

Abstract
Single-cell proteomics can provide unique insights into biological processes by resolving heterogeneity that is obscured by bulk measurements. Gains in the overall sensitivity and proteome coverage through improvements in sample processing and analysis increase the information content obtained from each cell, particularly for less abundant proteins. Here we report on improved single-cell proteome coverage through the combination of the previously developed nanoPOTS platform with further miniaturization of liquid chromatography (LC) separations and implementation of an ultrasensitive latest generation mass spectrometer. Following nanoPOTS sample preparation, protein digests from single cells were separated using a 20 μm i.d. in-housepacked nanoLC column. Separated peptides were ionized using an etched fused-silica emitter capable of stable operation at the ~20 nL/min flow rate provided by the LC separation. Ultrasensitive LC–MS analysis was achieved using the Orbitrap Eclipse Tribrid mass spectrometer. An average of 362 protein groups were identified by tandem mass spectrometry (MS/MS) from single HeLa cells, and 874 protein groups were identified using the Match Between Runs feature of MaxQuant. This represents an >70% increase in label-free proteome coverage for single cells relative to previous efforts using larger bore (30 μm i.d.) LC columns coupled to a previous-generation Orbitrap Fusion Lumos mass spectrometer.
Graphical Abstract
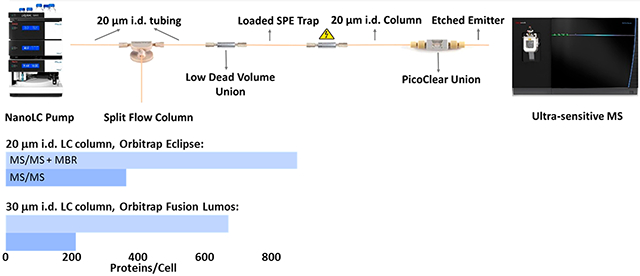
Measuring global protein expression within single cells promises to advance our understanding of the roles of various cell types contributing to normal and disease processes.1-5 However, extending proteomic analyses to single cells poses an enormous analytical challenge, as typical single mammalian cells range from 10 to 20 μm in diameter6 and contain just a few hundred picograms or less of total protein.7,8 Various methods have been developed to measure proteins within single cells based on, e.g., mass cytometry,9 immunofluorescence,10 and single-cell Western blotting,11 but these methods are limited to a small number of preselected target proteins for a given sample. In contrast, mass spectrometry (MS)-based proteomics enables antibody-free quantification of thousands of proteins12-16 but has historically been limited to the analysis of bulk samples. However, given the strong motivation to increase proteome coverage and reduce sample input requirements, consistent improvements have been made over the past 2 decades to increase the efficiency of separations,17,18 ionization,19 and ion handling within the mass spectrometer.20 As a result of these advances, the ability to analyze samples containing protein amounts at or near the level of single cells has been demonstrated.18,21,22 Still, until recently, global proteome profiling had not been demonstrated for single cells due primarily to limitations in sample processing and delivery of single-cell samples to the analytical platform.
Two recent developments have combined to bridge the remaining gap to enable profiling of hundreds of proteins from single mammalian cells. New approaches have been developed to dramatically reduce the analyte losses incurred during sample preparation such that the amount of required biological starting material can more closely match the detection limits of the capillary electrophoresis (CE) or liquid chromatography–mass spectrometry (LC–MS) analysis.22-24 To this end, we recently developed nanoPOTS (nanodroplet processing in one pot for trace samples),25 which combines nanopipetting using a robotic nanoliter liquid handling platform with microfabricated glass nanowell chips to reduce total surface exposure to <1 mm2 and total processing volumes to ~200 nL. Evaporation is minimized in the platform through a combination of a humidified chamber and a reversibly sealing cover. NanoPOTS has enabled highly reproducible label-free profiling of ~670 protein groups [based on both tandem mass spectrometry (MS/MS) identifications and transferred identifications from MS1-level feature matching] from single HeLa cells26 and has been applied to a variety of nanoscale27-31 and single-cell studies.32,33 Concurrently, Budnik et al. developed SCoPE-MS (single-cell proteomics by mass spectrometry), wherein isobarically labeled single cells were analyzed along with a labeled carrier sample containing hundreds of cells.34 The combined signal from the carrier and single cells enabled peptide identification, and peptides from each single cell were differentiated based on their corresponding reporter ion intensities. It should be noted that isobaric labeling strategies and efforts to miniaturize sample preparation are not mutually exclusive. For example, Specht et al.35 have improved the SCoPE-MS approach through the reduction of processing volumes to a few microliters before pooling samples for analysis. Similarly, we have implemented tandem mass tag (TMT) labeling within the nanoPOTS platform,36 and with the added sensitivity that nanoPOTS provides, we demonstrated the ability to differentiate cell types even when no carrier channel was used.
As with improvements in sample processing efficiency, both label-free and multiplexed workflows will benefit from added analytical sensitivity through further advances in separation and MS. To this end, here we have evaluated the coverage achievable for single cells by combining nanoPOTS with ultranarrow-bore (20 μm i.d.) packed-column LC separations and the latest generation Orbitrap Eclipse Tribrid MS (Eclipse MS), which offers improved sensitivity due to improvements in vacuum management within the instrument and better ion optics, such as a new segmented quadrupole. As a result of these improvements, we have achieved a cumulative gain in proteome coverage of >70% based on MS/MS identifications (without MS1-level feature matching) to 362 protein groups versus 211 reported previously.26 When matched features from the match between runs (MBR) algorithm37 were included, nearly 900 protein groups were identified on average from each HeLa cell. This is the deepest proteome coverage reported to date for single mammalian cells using a label-free workflow, and it points to additional gains to be realized through further miniaturization of the separation and improvements in ion transmission in the mass spectrometer.
EXPERIMENTAL SECTION
Material and Sample Preparation.
Dithiothreitol (DTT) and iodoacetamide (IAA) were products of Thermo Fisher Scientific (Waltham, MA) and were freshly prepared in 50 mM ammonium bicarbonate buffer before use. Water with 0.1% formic acid (v/v, Optima LC/MS grade), which served as mobile phase A, acetonitrile with 0.1% formic acid (v/v, Optima LC/MS grade), which served as mobile phase B, and Pierce HeLa protein digest standard were also from Thermo Fisher. MS-grade trypsin and Lys-C were from Promega (Madison, WI). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Cell Culture.
HeLa cells (ATCC, Manassas, VA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; VWR, Radnor, PA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were incubated at 37 °C in an aerobic environment with 5% CO2 and split every 2 days. Cultured HeLa cells were collected into a 10 mL centrifuge tube and centrifuged at 200g for 5 min to gently pellet the cells. After washing with PBS buffer three times, the cells were resuspended in 1 mL of PBS buffer, and then diluted to a concentration of ~10 cells/μL.
Microchip Fabrication and the NanoPOTS System.
The nanoPOTS sample processing platform was used as described previously.25,26 Briefly, an in-house-built robotic nanopipetting system with a pulled fused-silica capillary serving as a pipet tip was used to dispense reagents into 1.2 mm diameter nanowells that were fabricated on a glass microchip. After loading samples into nanowells, each subsequent step involved reagent dispensing and reaction incubation until the trypsin-digested peptides were aspirated from the nanowell into a fused-silica capillary using the same platform. Samples were then loaded to a trap column for further LC–MS analysis. One key modification was made to the nanoPOTS platform: the glass slide holder that immobilized nanoPOTS chips onto the robotic nanopipetting platform during dispensing operations was mounted onto an SVM340 inverted video microscope (LabSmith, Livermore, CA), which enabled single cells to be identified and selected for analysis as described below.
NanoPOTS Cell Isolation and Sample Preparation.
A 200 μL aliquot of cell suspension was dispensed onto an unpatterned, BSA-coated glass slide, mounted onto the slide holder in the nanopipetting robot, and imaged using the LabSmith microscope. Single cells were aspirated from the glass slide surface along with ~6 nL of PBS using a 360 μm o.d./200 μm i.d. capillary (Polymicro Technologies, Phoenix, AZ) with an ~30 μm i.d. pulled tip, and then dispensed into a nanowell on the nanoPOTS chip located in an adjacent position on the slide holder for processing (Figure 1A). After the single cells were isolated into nanowells, they were processed for proteomic analysis as described previously.25 Digested peptide samples were collected from nanowells and stored in a section of fused-silica capillary (200 μm i.d./360 mm o.d.), with both ends sealed with Parafilm, and stored at 4 °C until further use. For blank samples, an equivalent volume of cell-free PBS buffer from the cell suspension supernatant was collected and dispensed into nanowells and processed following the same protocol. For multicell samples (20 and 100 cells), a specific volume of cell suspension was dispensed into the nanowells and cells were counted under a microscope prior to processing using the same procedure as for single cells.
Figure 1.

Single-cell proteomics workflow. (A) A cell suspension is dispensed onto a slide, and a single cell is aspirated from the bulk solution and dispensed into a nanowell for further processing. (B) NanoPOTS-prepared samples are loaded onto an SPE column, separated using a 20 μm i.d. nanoLC column, and detected using the Orbitrap Eclipse MS.
Column Packing and Emitter Connection.
Jupiter 300 C18-bonded 3 μm porous particles (Phenomenex, Torrance, CA) were used as packing media. The 30 μm i.d. columns were packed in PicoFrit self-pack columns with a 10 μm i.d. integrated emitter (New Objective, Woburn, MA). The 20 μm i.d. columns were packed in fused-silica capillaries (Polymicro Technologies, Phoenix, AZ) with homemade sol–gel frits38 on one end. The frits were fabricated using Frit Kit (Next Advance Inc., Troy, NY) as described briefly here. Kasil 1, Kasil 1624, and formamide were mixed in 1:3:1 proportion in a microcentrifuge tube. One end of a 20 μm i.d. capillary was rapidly dipped into the vial and then incubated in an oven at 100 °C for 4 h. After incubation, the capillary was trimmed to leave an ~2 mm frit.
For column packing, an MS-188 Haskel pump (Burbank, CA) was employed to provide the packing pressure. A PEEK tubing sleeve (380 μm i.d., Upchurch Scientific, Oak Harbor, WA) was used to seal the empty fused-silica capillary column in a stainless-steel vessel made from a Swagelok union (Solon, OH), into which the packing particles and a magnetic stir bar were placed. Mobile phase B served as the slurry solvent. The particles were suspended in the slurry solvent using a magnetic stirrer, and then slowly delivered into the capillary by gradually increasing the pressure from 500 to 8000 psi. After filling 60 cm of the column, packing was stopped and the column was conditioned at 8000 psi for ~10 min in an ultrasonic bath, and then left overnight to depressurize. The fritted end of the column was polished using a capillary polishing station (ESI Source Solutions, Woburn, MA) and connected with a homeetched nanoelectrospray ionization (nanoESI) emitter19 (360 μm o.d., 10 μm i.d.) using a PicoClear Union (New Objective, Woburn, MA) (Figure S1). The solid-phase extraction (SPE) trap columns were slurry-packed with Jupiter 300 C18-bonded 3 μm porous particles into a dual fritted 5 cm long fused-silica capillary with 360 μm o.d. and 75 μm i.d.
LC–MS Detection.
Prior to separation, the sample was transferred from the storage capillary to the SPE trap column for desalting by infusing mobile phase A at a flow rate of 1 μL/min for 10 min using an UltiMate 3000 RSLCnano pump (Thermo Fisher). The SPE column was then connected to the LC column with a zero-dead-volume union (Valco, Houston, TX). The LC separation flow rate was ~50 nL/min for 30 μm i.d. columns and ~20 nL/min for 20 μm i.d. columns, which were split from 250 nL/min programmed flow provided by the LC pump, with a pressure of ~300 bar at the beginning of the runs for both columns. A home-packed 60 cm long, 75 μm i.d. column with 3 μm packing material served as the split column. A linear 100 min gradient of 8–22% mobile phase B was used for separation. An additional 20 min gradient of 22–45% mobile phase B was used to elute hydrophobic peptides, and the gradient was then ramped to 90% mobile phase B over 5 min and held for 5 min to wash the column. Finally, the gradient was ramped to 2% mobile phase A over 5 min and held for 15 min to re-equilibrate the column.
Orbitrap Fusion Lumos and Eclipse mass spectrometers were employed for data acquisition. Separated peptides were electrosprayed using a potential of 2.0 kV applied at the Nanospray Flex™ source, and the ion transfer tube was set at 150 °C for desolvation. The ion funnel rf level was 30, the MS1 Orbitrap resolution was set at 120 000 (at m/z 200), and the MS1 AGC target and the maximum injection time were set at 1 × 106 and 246 ms. The MS2 Orbitrap resolution as well as AGC target were varied depending on the expected total amount of protein within the samples to be analyzed, as described below. Data-dependent acquisition mode was used to trigger precursor isolation and sequencing. Precursor ions with charges of +2 to +6 were isolated for MS2 sequencing. The MS2 isolation window was 1.6 Da, the AGC target was set to 1 × 105, the dynamic exclusion time was set at 50 s, and a mass tolerance of ±10 ppm was utilized. For digests from 100 and 20 prepared HeLa cells, as well as 2 and 0.5 ng aliquots of commercial HeLa digest, the MS2 resolution and maximum injection time were set to 120 000 (at m/z 200) and 246 ms and the signal intensity threshold was set to 2 × 104. For single cells and 0.2 ng QC HeLa digest samples, the MS2 resolution and maximum injection time were set to 240 000 and 500 ms and the signal intensity threshold was set to 8 × 103. The long injection times enabled the accumulation of sufficient peptide ions for a measurable signal from these trace samples. For the Orbitrap Fusion Lumos MS, precursors were fragmented by higher energy collision-induced dissociation (HCD) with a normalized collision energy of 30%; for the Eclipse MS, the HCD normalized collision energy was 35%.
Data Analysis.
All raw files were processed using MaxQuant (version 1.6.3.3) for feature detection, database searching, and protein/peptide quantification. MS/MS spectra were searched against the UniProtKB/Swiss-Prot human database (downloaded on October 26, 2018, containing 20 397 reviewed sequences). N-Terminal protein acetylation and methionine oxidation were selected as variable modifications. Carbamidomethylation of cysteine residues was set as a fixed modification. The peptide mass tolerances of the first search and main search (recalibrated) were <30 and 5 ppm, respectively. The minimum peptide length was six amino acids, and the maximum peptide mass was 4600 Da. Only two missed cleavages were allowed for each peptide. The second peptide search was activated to identify coeluting and cofragmented peptides from one MS/MS spectrum. Both peptides and proteins were filtered with a maximum false discovery rate (FDR) of 0.01. The MBR feature,37 with a matching window of 0.7 min and an alignment window of 20 min, was activated. Label-free quantitation (LFQ) calculations were performed separately in each parameter group containing similar cell loadings. Both unique and razor peptides were selected for protein quantification. Other unmentioned parameters were the MaxQuant default settings. The extracted data were further processed and visualized with Microsoft Excel. Potential contaminants and reverse sequences were filtered out. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository39 with the data set identifier PXD016921.
RESULTS AND DISCUSSION
Reducing the bore of an LC column while holding other chromatographic parameters constant can maintain the separation efficiency while decreasing the volumetric flow rate. For nanoLC–ESI-MS, this has the dual benefits of increasing both ionization efficiency and the eluting peptide concentrations. Increased ionization efficiency leads to greater ion flux, and increased eluting peptide concentrations maintain the analyte flux while reducing solvent-associated chemical noise.38 While miniaturizing the LC column inner diameter from 30 to 20 μm may seem straightforward, there are several implications that must be carefully considered. For example, slurry-packing of narrow-bore capillaries requires additional care at reduced column diameters. Conventional sample loading through a standard sample loop would lead to excessive sample injection times and potential analyte losses due to the low flow rate (~20 nL/min) and the large flow path (with potentially adsorptive surfaces) from the sample loop to the LC column. In addition, most commercially available column/ESI emitter combinations are unable to support a stable electrospray at such reduced flow rates.
To slurry-pack the 20 μm i.d. column, ultrasonication and agitation of the column by frequent tapping was applied. To address the long sample loading times, we manually inserted a sample-containing SPE column at the head of the nanoLC column for each analysis as for previous studies. While this requires depressurizing the LC system in between runs, it effectively minimizes sample loading times and reduces the potential for sample losses to the surfaces of transfer tubing during loading. However, nanoliter autosampling capabilities are of interest and are under development. To provide robust and stable electrosprays at the low flow rates provided by the 20 μm i.d. LC columns, we coupled our fritted columns with in-house-prepared chemically etched nanoESI emitters. These etched emitters have proven capable of supporting stable electrosprays at very low flow rates (including picoliters per minute in some cases).40 Dead volume was avoided between the LC column and the emitter by carefully polishing the abutting ends and verifying the zero-volume connection through the transparent PicoClear union (Figure S1A). Thus, the only extracolumn broadening occurs due to diffusion and Taylor dispersion through the 10 μm i.d., ~5 cm long emitter. This was found to be negligible using longitudinal peak broadening calculations41 as well as experimentally. For example, Figure S1B compares representative peaks from a peptide that elutes early in the gradient (m/z 408.73) and a later eluting peptide (m/z 655.86) from a 20 μm i.d. column with an etched emitter and a 30 μm i.d. column with an integrated emitter using the same gradient elution profile and packing material. As such, the 10 μm i.d. chemically etched emitter provides electrospray stability at the reduced flow rates of the narrow-bore LC columns without sacrificing separation performance, and it also enables facile replacement of the emitter.
The impact of miniaturized LC separations on proteome coverage was evaluated initially by analyzing 0.2, 0.5, and 2 ng aliquots of Pierce HeLa Digest Standard corresponding to approximately 1–10 cells on both 30 and 20 μm i.d. columns. As shown in Figure 2, peptide and proteome coverage respectively increased by ~50% and ~20% on average for the 20 μm i.d. separations. The largest gains were realized for the smallest samples (0.2 ng), which increased by 75% and 32% at the peptide and protein levels, respectively. This indicates significant promise of narrow-bore separations for analysis of single cells and other ultratrace samples, and additional gains will likely be achieved through further miniaturization.
Figure 2.

Performance comparison for 30 and 20 μm i.d. columns using the Orbitrap Fusion Lumos. Average number of unique peptides (A) and protein groups (B) identified from 0.2, 0.5, and 2 ng of HeLa tryptic digest without MS1-level feature matching. Error bars represent standard deviations from three technical replicates.
Single-Cell Analysis.
We next compared proteome coverage for nanoPOTS-prepared single HeLa cells. In addition to single cells, samples comprising 20 and 100 cells were prepared and analyzed to serve as a reference library for identifications using MBR; 6 nL of cell-free supernatant was also prepared and analyzed to serve as a blank control.
The single HeLa cell proteome coverage increased as a result of LC column miniaturization. Figure 3 shows a comparison of unique peptides and protein groups identified from single HeLa cells (without matching) by a 30 μm i.d. column26 and a 20 μm i.d. column, both using the Orbitrap Fusion Lumos. An average of 792 peptides and 211 protein groups were identified using the 30 μm i.d. column, while 939 unique peptides and 297 protein groups were identified on average from single HeLa cells using the 20 μm i.d. column, indicating respective gains of ~19% and ~41% at the peptide and protein levels.
Figure 3.

Unique peptides (A) and protein groups (B) identified from analysis of single HeLa cells based on MS/MS identification by a 30 μm i.d. LC column with the Orbitrap Fusion Lumos MS, 20 μm i.d. LC column with the Orbitrap Fusion Lumos MS, and 20 μm i.d. LC column with the Orbitrap Eclipse MS. Error bars represent standard deviations from three biological replicate measurements of single HeLa cells.
The Orbitrap Eclipse mass spectrometer offers improved sensitivity due to improvements in transmission of ions across the different vacuum regions of the mass spectrometer, diminishing ion losses and therefore improving sensitivity. We expected to observe increased proteome coverage when the Orbitrap Eclipse Tribrid mass spectrometer was employed. Indeed, 1277 unique peptides and 362 protein groups were identified on average for single HeLa cells, indicating a respective gain of ~36% and ~20% at the peptide and protein levels (Figure 3). The relative standard deviation (RSD) of the protein groups identified by MS/MS from three replicates is ~10%. The variation may be due to the manual sample loading and the fact that the single cells that were analyzed were not identical. MS/MS-based protein group identifications were compared for the three conditions presented in Figure 3 using the Student t test, which yielded p-values of 0.04, 0.02, and 0.01 for comparisons between LC columns with the same mass spectrometer (Lumos), between different mass spectrometers (Lumos and Eclipse) with the same 20 μm i.d. column, and for the combined LC and MS improvements, respectively. This provides a measure of statistical significance for improved coverage resulting from column miniaturization and the latest generation Orbitrap Eclipse MS.
Samples containing 20 and 100 HeLa cells were analyzed in addition to single cells to facilitate MBR feature matching, and the peptide and protein coverages are shown in Figure 4. Importantly, only 6 and 88 protein groups were identified from the blank samples without and with MBR, respectively, indicating minimal cross contamination and false identifications. By contrast, an average of 362 protein groups were identified from single cells without MBR, which increased to 874 with MBR. In addition to measuring single HeLa cells of typical size (<20 μm diameter), a large HeLa cell (~25 μm diameter) was intentionally selected for analysis. For this larger cell, 704 protein groups were identified without matching, which increased to 1292 protein groups with MBR. Among the 1372 total protein groups found in the four single HeLa cells, 611 were common to all of the cells. While only 19, 10, and 26 protein groups were uniquely detected in each of the cells of typical size, 254 protein groups were uniquely identified in the larger cell (single cell 4). Gene ontology analysis using DAVID (https://david.ncifcrf.gov/tools.jsp) indicated most of the proteins unique to the larger cell were related to the cytoplasm. Protein intensities are provided in the Supporting Information as a Microsoft Excel file, and as with previous work,25 label-free intensities span a dynamic range of 4 orders of magnitude and are biased toward the most abundant proteins.
Figure 4.

(A) Number of unique peptides and (B) protein groups identified from analysis of 100 HeLa cells, 20 HeLa cells, four single HeLa cells (SC-1, SC-2, SC-3, SC-4), and zero cells (blank) using the Orbitrap Eclipse without and with MBR identifications. (C) Venn diagram showing unique protein expression in four single HeLa cells. Note that SC-1–3 are average-sized HeLa cells (<20 μm diameter) and SC-4 is a large cell (~25 μm diameter).
CONCLUSIONS
In this study, we have achieved the greatest coverage to date for label-free single-cell proteomics through the combination of nanoPOTS sample preparation, further miniaturized LC separations using 20 μm i.d. packed columns, and the latest generation Orbitrap Eclipse Tribrid MS instrument. The use of a 20 μm i.d. column with a narrow-bore chemically etched emitter provided stable operation with enhanced sensitivity at ~20 nL/min flow rate and made it possible to replace damaged emitters without discarding the entire column. Decreasing the LC column diameter from 30 to 20 μm resulted in an average increase of coverage of ~50% and ~20% at the peptide and protein levels. An additional >20% gain was achieved with the Orbitrap Eclipse Tribrid MS relative to the Orbitrap Fusion Lumos Tribrid MS, resulting in identification of >360 and 870 protein groups without and with feature matching, respectively. It is likely that significant improvements can be achieved through further miniaturization of nanoPOTS preparation and the LC separation; for example, we have previously demonstrated substantial additional sensitivity gains by reducing ESI flow rates to ~1 nL/min,40 and the ongoing improvements in MS instrumentation will additionally contribute to enabling thousands of proteins to be quantified from single cells. Further automation of the workflow will also be crucial for making such analyses routine.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH Grants R21 EB020976 and R33 CA225248. Part of this research was performed using EMSL (grid.436923.9), a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at PNNL.
Footnotes
SI Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b04631.
Evaluation of peak broadening and unique peptides and protein groups identified from single HeLa cells (PDF) Protein lists with intensities (XLSX)
The authors declare no competing financial interest.
Contributor Information
Yongzheng Cong, Brigham Young University, Provo, Utah.
Yiran Liang, Brigham Young University, Provo, Utah.
Khatereh Motamedchaboki, Thermo Fisher Scientific, San Jose, California.
Romain Huguet, Thermo Fisher Scientific, San Jose, California.
Thy Truong, Brigham Young University, Provo, Utah.
Rui Zhao, Pacific Northwest National Laboratory, Richland, Washington.
Yufeng Shen, CoAnn Technologies, LLC, Richland, Washington.
Daniel Lopez-Ferrer, Thermo Fisher Scientific, San Jose, California.
Ying Zhu, Pacific Northwest National Laboratory, Richland, Washington.
Ryan T. Kelly, Brigham Young University, Provo, Utah, and Pacific Northwest National Laboratory, Richland, Washington.
REFERENCES
- (1).Wang D; Bodovitz S Trends Biotechnol. 2010, 28 (6), 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lombard-Banek C; Moody SA; Nemes P Angew. Chem., Int. Ed 2016, 55 (7), 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Levy E; Slavov N Essays Biochem. 2018, 62 (4), 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Marx V Nat. Methods 2019, 16 (9), 809–812. [DOI] [PubMed] [Google Scholar]
- (5).Couvillion SP; Zhu Y; Nagy G; Adkins JN; Ansong C; Renslow RS; Piehowski PD; Ibrahim YM; Kelly RT; Metz TO Analyst 2019, 144 (3), 794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fessenden M Nature 2016, 540 (7631), 153–155. [DOI] [PubMed] [Google Scholar]
- (7).Wisniewski JR; Hein MY; Cox J; Mann M Mol. Cell. Proteomics 2014, 13 (12), 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Milo R; Phillips R Cell Biology by the Numbers; Garland Science, Taylor & Francis Group: New York, 2015; p xlii. [Google Scholar]
- (9).Bendall SC; Simonds EF; Qiu P; Amir E.-a. D.; Krutzik PO; Finck R; Bruggner RV; Melamed R; Trejo A; Ornatsky OI; Balderas RS; Plevritis SK; Sachs K; Pe’er D; Tanner SD; Nolan GP Science 2011, 332 (6030), 687–696.21551058 [Google Scholar]
- (10).Thul PJ; Akesson L; Wiking M; Mahdessian D; Geladaki A; Ait Blal H; Alm T; Asplund A; Bjork L; Breckels LM; Backstrom A; Danielsson F; Fagerberg L; Fall J; Gatto L; Gnann C; Hober S; Hjelmare M; Johansson F; Lee S; Lindskog C; Mulder J; Mulvey CM; Nilsson P; Oksvold P; Rockberg J; Schutten R; Schwenk JM; Sivertsson A; Sjostedt E; Skogs M; Stadler C; Sullivan DP; Tegel H; Winsnes C; Zhang C; Zwahlen M; Mardinoglu A; Ponten F; von Feilitzen K; Lilley KS; Uhlen M; Lundberg E Science 2017, 356 (6340), eaal3321. [DOI] [PubMed] [Google Scholar]
- (11).Kang CC; Yamauchi KA; Vlassakis J; Sinkala E; Duncombe TA; Herr AE Nat. Protoc 2016, 11 (8), 1508–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fujii K; Nakamura H; Nishimura T Expert Rev. Proteomics 2017. 14 (4), 373–386. [DOI] [PubMed] [Google Scholar]
- (13).Kreimer S; Belov AM; Ghiran I; Murthy SK; Frank DA; Ivanov AR J. Proteome Res. 2015, 14 (6), 2367–2384. [DOI] [PubMed] [Google Scholar]
- (14).Doerr A Nat. Methods 2013, 10 (1), 23. [DOI] [PubMed] [Google Scholar]
- (15).Ankney JA; Muneer A; Chen X Annu. Rev. Anal. Chem 2018. 11 (1), 49–77. [DOI] [PubMed] [Google Scholar]
- (16).Aebersold R; Mann M Nature 2003, 422 (6928), 198–207. [DOI] [PubMed] [Google Scholar]
- (17).Shen Y; Zhao R; Berger SJ; Anderson GA; Rodriguez N; Smith R D. Anal. Chem 2002, 74 (16), 4235–4249. [DOI] [PubMed] [Google Scholar]
- (18).Sun L; Zhu G; Zhao Y; Yan X; Mou S; Dovichi NJ Angew. Chem., Int. Ed 2013, 52 (51), 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kelly RT; Page JS; Luo QZ; Moore RJ; Orton DJ; Tang KQ; Smith RD Anal. Chem 2006, 78 (22), 7796–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kelly RT; Tolmachev AV; Page JS; Tang K; Smith RD Mass Spectrom. Rev 2009, 29 (2), 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhu Y; Zhao R; Piehowski PD; Moore RJ; Lim S; Orphan VJ; Pasa-Tolic L; Qian WJ; Smith RD; Kelly RT Int. J. Mass Spectrom 2018, 427, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhu Y; Piehowski PD; Kelly RT; Qian WJ Expert Rev. Proteomics 2018, 15 (11), 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X Anal. Chem 2018, 90 (23), 14003–14010. [DOI] [PubMed] [Google Scholar]
- (24).Li ZY; Huang M; Wang XK; Zhu Y; Li JS; Wong CCL; Fang Q Anal. Chem 2018, 90 (8), 5430–5438. [DOI] [PubMed] [Google Scholar]
- (25).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian WJ; Kelly RT Nat. Commun 2018, 9 (1), 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; Ansong C; Kelly RT Angew. Chem., Int. Ed 2018, 57 (38), 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Piehowski PD; Zhu Y; Bramer LM; Stratton KG; Zhao R; Orton DJ; Moore RJ; Yuan J; Mitchell HD; Gao Y; Webb-Robertson B-JM; Dey SK; Kelly RT; Burnum-Johnson KE Nat. Commun 2020, 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Anal. Bioanal. Chem 2019, 411 (19), 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liang Y; Zhu Y; Dou M; Xu K; Chu RK; Chrisler WB; Zhao R; Hixson KK; Kelly RT Anal. Chem 2018, 90 (18), 11106–11114. [DOI] [PubMed] [Google Scholar]
- (30).Zhu Y; Dou M; Piehowski PD; Liang Y; Wang F; Chu RK; Chrisler WB; Smith JN; Schwarz KC; Shen Y; Shukla AK; Moore RJ; Smith RD; Qian WJ; Kelly RT Mol. Cell. Proteomics 2018, 17 (9), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dou M; Zhu Y; Liyu A; Liang Y; Chen J; Piehowski PD; Xu K; Zhao R; Moore RJ; Atkinson MA; Mathews CE; Qian WJ; Kelly RT Chem. Sci 2018, 9 (34), 6944–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhu Y; Podolak J; Zhao R; Shukla AK; Moore RJ; Thomas GV; Kelly RT Anal. Chem 2018, 90 (20), 11756–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhu Y; Scheibinger M; Ellwanger DC; Krey JF; Choi D; Kelly RT; Heller S; Barr-Gillespie PG eLife 2019, 8, e50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Budnik B; Levy E; Harmange G; Slavov N Genome Biol. 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Specht H; Emmott E; Perlman DH; Koller T; Slavov N bioRxiv 2019, 665307. [Google Scholar]
- (36).Dou M; Clair G; Tsai CF; Xu K; Chrisler WB; Sontag RL; Zhao R; Moore RJ; Liu T; Pasa-Tolic L; Smith RD; Shi T; Adkins JN; Qian WJ; Kelly RT; Ansong C; Zhu Y Anal. Chem 2019, 91, 13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tyanova S; Temu T; Cox J Nat. Protoc 2016, 11 (12), 2301–2319. [DOI] [PubMed] [Google Scholar]
- (38).Maiolica A; Borsotti D; Rappsilber J Proteomics 2005, 5 (15), 3847–3850. [DOI] [PubMed] [Google Scholar]
- (39).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Marginean I; Tang KQ; Smith RD; Kelly RT J. Am. Soc. Mass Spectrom 2014, 25 (1), 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vandeemter JJ; Zuiderweg FJ; Klinkenberg A Chem. Eng. Sci 1956, 5 (6), 271–289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.