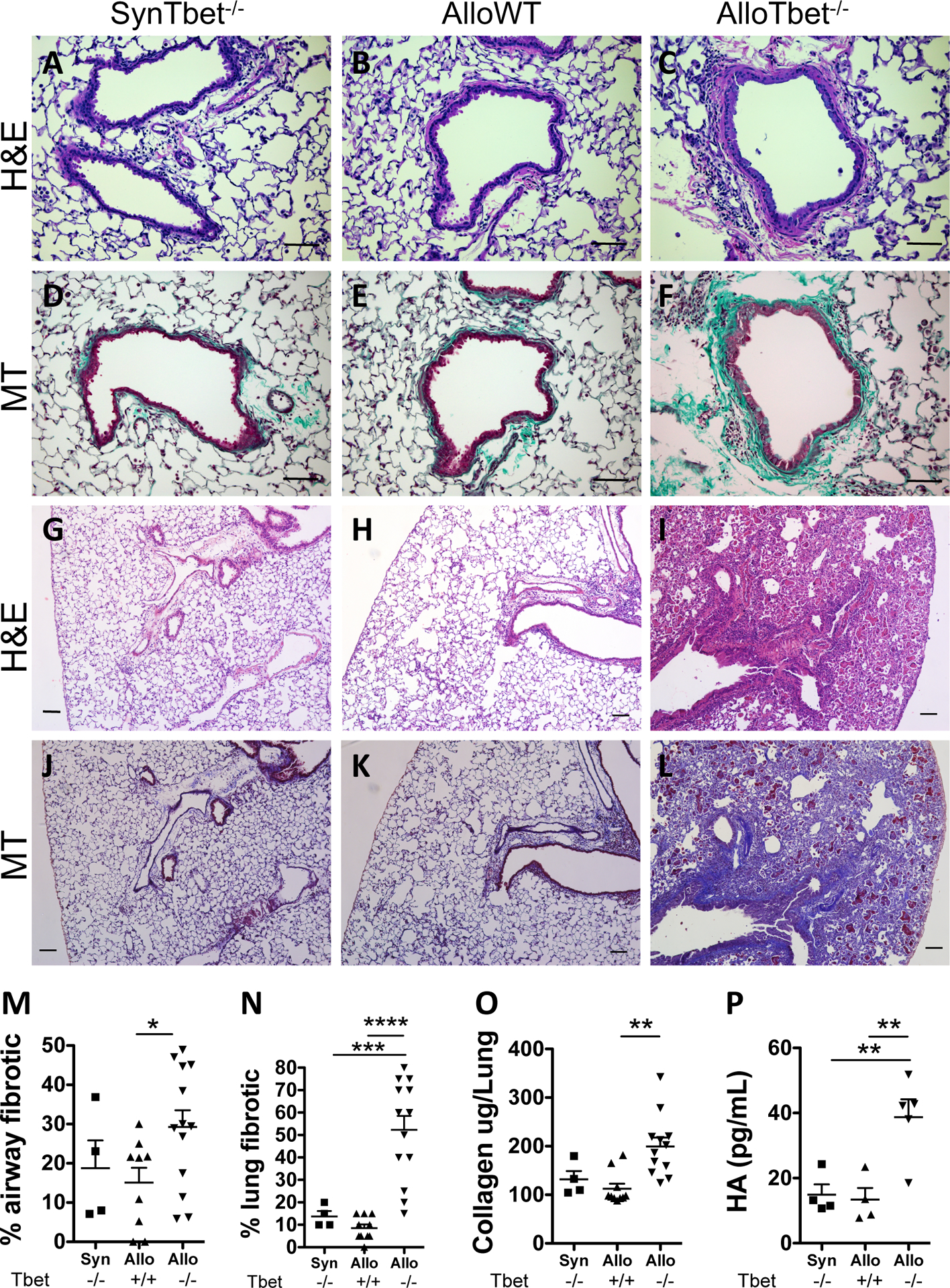
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) (n=13) developed prominent peri-airway fibrosis (C,F) and parenchymal fibrosis (I,L). In contrast, control B10.BR mice that received an allogeneic HCT from WT donors (AlloWT) (n=9) (B,E) or C57BL/6 mice that received a syngeneic HCT from Tbet−/− donors (SynTbet−/−) (n=4) (A,D) demonstrated minimal peri-airway (Figure 1A,B,D,E) and parenchymal collagen (G,H,J,K). Representative H&E (A-C, G-I) and Masson-Trichrome (D-F, J-L) images are shown: 200x (A-F), 100X (G-L) (Scale bar=100μm). Blinded histologic assessment showed a significant increase in the percent of fibrotic airways in AlloTbet−/− mice compared to AlloWT (M). The percentage of the lung parenchyma affected by fibrosis was also significantly greater in AlloTbet−/− compared to AlloWT or SynTbet−/− (N). Quantification of total lung collagen content further demonstrated higher levels in AlloTbet−/− compared to control groups (O). Finally, accumulation of hyaluronan (HA), a glycosaminoglycan component of the extracellular matrix, was greater in the BAL of AlloTbet−/− (n=5) mice compared to controls (n=4 for AlloWT and SynTbet−/−) (P). As previously published by our group17, AlloTbet−/− mice had increased numbers of CD4+IL-17+ cells and reduced CD4+IFN-γ+ cells in their BAL (Supplemental Figure 1). Quantitative data represent the average +SEM and * = p<0.05; ** = p<0.005; *** = p<0.0005. Pathology and hydroxyproline data have been replicated in 3 independent experiments.
RESULTS
Allogeneic HCT with Tbet−/− cells leads to airway and parenchymal lung fibrosis in the setting of IL-17A+ T cell accumulation:
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) developed prominent peri-airway fibrosis (Figure 1C,F) and parenchymal fibrosis (Figure 1I,L). In contrast, control B10.BR mice that received an allogeneic HCT from WT donors (AlloWT) (Figure 1B,E) or C57BL/6 mice that received a syngeneic HCT from Tbet−/− donors (SynTbet−/−) (Figure 1A,D) demonstrated minimal peri-airway (Figure 1A,B,D,E) and parenchymal fibrotic tissue (Figure 1G,H,J,K). Blinded histologic assessment showed a significant increase in the percent of fibrotic airways in AlloTbet−/− mice compared to AlloWT (Figure 1M). The percentage of the lung parenchyma affected by fibrosis was also significantly greater in AlloTbet−/− compared to AlloWT or SynTbet−/− (Figure 1N). Quantification of total lung collagen content further demonstrated higher levels in AlloTbet−/− compared to control groups (Figure 1O). Finally, accumulation of hyaluronan, a glycosaminoglycan component of the extracellular matrix, was greater in the BAL of AlloTbet−/− mice compared to controls (Figure 1P). As previously published by our group,17 AlloTbet−/− mice had increased numbers of CD4+IL-17+ cells and reduced CD4+IFN-γ+ cells in their BAL (Supplemental Figure 1).
Treatment with anti-IL-17A antibody decreases airway and parenchymal fibrosis in AlloTbet−/− mice:
AlloTbet−/− were treated with intraperitoneal injections of weekly anti-IL-17A antibody (AlloTbet−/−αIL17A) vs. IgG isotype control (AlloTbet−/−) (starting 2 weeks after HCT). An additional control group included mice that received an allogeneic HCT from WT donors (AlloWT) and were treated with IgG isotype injections. Treatment with anti-IL-17A led to reduced airway (Figure 2C) and parenchymal lung fibrosis (Figure 2F) in the AlloTbet−/− mice compared to the IgG-treated AlloTbet−/− group (Figure 2B,E). This level of fibrosis was similar to the IgG-treated AlloWT group (Figure 2A,D). Semi-quantitative grading showed a statistical reduction of parenchymal fibrosis (Figure 2G) and airway fibrosis (Figure 2H) in AlloTbet−/−αIL17A compared to the isotype-treated mice. In spite of a trend towards reduced lung collagen content (Figure 2I) as well as BAL hyaluronan (HA) (Figure 2J), these changes were not statistically significant.
Figure 2. Treatment with anti-IL-17A antibody reduces airway and parenchymal fibrosis in mice that received allogeneic HCT with Tbet−/− cells:
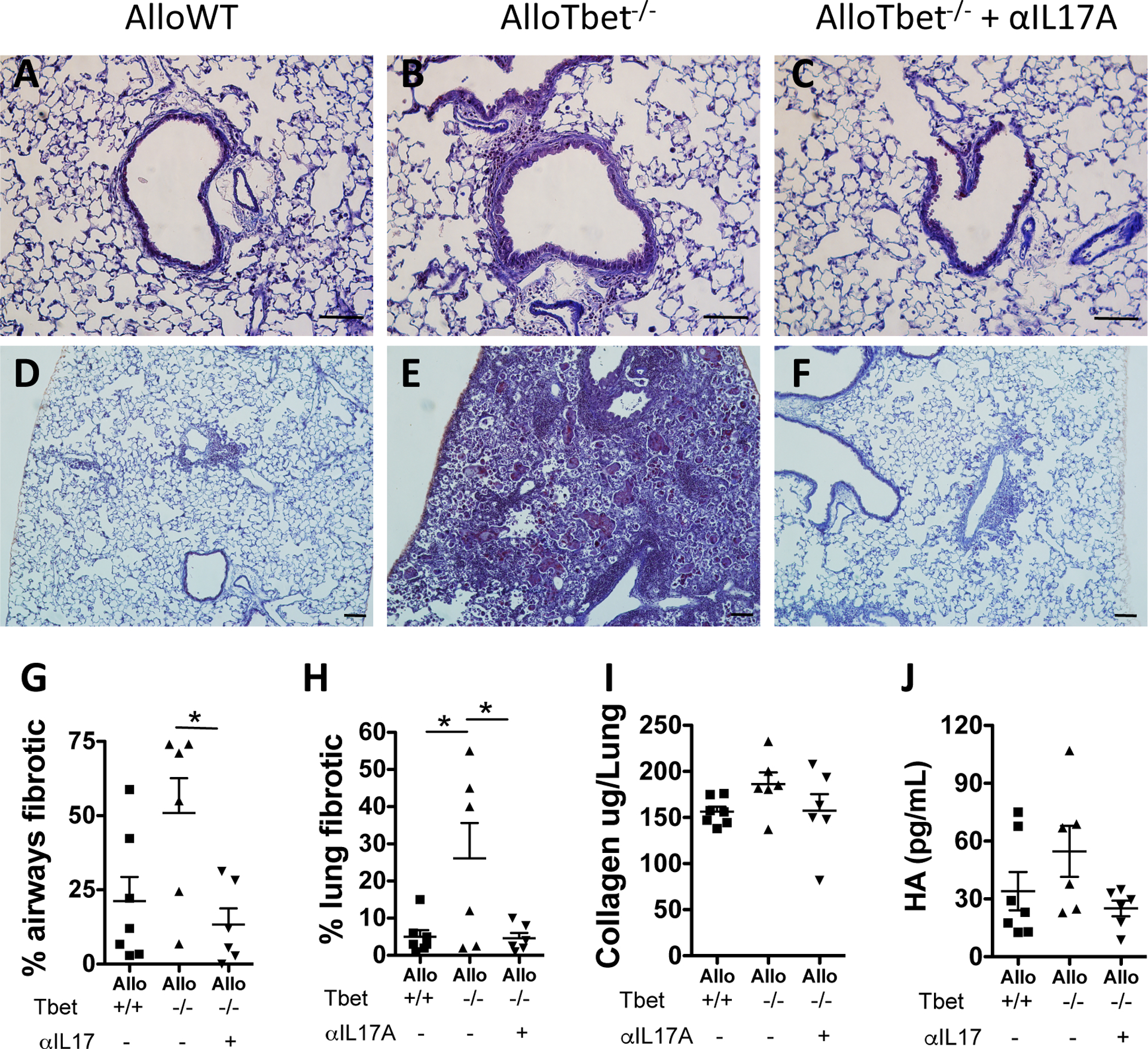
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) were treated with anti-IL-17A antibody (n=6) or isotype IgG (n=6). Control B10.BR recipient mice received an allogeneic HCT from WT donors (AlloWT) and were treated with isotype IgG (n=7). Anti-IL-17A-treated AlloTbet−/− mice had significantly reduced airway (Figure 2 C,G), and parenchymal (Figure 2F,H) fibrosis compared to the IgG-treated (B,E) and AlloWT (A,D) controls. Representative Masson-Trichrome (A-F) images are shown: 200x (A-C), 100X (D-F) (Scale bar=100μm). Collagen lung content (I) and hyaluronan (HA) in the BAL (J) were not significantly changed in this experiment. Quantitative data represent the average +SEM and * = p<0.05. Data have been replicated in 2 independent experiments.
Treatment with anti-IL-17A antibody decreases pulmonary neutrophils and neutrophil chemoattractants in AlloTbet−/− mice:
As IL-17A is known to mediate neutrophilic inflammation in other disease states, we were interested in assessing neutrophils and non-IL-17A neutrophil chemoattractants in our model. Treatment with anti-IL-17A in AlloTbet−/− mice, compared to isotype, led to reduced total cell numbers (Figure 3B) and neutrophil numbers (Figure 3D) in the BAL. The percentage of neutrophils among all BAL leukocytes was, however, not significantly different between anti-IL-17A-treated and isotype-treated AlloTbet−/− (Figure 3A,C). The reduction of neutrophil numbers in lungs of AlloTbet−/−αIL17A mice was associated with decreased levels of CXCL5, CXCL1, IL-6, MIP-1a, and MIP-1b (Figure 3E), but no significant changes in G-CSF and GM-CSF (Figure 3E).
Figure 3. Treatment with anti-IL-17A antibody decreases neutrophils and pro-inflammatory cytokines in mice that received allogeneic HCT with Tbet−/− cells:
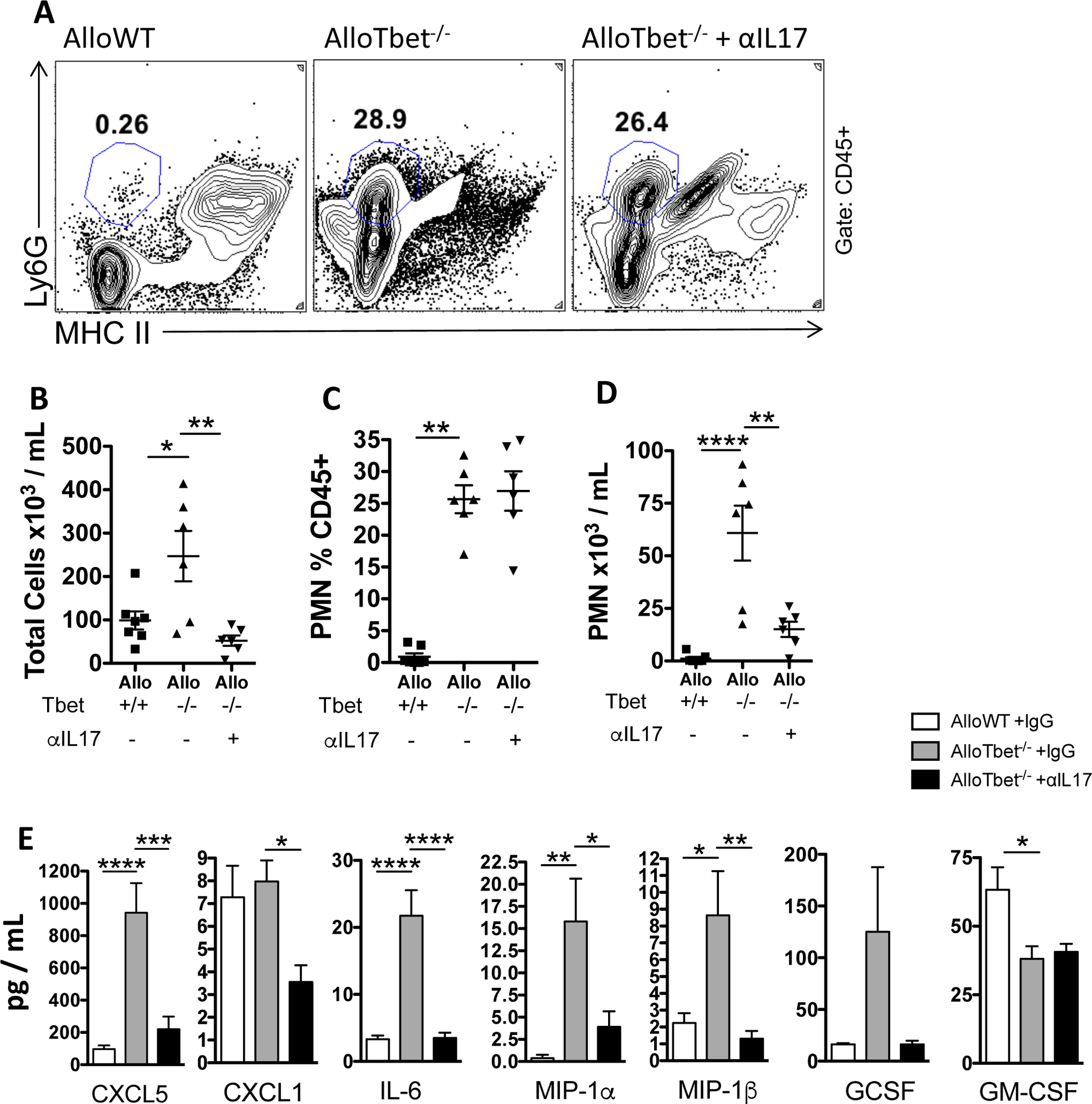
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) were treated with anti-IL-17A antibody (n=6) or isotype IgG (n=6). Control B10.BR recipient mice received an allogeneic HCT from WT donors (AlloWT) and were treated with isotype IgG (n=7). Anti-IL-17A-treated AlloTbet−/− mice showed no change in neutrophil frequency among all leukocytes (A,C) but did have a significant reduction in neutrophil numbers in the BAL (D), along with a significant reduction in overall leukocytes (B), compared to isotype-treated controls. Anti-IL-17A-treated AlloTbet−/− mice had reduced levels of CXCL5, CXCL1, IL-6, MIP-1a, MIP-1b but no significant change in G-CSF and GM-CSF in the BAL, compared to isotype-treated controls (E). Quantitative data represent the average +SEM and * = p<0.05; ** = p<0.005; *** = p<0.0005; **** = p<0.00005. Data have been replicated in 2 independent experiments.
Anti-IL-17A treatment in AlloTbet−/− mice results in reduction in inflammatory monocytes and macrophages as well as CCL2:
The lung fibrosis phenotype of AlloTbet−/− mice was associated with the presence of abundant large macrophages within airspaces (Figure 4B), which were not seen in AlloWT mice (Figure 4A). Treatment with anti-IL-17A decreased such macrophages within lung histology specimens (Figure 4C). Flow cytometric assessment of BAL cells showed that Gr1+CD11b+ macrophages (Figure 4D,E,F) and Gr1+ monocytes (Figure 4D,G,H) were significantly more frequent and abundant in AlloTbet−/− lungs compared to AlloWT and treatment with anti-IL-17A reduced both their percentages and their numbers (Figure 4D–H). Anti-IL-17A treatment in AlloTbet−/− mice reduced levels of CCL2 (Figure 4I). Levels of IL-1α, IL-12(p40), and TNF-α were not significantly changed by IL-17A antibody treatment (Figure 4I).
Figure 4. Treatment with anti-IL-17A antibody decreases inflammatory monocytes and macrophages as well as CCL2 in mice that received allogeneic HCT with Tbet−/− cells:
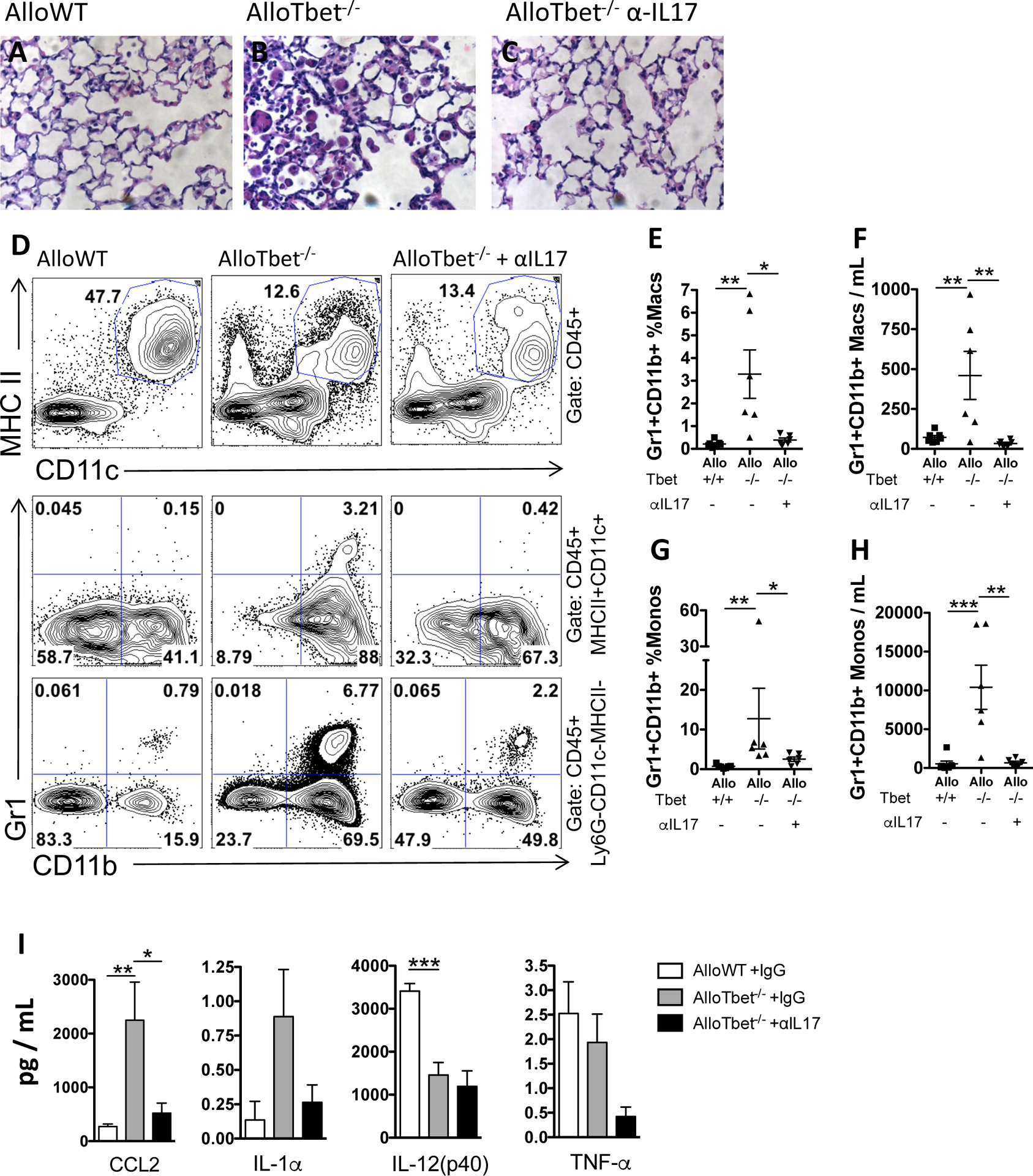
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) were treated with anti-IL-17A antibody (n=6) or isotype IgG (n=6). Control B10.BR recipient mice received an allogeneic HCT from WT donors (AlloWT) and were treated with isotype IgG (n=7). AlloTbet−/− mice showed increased parenchymal macrophages on H&E stained lung histology sections (B) compared to AlloWT mice (A). Anti-IL-17A-treatment reduced the parenchymal macrophages in AlloTbet−/− mice (C). Flow cytometric gating in (D) shows CD11c+MHCII+ macrophages (this gate also includes a small proportion of dendritic cells) among all CD45+ leukocytes (first row), Gr1+CD11b+ inflammatory macrophages among all CD45+CD11c+MHCII+ macrophages (second row), and Gr1+ inflammatory monocytes among all CD45+Ly6G−CD11c−MHCII−CD11b+ monocytes (third row). Gr1+CD11b+ macrophages (D,E,F) and Gr1+ monocytes (D,G,H) were increased in the BAL of AlloTbet mice but significantly reduced by anti-IL-17A treatment, both in terms of frequency (D,E,G) as well as total numbers (F,H). BAL levels of CCL2 were significantly reduced in IL-17A-treated AlloTbet mice while levels of IL-1a, IL-17(p40), and TNF-a were not significantly changed (I). Quantitative data represent the average +SEM and * = p<0.05; ** = p<0.005; *** = p<0.0005; **** = p<0.00005. Data have been replicated in 2 independent experiments.
IL-17A+ CD4+ T cells are reduced in lungs of AlloTbet−/− mice treated with anti-IL-17A antibody with minimal changes in other T cells or T cell-related cytokines:
T cells were quantified in the BAL of AlloWT mice and AlloTbet−/− mice treated with anti-IL-17A or isotype injections. Percentages and total numbers of CD4+ (Figure 5C) or CD8+ (Figure 5D) T cells were not significantly changed by anti-IL-17A, although a trend towards reduced total T cells was seen (Figure 5B). Treatment with anti-IL-17A reduced the percentage and number of CD4+ T cells that expressed IL-17A (Figure 5A,E,G). IL-17A-producing CD8+ T cells (Figure 5A,F,H) and IFN-γ-producing CD4+ (Figure 5A,I) or CD8+ (Figure 5A,J) T cells were not significantly affected by anti-IL17A treatment. BAL levels of cytokines commonly produced by T cells, including IFN-γ, IL-5, IL-9, IL-13, and IL-10 did not change with anti-IL-17A administration (Figure 5K). The T cell chemotactic protein CCL5 also did not change with anti-IL-17A treatment (Figure 5K). Treatment with anti-IL-17A did decrease the BAL concentration of IL-4 in AlloTbet−/− mice (Figure 5K). IL-17A levels remained elevated in AlloTbet−/−αIL17A mice (Figure 5K); this finding was confirmed with a specific IL-17A ELISA (Supplemental Figure 2A). In contrast, IL-17a transcripts were not elevated in the lung tissue of AlloTbet−/−αIL17A mice compared to isotype-treated controls (Supplemental Figure 2B).
Figure 5. Treatment with anti-IL-17A antibody decreases IL-17A+CD4+ T cells in mice that received allogeneic HCT with Tbet−/− cells:
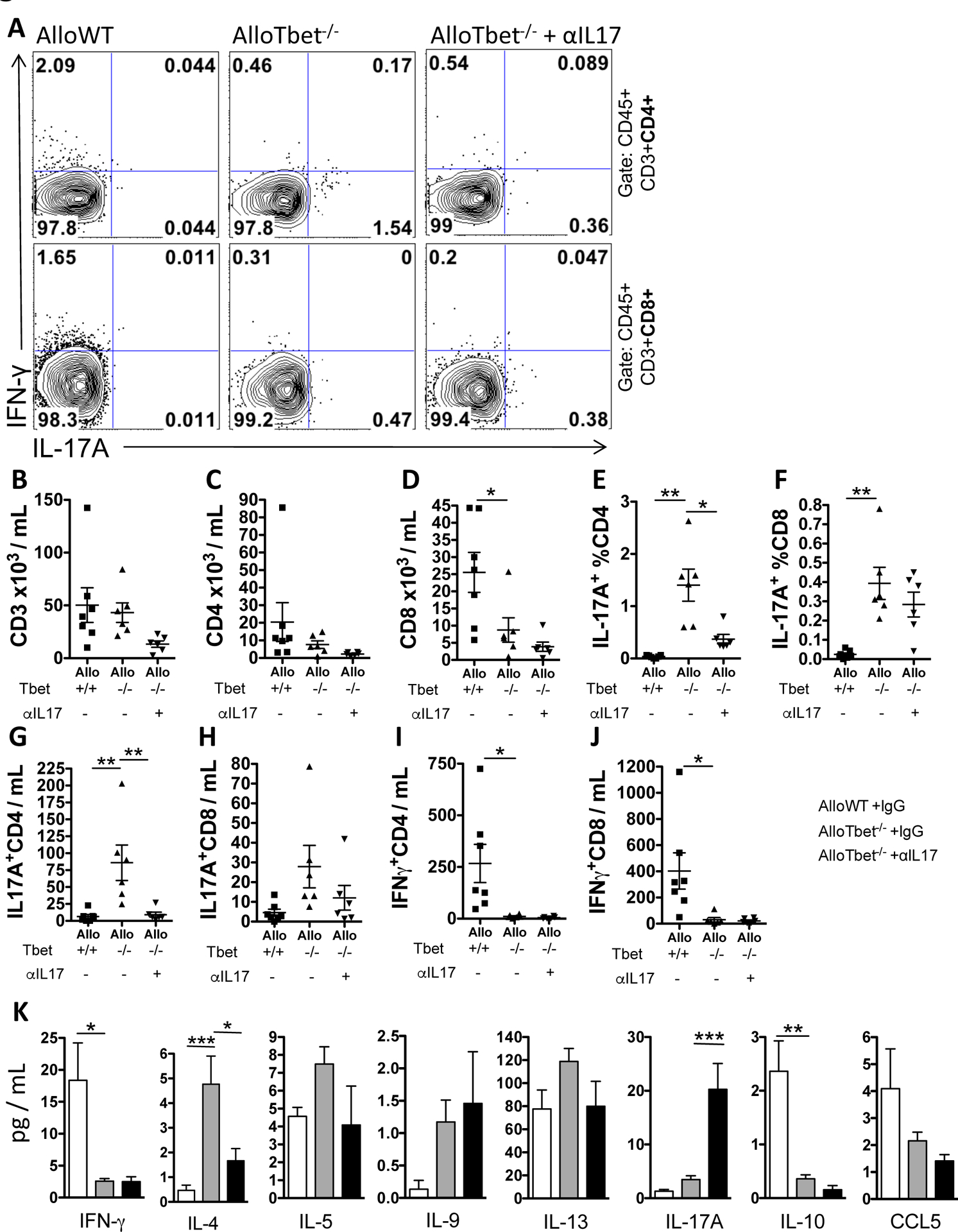
B10.BR recipient mice that received an allogeneic HCT from Tbet−/− donors (AlloTbet−/−) were treated with anti-IL-17A antibody (n=6) or isotype IgG (n=6). Control B10.BR recipient mice received an allogeneic HCT from WT donors (AlloWT) and were treated with isotype IgG (n=7). Anti-IL-17A-treated AlloTbet−/− mice showed reduced IL-17A+CD4+ T cells bot by frequency (A) and total numbers in the BAL (E). There were no significant differences in total T cells (B), CD4+ T cells (C), CD8+ T cells (D), IL-17A+CD8+ T cells (G), or IFN-γ+ T cells (F,H). T cell-related cytokines were measured in the BAL (I). Anti-IL-17A reduced levels of IL-4 and increased levels of IL-17A in the BAL of AlloTbet mice. There were no significant changes in IFN-γ, IL-5, IL-9, Il-13, IL-10, or CCL5 between anti-IL-17A-treated AlloTbet and isotype-treated mice. Quantitative data represent the average +SEM and * = p<0.05; ** = p<0.005; *** = p<0.0005. Data have been replicated in 2 independent experiments.
DISCUSSION
Using a model of cpGVHD with alloimmune-dependent lung fibrosis, we demonstrate that treatment with anti-IL-17A antibody modestly decreases airway and parenchymal lung fibrosis in conjunction with a marked reduction in neutrophils, neutrophil chemoattractants, inflammatory macrophages and monocytes, and Th17 cells.
Our model of cpGVHD employs a low-dose HCT, resulting in very little systemic or lung GVHD at baseline, as shown in our prior studies18,25,26 and as seen in the AlloWT control group in this study. In the context of this low-dose HCT, transfer of Tbet−/− donor cells leads to increased Th17 polarization with augmented IL-17A production and airway fibrosis, as outlined in our prior publication.17 In this present study, we broadened the analysis of the cpGVHD phenotype with semi-quantitative grading of the fibrotic pathology and a more robust evaluation of the parenchymal fibrosis. Airway fibrosis in this model manifests mainly as peribronchial and peribronchiolar collagen deposition, features commonly seen in human cpGVHD, without however replicating the airway lumen obliteration seen in human OB. While airway fibrosis, including OB, has been noted in HCT recipients since the 1980s,27 lung parenchymal fibrosis has been only recently recognized as an important complication of HCT.28 Our mouse model thus recapitulates two key histologic hallmarks of human cpGVHD. The model is also reminiscent of lung transplant recipients who primarily develop peri-airway fibrosis and OB as a manifestation of chronic lung allograft dysfunction, but may also present with a more aggressive parenchymal fibrosis phenotype.29–33 Additionally, the fibrotic phenotype is associated with BAL accumulation of hyaluronan (HA), a component of the extracellular matrix, highlighting HA as a marker of fibrosis, consistent with HA accumulation in fibrotic lesions of chronic skin GVHD34 and with our previous study of BAL HA in lung transplant recipients with OB.35
Several studies have demonstrated the importance of Th17 and IL-17A in acute GVHD,36–38 including acute lung GVHD,39,40 and chronic skin and liver GVHD.5–7 The importance of Th17 cells has been further implied in a study showing reduced cpGVHD pathology in the setting of STAT3 inhibition.5 However, IL-17A, the major product of Th17 cells, has not been specifically studied in cpGVHD. For the first time, we show that blockade of IL-17A reduces airway and parenchymal lung fibrosis in a murine model of cpGVHD.
While blinded pathology scoring showed a significant reduction in the fibrotic phenotype, we detected only a trend towards reduced lung collagen and BAL HA contents after IL-17A blockade in our model. This discrepancy may reflect regional distribution of the disease, environmental variations between experiments, variability of the phenotype in transplanted animals, or simply the fact that the overall fibrosis reduction was modest. It is possible that higher doses of anti-IL-17A, more frequent administration, or combination with another blocking agent, may have had a stronger effect on the fibrotic phenotype.
The effect of IL-17A on alloimmune-dependent lung fibrosis seen in this study is consistent with studies of chronic allograft rejection after lung transplantation. Fan et al. showed that concurrent neutralization of both IL-17A and IL-17F reduced airway fibrosis after minor-mismatched mouse orthotopic lung transplantation.11 Subsequent studies using the same model revealed that both γδ and αβ T cells contribute to IL-17A production13,14 and treatment with anti-IL-17A12,15,41 or Halofuginone,41 a Th17 differentiation inhibitor, decreased fibrosis. Based on this combined data, we postulate that IL-17A may have an effect on fibroblasts themselves, stimulating myofibroblast differentiation or activation,42,43 or may augment fibrosis through increased inflammation and stimulation of downstream pro-fibrotic factors. Our study thus expands on the published literature by showing that IL-17A blockade reduces fibrosis in a cpGVHD model. Additionally, in contrast to the minor MHC mismatch used in prior lung transplant studies, our model has the advantage of assessing alloimmune-mediated lung fibrosis in the context of a major mismatch, which may have greater relevance to the clinical setting.
The cpGVHD in our mouse model is associated with increased lung neutrophils, reminiscent of pulmonary neutrophil accumulation in lung transplant recipients with OB.44 Based on our study, IL-17A mediates neutrophil recruitment to the lungs, likely through induction of pro-inflammatory cytokines. IL-17A blockade reduced known neutrophil chemoattractants CXCL5 and CXCL1, which are expressed by macrophages and airway epithelial cells in response to IL-17A.45 IL-17A neutralization further decreased IL-6, which mediates release of neutrophils from the bone marrow, and the neutrophil activating cytokines MIP-1α and MIP-1β.4 Interestingly, the profound neutrophil reduction in our IL-17A blockade experiments was associated with incomplete reduction in fibrosis. This is consistent with published data showing that neutrophil depletion does not decrease IL-17 levels or fibrosis in a model of γ-herpesvirus-induced pneumonitis.46 Therefore, in the specific case of alloimmune fibrosis, it remains unclear whether neutrophils are simply bystanders or whether they may have a role in the actual fibrosis pathogenesis.
IL-17A has been shown to affect activation of monocytes and macrophages. Th17 cells are also strong inducers of Th1 polarizing monocyte-derived dendritic cells.47 In our model of cpGVHD, we found that IL-17A neutralization decreased numbers of activated monocytes and macrophages. This reduction may have been mediated by decreased CCL2, a potent monocyte chemoattractant,18,23 known to increase after IL-17A stimulation.4 The effect of IL-17A on monocyte recruitment highlights its capacity to further augment innate immune responses and antigen presentation in an allogeneic milieu. Additionally, the decrease in activated macrophages seen after IL-17A blockade may account for the reduced neutrophil chemoattractants that these cells produce (G-CSF, GM-CSF, CXCL1, MIP-1α and MIP-1β).4 Inflammatory monocytes and macrophages have also been implicated as regulators in tissue repair and fibrosis through production of pro-fibrotic factors such as TNF-α.48 We found an accumulation of large macrophages in the fibrotic areas of AlloTbet−/− mice and postulate that these cells may participate in tissue remodeling in cpGVHD. It is notable, nevertheless, that several inflammatory components, such as CXCL5, MIP-1α, and CCL2, remain above the AlloWT baseline. It is possible that the combination of these, along with potentially other concurrent immune processes, continue to drive the residual fibrosis seen in the AlloTbet−/− mice treated with anti-IL-17A.
Anti-IL-17A treatment had minimal effects on overall lung T cells of AlloTbet−/− mice. Nevertheless, IL-17A neutralization reduced CD4+IL-17A+ Th17 cells, possibly through reduction of downstream pro-inflammatory cytokines, which contribute to the known auto-feedback loop that augments Th17 cells.49 As expected, IFN-γ was nearly undetectable in both AlloTbet−/− groups, with and without anti-IL-17A. Furthermore, IL-17A inhibition reduced IL-4 without significant effects on IL-5, IL-9, or IL-13. We wonder whether Th2 responses act synergistically with Th17 to augment cpGVHD fibrosis. The persistent Th2 cytokine elevation may be responsible for the incomplete fibrosis inhibition in the setting of IL-17A blockade. IL-13, in particular, has been previously implicated in pathogenesis of OB in a tracheal transplant model.50
IL-17A protein levels in the BAL actually increased in the setting of IL-17A blockade. This was in contrast to decreased pulmonary intracellular IL-17A and lung tissue IL-17A transcript in AlloTbet−/− mice treated with anti-IL-17A. This suggests that the anti-IL-17A treatment may have decreased clearance and prolonged the half-life of the already secreted IL-17A, while neutralizing its effects. This phenomenon has been previously described in the case of anti-IL-251 and anti-TNFα neutralizing antibodies.52–54
In conclusion, we show for the first time that specific IL-17A blockade in a model of cpGVHD reduces airway and lung parenchymal fibrosis, neutrophils, activated monocytes, macrophages, and related cytokines. This study adds to the literature that identifies Th17 and IL-17A as important contributors to alloimmune-dependent lung fibrosis and highlights the potential role for neutrophils, monocytes, and macrophages as potential mediators of this fibrosis.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Stephen Juvet for helpful input regarding figures and manuscript preparation.
Funding:
Funding was provided by NIH / NHLBI 1P50-HL084917-01 (SCCOR) (to SMP, TM, KG), NIH 1 K24 HL91140-01A2 (to SMP), Lung Transplant Foundation (to SMP), ISHLT Norman E. Shumway Career Development Award (to TM), and Steadman award (to WCM)
ABBREVIATIONS
- AlloTbet−/−
Allogeneic HCT with Tbet-knockout donors
- AlloWT
Allogeneic HCT from wild-type donors
- αIL17A
Anti-IL-17A antibody
- BAL
Bronchoalveolar lavage
- B10.BR
B10.BR-H2kH2-T18a/SgSnJ
- cpGVHD
Chronic pulmonary graft-versus-host disease
- GVHD
Graft-versus-host disease
- HCT
Hematopoietic cell transplant
- HA
Hyaluronan
- OB
Obliterative bronchiolitis
- SynTbet−/−
Syngeneic HCT from Tbet−/− donors
- Th17
T helper 17
- Tbet
T-box expressed in T cells
- Tbet−/−
Tbet-deficient
- WT
Wild-type
Footnotes
Disclosure:
The authors of this manuscript have no conflicts of interest to disclose.
REFERENCES
- 1.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet 2009;373:1550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooke KR, Yanik G. Acute lung injury after allogeneic stem cell transplantation: is the lung a target of acute graft-versus-host disease? Bone Marrow Transplant 2004;34:753–65. [DOI] [PubMed] [Google Scholar]
- 3.Hodge G, Hodge S, Yeo A, et al. BOS Is Associated With Increased Cytotoxic Proinflammatory CD8 T, NKT-Like, and NK Cells in the Small Airways. Transplantation 2017;101:2469–76. [DOI] [PubMed] [Google Scholar]
- 4.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity 2004;21:467–76. [DOI] [PubMed] [Google Scholar]
- 5.Flynn R, Paz K, Du J, et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016;127:2144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serody JS, Hill GR. The IL-17 differentiation pathway and its role in transplant outcome. Biol Blood Marrow Transplant 2012;18:S56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malard F, Bossard C, Brissot E, et al. Increased Th17/Treg ratio in chronic liver GVHD. Bone Marrow Transplant 2014;49:539–44. [DOI] [PubMed] [Google Scholar]
- 8.Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. The Journal of clinical investigation 2007;117:3498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanaudenaerde BM, De Vleeschauwer SI, Vos R, et al. The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2008;8:1911–20. [DOI] [PubMed] [Google Scholar]
- 10.Lemaitre PH, Vokaer B, Charbonnier LM, et al. IL-17A mediates early post-transplant lesions after heterotopic trachea allotransplantation in Mice. PloS one 2013;8:e70236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan L, Benson HL, Vittal R, et al. Neutralizing IL-17 prevents obliterative bronchiolitis in murine orthotopic lung transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2011;11:911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta PK, Wagner SR, Wu Q, Shilling RA. IL-17A Blockade Attenuates Obliterative Bronchiolitis and IFNgamma Cellular Immune Response in Lung Allografts. American journal of respiratory cell and molecular biology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Q, Gupta PK, Suzuki H, et al. CD4 T Cells but Not Th17 Cells Are Required for Mouse Lung Transplant Obliterative Bronchiolitis. Am J Transplant 2015;15:1793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta PK, Wagner SR, Wu Q, Shilling RA. Th17 cells are not required for maintenance of IL-17A-producing gammadelta T cells in vivo. Immunol Cell Biol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang R, Fang H, Chen R, Ochando JC, Ding Y, Xu J. IL-17A Is Critical for CD8+ T Effector Response in Airway Epithelial Injury After Transplantation. Transplantation 2018;102:e483–e93. [DOI] [PubMed] [Google Scholar]
- 16.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 2002;295:338–42. [DOI] [PubMed] [Google Scholar]
- 17.Gowdy KM, Nugent JL, Martinu T, et al. Protective role of T-bet and Th1 cytokines in pulmonary graft-versus-host disease and peribronchiolar fibrosis. American journal of respiratory cell and molecular biology 2012;46:249–56. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 18.Martinu T, Gowdy KM, Nugent JL, et al. Role of C-C motif ligand 2 and C-C motif receptor 2 in murine pulmonary graft-versus-host disease after lipopolysaccharide inhalations. American journal of respiratory cell and molecular biology 2014;51:810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol 2009;182:309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiriveedhi V, Takenaka M, Ramachandran S, et al. T regulatory cells play a significant role in modulating MHC class I antibody-induced obliterative airway disease. Am J Transplant 2012;12:2663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasquevich KA, Ibanez AE, Coria LM, et al. An oral vaccine based on U-Omp19 induces protection against B. abortus mucosal challenge by inducing an adaptive IL-17 immune response in mice. PloS one 2011;6:e16203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamson IY, Bowden DH. The pathogenesis of bleomycin-induced pulmonary fibrosis in mice. The American journal of pathology 1974;77:185–97. [PMC free article] [PubMed] [Google Scholar]
- 23.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 2008;180:2562–72. [DOI] [PubMed] [Google Scholar]
- 24.Nakano H, Lin KL, Yanagita M, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol 2009;10:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinu T, Kinnier CV, Gowdy KM, et al. Innate immune activation potentiates alloimmune lung disease independent of chemokine (C-X-C motif) receptor 3. J Heart Lung Transplant 2011;30:717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garantziotis S, Palmer SM, Snyder LD, et al. Alloimmune lung injury induced by local innate immune activation through inhaled lipopolysaccharide. Transplantation 2007;84:1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ralph DD, Springmeyer SC, Sullivan KM, Hackman RC, Storb R, Thomas ED. Rapidly progressive air-flow obstruction in marrow transplant recipients. Possible association between obliterative bronchiolitis and chronic graft-versus-host disease. Am Rev Respir Dis 1984;129:641–4. [PubMed] [Google Scholar]
- 28.von der Thusen JH, Hansell DM, Tominaga M, et al. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol 2011;24:1633–9. [DOI] [PubMed] [Google Scholar]
- 29.Mariani F, Gatti B, Rocca A, et al. Pleuroparenchymal fibroelastosis: the prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn Interv Radiol 2016;22:400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato M, Waddell TK, Wagnetz U, et al. Restrictive allograft syndrome (RAS): a novel form of chronic lung allograft dysfunction. J Heart Lung Transplant 2011;30:735–42. [DOI] [PubMed] [Google Scholar]
- 31.Ofek E, Sato M, Saito T, et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Mod Pathol 2013;26:350–6. [DOI] [PubMed] [Google Scholar]
- 32.Gauthier JM, Ruiz-Perez D, Li W, et al. Diagnosis, Pathophysiology and Experimental Models of Chronic Lung Allograft Rejection. Transplantation 2018;102:1459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada Y, Windirsch K, Dubs L, et al. Chronic Airway Fibrosis in Orthotopic Mouse Lung Transplantation Models-An Experimental Reappraisal. Transplantation 2018;102:e49–e58. [DOI] [PubMed] [Google Scholar]
- 34.Fleming JN, Shulman HM, Nash RA, et al. Cutaneous chronic graft-versus-host disease does not have the abnormal endothelial phenotype or vascular rarefaction characteristic of systemic sclerosis. PloS one 2009;4:e6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd JL, Wang X, Sugimoto S, et al. Hyaluronan contributes to bronchiolitis obliterans syndrome and stimulates lung allograft rejection through activation of innate immunity. Am J Respir Crit Care Med 2014;189:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson JS, Chu Y, Glass JF, Brown SA. Absence of IL-23p19 in donor allogeneic cells reduces mortality from acute GVHD. Bone Marrow Transplant 2010;45:712–22. [DOI] [PubMed] [Google Scholar]
- 37.Iclozan C, Yu Y, Liu C, et al. T helper17 cells are sufficient but not necessary to induce acute graft-versus-host disease. Biol Blood Marrow Transplant 2010;16:170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng H, Tian J, Li Z, et al. TH17 Cells Are Critical for Skin-Specific Pathological Injury in Acute Graft-Versus-Host Disease. Transplant Proc 2012;44:1412–8. [DOI] [PubMed] [Google Scholar]
- 39.Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In vitro-differentiated TH17 cells mediate lethal acute graft-versus-host disease with severe cutaneous and pulmonary pathologic manifestations. Blood 2009;113:1365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uryu H, Hashimoto D, Kato K, et al. alpha-Mannan induces Th17-mediated pulmonary graft-versus-host disease in mice. Blood 2015;125:3014–23. [DOI] [PubMed] [Google Scholar]
- 41.Oishi H, Martinu T, Sato M, et al. Halofuginone treatment reduces interleukin-17A and ameliorates features of chronic lung allograft dysfunction in a mouse orthotopic lung transplant model. J Heart Lung Transplant 2016;35:518–27. [DOI] [PubMed] [Google Scholar]
- 42.Lei L, Zhao C, Qin F, He ZY, Wang X, Zhong XN. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin Exp Rheumatol 2016;34 Suppl 100:14–22. [PubMed] [Google Scholar]
- 43.Zhou X, Loomis-King H, Gurczynski SJ, et al. Bone marrow transplantation alters lung antigen-presenting cells to promote TH17 response and the development of pneumonitis and fibrosis following gammaherpesvirus infection. Mucosal immunology 2016;9:610–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elssner A, Vogelmeier C. The role of neutrophils in the pathogenesis of obliterative bronchiolitis after lung transplantation. Transplant infectious disease : an official journal of the Transplantation Society 2001;3:168–76. [DOI] [PubMed] [Google Scholar]
- 45.Chen K, Eddens T, Trevejo-Nunez G, et al. IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe 2016;20:596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gurczynski SJ, Procario MC, O’Dwyer DN, Wilke CA, Moore BB. Loss of CCR2 signaling alters leukocyte recruitment and exacerbates gamma-herpesvirus-induced pneumonitis and fibrosis following bone marrow transplantation. American journal of physiology 2016;311:L611–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson MG, Alonso MN, Yuan R, et al. Th17 cells induce Th1-polarizing monocyte-derived dendritic cells. J Immunol 2013;191:1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016;44:450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007;448:484–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keane MP, Gomperts BN, Weigt S, et al. IL-13 is pivotal in the fibro-obliterative process of bronchiolitis obliterans syndrome. Journal of immunology 2007;178:511–9. [DOI] [PubMed] [Google Scholar]
- 51.Phelan JD, Orekov T, Finkelman FD. Cutting edge: mechanism of enhancement of in vivo cytokine effects by anti-cytokine monoclonal antibodies. J Immunol 2008;180:44–8. [DOI] [PubMed] [Google Scholar]
- 52.Mann DL, Bozkurt B, Torre-Amione G, Soran OZ, Sivasubramanian N. Effect of the soluble TNF-antagonist etanercept on tumor necrosis factor bioactivity and stability. Clin Transl Sci 2008;1:142–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schulz M, Dotzlaw H, Neeck G. Ankylosing spondylitis and rheumatoid arthritis: serum levels of TNF-alpha and Its soluble receptors during the course of therapy with etanercept and infliximab. Biomed Res Int 2014;2014:675108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madhusudan S, Foster M, Muthuramalingam SR, et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2004;10:6528–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.