

Graphical abstract
Keywords: G protein coupled receptors, Adenosine A2A receptor, Functional selectivity, G protein binding, β-Arrestin recruitment, Molecular dynamic simulations
Abstract
Biased agonism, the ability of agonists to differentially activate downstream signaling pathways by stabilizing specific receptor conformations, is a key issue for G protein-coupled receptor (GPCR) signaling. The C-terminal domain might influence this functional selectivity of GPCRs as it engages G proteins, GPCR kinases, β-arrestins, and several other proteins. Thus, the aim of this paper is to compare the agonist-dependent selectivity for intracellular pathways in a heterologous system expressing the full-length (A2AR) and a C-tail truncated (A2AΔ40R lacking the last 40 amino acids) adenosine A2A receptor, a GPCR that is already targeted in Parkinson’s disease using a first-in-class drug. Experimental data such as ligand binding, cAMP production, β-arrestin recruitment, ERK1/2 phosphorylation and dynamic mass redistribution assays, which correspond to different aspects of signal transduction, were measured upon the action of structurally diverse compounds (the agonists adenosine, NECA, CGS-21680, PSB-0777 and LUF-5834 and the SCH-58261 antagonist) in cells expressing A2AR and A2AΔ40R. The results show that taking cAMP levels and the endogenous adenosine agonist as references, the main difference in bias was obtained with PSB-0777 and LUF-5834. The C-terminus is dispensable for both G-protein and β-arrestin recruitment and also for MAPK activation. Unrestrained molecular dynamics simulations, at the μs timescale, were used to understand the structural arrangements of the binding cavity, triggered by these chemically different agonists, facilitating G protein binding with different efficacy.
1. Introduction
Adenosine triphosphate (ATP) is the main energy-transfer molecule and adenosine is one of its main metabolites. Adenosine receptors appeared early in evolution, as sensors of ATP decay, because excess of adenosine correlates with ATP depletion. They are considered as the most ancient within the class A (or rhodopsin-like) G-protein-coupled receptor (GPCR) family. Moreover, phylogenetic studies suggest that adenosine receptors were the first to start diverging [1] from the MECA receptor cluster [2], which is formed by the melanocortin, endothelial differentiation sphingolipid, and cannabinoid receptors. There are four identified mammalian adenosine receptors (humans included) whose cognate heterotrimeric G proteins are Gi (A1 and A3) or Gs/Golf (A2A and A2B) [3].
The crystal structure of the A2A receptor (A2AR) was one of the first reported for the G-protein-coupled receptor (GPCR) family [4]. Today, there are nearly fifty structures of A1R and A2AR bound to agonists, antagonists, and/or to G proteins [5]; all of them displaying the common features of class A GPCRs [6]. Unfortunately, the N- and C-terminal domains, which are highly variable in sequence, length, and structure [7], have been removed for crystallization purposes. The long C-terminal domain of A2AR (about 122 amino acids) is known to engage G proteins, GPCR kinases, arrestins and several other proteins [8], [9]. The C-terminus is also responsible for the constitutive activity of the receptor [10], influences the quaternary structure of heteromers [11], [12], and is involved in differential intracellular signaling [13].
A2AR offers numerous possibilities for therapeutic applications [14], [15], [16]. A2AR antagonists show promise, among others, in Alzheimer’s and Parkinson’s diseases, attention-deficit hyperactivity disorder, depression, and anxiety. Istradefylline, one of the most studied antagonists, is safe and efficacious in Parkinson and was approved in Japan as Nouriast™ and by the US Food and Drug Administration as Nourianz™ [17]. A2AR agonists could be used in Niemann Pick type C disease, autism-spectrum disorders, and schizophrenia. Regadenoson, a selective A2AR agonists that is a coronary vasolidator, was approved in the United States as Lexiscan™.
These facts open the possibilities for more drug approvals, both agonists and antagonists, related to adenosine receptors. Thus, the aim of this work was double. First, we wanted to analyze the agonist-dependent selectivity for intracellular pathways, known as functional selectivity or biased agonism, using structurally diverse compounds in a heterologous system expressing A2AR. Biased agonists might offer attractive therapeutic properties relative to their unbiased counterpart, circumstance that remains to be validated in clinical trials [18]. In fact, adenosine receptor biased agonists could be used for cardioprotection without bradycardia as a serious adverse effect [19], [20]. Second, we analyzed in living cells the effect of the C-terminal domain of A2AR in ligand binding and in agonist-induced signaling.
2. Materials and methods
2.1. Reagents
Adenosine, NECA, CGS-21680, PSB-0777, LUF-5834 and SCH-58261 were purchased from Tocris Biosciences (Bristol, UK). HEPES was purchased from SigmaAldrich (St. Louis, MO, US). Stock solutions were prepared in DMSO. Aliquots of these stock solutions were kept frozen at −20 °C until use.
2.2. Cell culture and transient transfection
Human embryonic kidney 293T (HEK-293T) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Paisley, Scotland, UK) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin/streptomycin, MEM non-essential amino acids solution (1/100) and 5% (v/v) heat inactivated fetal bovine serum (FBS) [all supplements were from Invitrogen, (Paisley, Scotland, UK)]. Cells were incubated in a humid atmosphere of 5% CO2 at 37 °C. 24 h after seeding in 6-well or 96-well (for ERK phosphorylation assay) plates, cells were transiently transfected with cDNA coding for wild-type A2AR or A2AΔ40R (amino acids 372–412 on the C-terminal domain were deleted [21]) with the PEI (PolyEthylenImine, SigmaAldrich) method as previously described [13], [22]. Truncation of this part of the C-tail does not remove the proposed phosphorylation codes (PxPxxP/E/D or PxxPxxP/E/D, where P represents phospho-serine or phospho-threonine) for arrestin recruitment [23]. This phosphorylation code starts 33 amino acids after P7.50 of the NPxxY motif in human rhodopsin, whereas truncation of A2AR starts 87 amino acids after P7.50. Cells were incubated for 4 to 5 h with the cDNA of interest, PEI and NaCl in serum-free medium. After that, the serum-free medium was replaced by complete culture medium and cells were incubated for 48 h at 37 °C in 5% CO2 humid atmosphere. The sequences in the plasmids were those coding for human receptors. It should be noted that within a given experimental session, for instance of determination of cAMP levels, all agonists (plus/minus antagonist when indicated) were tested in the same batch of transfected cells.
2.3. Homogeneous HTRF binding assays
HEK-293T cells growing in 6-well plates were transiently transfected with 1 µg of plasmid encoding for pHALO-tagged human A2AR (pHALO-A2AR, Cisbio Bioassays, Codolet, France) or 1 µg of plasmid encoding for pHALO-tagged human A2AΔ40R and incubated at 37 °C in a 5% CO2 humid atmosphere (24 h).
2.4. Covalent labeling of cells expressing pHALO-tagged receptors
48 h post transfection culture medium was removed, cells were washed with PBS and incubated with 100 nM of HALO-Lumi4Tb, a terbium derivative of O6-benzylguanine (SSNPTBC, Cisbio Bioassays, Codolet, France) - previously diluted in TagLite Buffer (TLB) (LABMED, Cisbio Bioassays, Codolet, France) for 1 h at 37 °C under 5% CO2 humid atmosphere. After that, cells were washed with PBS to remove the excess of HALO-Lumi4Tb, detached with Versene (Gibco Life Technologies, Paisley, Scotland, UK), centrifuged at 1,500 rpm for 5 min and resuspended in TLB. Densities of 10,000 cells/well were used to carry out binding assays in white opaque 384-well plates.
2.5. Non-radioactive homogeneous time-resolved FRET-based binding assays
Saturation binding experiments were performed by incubating cells expressing Tb-labeled HALO-A2AR with increasing concentrations of the A2AR antagonist SCH-442416, conjugated to a fluorescent probe developed by Cisbio (red A2AR ligand, purchased from Cisbio Bioassays, Codolet, France). The unspecific signal was obtained by incubating cells expressing Tb-labeled HALO-A2AR with increasing concentrations of red A2AR ligand in the presence of 10 µM unlabeled SCH-58261. Both, fluorescent ligands and unlabeled compounds, were diluted in TLB. 10 µl of labeled cells (10,000 cells/well) were loaded onto 384-well white plates and 5 µl unlabeled SCH-58261 (10 µM final concentration) or TLB were added, followed by the addition of 5 µl of increasing concentrations (0–60 nM range) of red A2AR ligand. Plates were incubated protected from light for 2 h at room temperature before time-resolved fluorescence resonance energy transfer (TR-FRET) signal reading. The specific binding was calculated by subtracting the unspecific binding from the total binding.
For competition binding assays, HEK-293T cells transiently expressing Tb-labeled HALO-A2AR, or A2AΔ40R, were incubated with 20 nM fluorophore-conjugated A2AR ligand, in the presence of increasing concentrations (0–10 µM range) of agonists (or antagonists when indicated). Plates contained 10 µl of labeled cells, and 5 µl of tested compounds or TLB were added prior to the addition of 5 µl of the red A2AR ligand. Plates were then incubated for at least 2 h at room temperature prior to TR-FRET signal detection. Detailed description of the HTRF assay is found elsewhere [24].
2.6. Signal detection and data analysis
Signal was detected using a PHERAstar FS (BMG Lab technologies, Offenburg, Germany) microplate reader equipped with a FRET optic module allowing donor excitation at 337 nm and signal collection at both 665 and 620 nm. A frequency of 10 flashes/well was selected for the xenon flash lamp excitation. The signal was collected at both 665 and 620 nm using the following time-resolved settings: delay, 150 ms; integration time, 500 ms. HTRF ratios were obtained by dividing the acceptor signal (665 nm) by the donor signal (620 nm) and multiplying this value by 10,000. The 10,000-multiplying factor is used solely for the purpose of easier data handling.
Data were analyzed using Prism 7 software (GraphPad Software, Inc., San Diego, CA), and competition data were fitted by non-linear regression to a one site-fit logIC50, competition curves were -in all cases- monophasic. KD (dissociation constant) values of the fluorescent ligand were obtained from the specific binding saturation curves. Note that Bmax values obtained from HTRF saturation curves do not reflect absolute values of receptor binding sites. Ki values were determined from competition binding assays by using the calculated IC50 and KD values and the Cheng-Prusoff equation [25].
2.7. cAMP determination
HEK-293T cells were grown in 6-well plates and transiently transfected with cDNAs for A2AR or for A2AΔ40R as described in 2.2. Two hours before initiating the experiment, medium was replaced by serum-free DMEM medium. Then cells were detached, isolated by centrifugation (5 min at 1,500 rpm) and resuspended in DMEM containing 50 µM zardaverine (phosphodiesterase inhibitor) to prevent degradation of cAMP, and 5 mM HEPES (pH 7.4). Cells were placed in white 384-well plates (PerkinElmer) (6,000 cells/well) and incubated with antagonists or vehicle for 15 min before treatment with agonist or vehicle for 15 min. Finally, cAMP-Eu and the fluorescent antibody were added. Readings were performed after one hour incubation at 25 °C. Homogeneous time-resolved fluorescence energy transfer (HTRF) measures were performed using the Lance Ultra cAMP kit (PerkinElmer, Waltham, MA, US). Fluorescence at 665 nm was measured in a PHERAstar Flagship plate reader equipped with an HTRF optical module (BMG Lab technologies, Offenburg, Germany).
2.8. ERK1/2 phosphorylation assays
HEK-293T cells were grown on transparent Biocat Poly-D-Lysine 96-well plates (Deltalab) and kept at the incubator for 24 h. Then cells were transiently transfected with cDNA coding for A2AR or A2AΔ40R and incubated for 48 h at 37 °C in a 5% CO2 humid atmosphere. Supplementary Fig. S1A shows that treatment with CGS-21680 in non-transfected HEK-293T cells is not significantly different to the basal condition. Thus, the expression level of A2AR in non-transfected HEK-293T cells is negligible. 2 h before initiating the experiment, the medium was substituted by serum-free DMEM. Cells were stimulated at 25 °C for 10 min with vehicle or agonists (Supplementary Fig. S1D). ERK phosphorylation was measured at “short” times to detect G-protein mediated signal. After that, cells were washed twice with cold PBS before the addition of lysis buffer (30 µl/well; Perkin Elmer, Waltham, MA, US) and incubated for 15 min at 25 °C on a Heidolph Titramax 100 shaker. 10 µl of each cell lysate was transferred to white ProxiPlate 384-well microplates (PerkinElmer; Waltham, MA, USA). ERK1/2 phosphorylation was determined using AlphaScreen®SureFire® kit (Perkin Elmer, Waltham, MA, US): 5 µl/well of acceptor beads were added. Plates, protected from light, were incubated for 2 h at 25 °C. Finally, 5 μl/well of donor beads were added and plates, protected from light, were incubated for 2 h before analysis. Fluorescence was determined using an EnSpire® Multimode Plate Reader (PerkinElmer, Waltham, MA, USA). The value of reference (100%) was that achieved in the absence of any treatment (basal). The effect of ligands was given in percentage respect to the basal value.
2.9. β-Arrestin 2 recruitment
HEK-293T cells were transiently transfected with 0.625 µg of cDNA coding for β-arrestin 2-Rluc and 2 µg of cDNA coding for A2A-YFP or with 1.5 µg of cDNA coding for β -arrestin 2-Rluc and 4 µg of cDNA coding for A2AΔ40R-YFP. BRET experiments were performed 48 h after transfection. Cells were detached using HBSS containing 0.1% glucose, centrifuged for 5 min at 3,200 rpm and resuspended in the same buffer. Protein concentration was quantified by using the Bradford assay kit (Bio-Rad, Munich, Germany). Hereafter, YFP fluorescence at 530 nm was quantified in a FluoStar Optima Fluorimeter (BMG Labtechnologies, Offenburg, Germany) to quantify receptor-YFP expression. To measure β -arrestin recruitment, cells (20 µg of protein) were distributed in 96-well microplates (Corning 3600, white plates with white bottom) and were incubated for 10 min with antagonists. Cells were then stimulated with agonists prior to the addition of 5 µM Coelenterazine H (Molecular Probes, Eugene, OR). BRET between β-arrestin 2-Rluc and receptor-YFP was determined and quantified at 5 min after adding coelenterazine H. This time of response was selected from time-response curves (Supplementary Fig. S1C). The readings were collected using a Mithras LB 940 (Berthold Technologies, Bad Wildbad, GE), which allows the integration of the signals detected in the short-wavelength filter (485 nm) and the long wavelength filter (530 nm). To quantify protein-RLuc expression, luminescence readings were also collected 10 min after the addition of 5 µM coelenterazine H.
2.10. Dynamic mass redistribution assays (DMR)
Cell mass redistribution induced upon receptor activation was detected by illuminating with polychromatic light the underside of a biosensor and measuring the changes in the wavelength of the reflected monochromatic light, that is a sensitive function of the index of refraction. The magnitude of the wavelength shift (in picometers) is directly proportional to the amount of mass redistribution. 48 h before the assay, HEK-293T cells were transiently transfected with 2 µg of cDNA coding for A2AR or A2AΔ40R. HEK-293T cells were seeded in 384-well sensor microplates to obtain 70–80% confluent monolayers constituted by approximately 10,000 cells/well. Prior to the assay, cells were washed twice with assay buffer (HBSS with 20 mM HEPES, pH 7.15 and 1% BSA) (SigmaAldrich, St. Louis, MO, US) and incubated for 2 h with assay-buffer containing 0.1% DMSO (24 °C, 30 µl/well). Hereafter, the sensor plate was scanned, and a baseline optical signature was recorded for 10 min before adding 10 µl of the selective antagonists for 30 min, followed by the addition of 10 µl of the selective agonists; all test compounds were diluted in assay buffer. Then, DMR responses were monitored for at least 5000 s in an EnSpire® Multimode Plate Reader (PerkinElmer, Waltham, MA, USA) by a label-free technology. Results were analyzed using EnSpire Workstation Software v 4.10.
2.11. Calculation of bias factor
Bias factor (bias) was calculated with the following formulas adapted from [26] in which τ represents the agonist efficacy, and KA the agonist affinity [27].
log (τ/KA) is defined as the transduction coefficient or synonymously as logR, a parameter that can be used to compare agonist activity between different systems. The transduction coefficient is a measure of the ability of a ligand to activate the receptor [28].
The pathway of reference j1 was cAMP determination for Gs-coupling, whereas the other pathways (ERK1/2 phosphorylation, ß-arrestin 2 recruitment, or DMR) were j2. τ denotes the maximum value in each response and KA is the antilogarithm of half maximal effective concentration (EC50 if the agonist provides an increase response, IC50 if the agonist provides a reduction of the response induced by another reagent).
2.12. Computational methods
Preparation of protein structure and ligand parametrization. Crystal structures of the A2AR in its active intermediate state in complex with the adenosine (PDB id 2YDO), NECA (2YDV) and CGS-21680 (4UHR) agonists, and the crystal structure of A2AR in its inactive state in complex with the ZM241385 antagonist (PDB id 4EIY) were obtained from the Protein Data Bank (rcsb.org). Fusion proteins were removed and stabilizing mutations were mutated to the native sequence using MODELLER v9.12 [29]. Parameters for adenosine, NECA, CGS-21680, PSB-0777, LUF-5834 and SCH-58261 were obtained from the general Amber force field (GAFF) and HF/6-31G*//HF/6-31G*-derived RESP atomic charges calculated with Gaussian09.
Molecular docking. The PSB-0777 and LUF-5834 agonists were docked into the active intermediate state (2YDV) and the SCH-58261 antagonist was docked into the inactive state (4EIY) using the Molecular Operating Environment (MOE) software (Chemical Computing Group Inc., Montreal, Quebec, Canada). One hundred docking solutions per ligand were generated by the triangle matcher algorithm into the active site of the receptor structures. Top-ranking solutions were inspected and conformations in which the central moiety of PSB-0777 and the aminopyridine group of LUF-5834 were located in the same region as the adenine moiety of adenosine and NECA were selected (Supplementary Fig. S2). The binding pose of SCH-58261 was selected in such a manner that the orientation of its pyrazolo-triazolo-pyrimidine central moiety was similar to the bicyclic triazolotrazine core of the highly potent selective antagonist ZM241385 found in the 4EIY structure (Supplementary Fig. S2).
Molecular dynamics (MD) simulations. The complexes of adenosine, NECA, CGS-21680, PSB-0777 and LUF-5834 with the intermediate active structure of A2AR and the complex of SCH-58261 with the inactive structure of A2AR (see above) were embedded in a pre-equilibrated lipid bilayer box containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), water molecules (TIP3P) and monoatomic Na + and Cl− ions (0.2 M). Assignment of ionization states and hydrogens at physiological pH for the selected structures was conducted with the Protonate3D method [30] as implemented in MOE. D2.50 was deprotonated (negatively charged) for antagonist-bound receptor simulations and protonated (neutral) for agonist-bound receptor simulations [31], whereas H264 was protonated (positively charged) in all simulations [32]. A sodium ion was placed, near the negatively charged D2.50, in the inactive, antagonist-bound, structure of A2AR [33], [34]. Molecular systems were subject to a 1000 cycles of energy minimization, followed by 20 ns of gradual relaxation of positional restraints (corresponding to 100, 50, 25 and 10 kJ.mol−1nm−2) at protein backbone coordinates before the production phase in order to hydrate the receptor cavities and allow lipids to pack around the protein. After equilibration, unrestrained MD simulation (3 replicas of 1 μs giving a total of 3 μs of sampling time) were produced for each ligand-receptor complex at a constant temperature of 300 K using separate v-rescale thermostats for the receptor, ligands, lipids and solvent molecules. A time step of 2.0 fs was used for the integration of equations of motions. All bonds and angles were kept frozen using the LINCS algorithms. Lennard-Jones interactions were computed using a cutoff of 10 Å, and the electrostatic interactions were treated using PME with the same real-space cutoff under periodic boundary conditions. MD simulations were performed using GROMACS v5.0.7. The AMBER99SB force field as implemented in GROMACS, Berger parameters for POPC lipids, and the GAFF parameters for the ligands (see above) were used for the MD simulations, attending the performance of this protocol on membrane-protein systems [35].
2.13. Statistical analysis
Experimental data was managed and analysed with GraphPad Prism software version 7 (San Diego, CA, USA) or IBM SPSS Statistics version 25.0 (IBM Corp., NY, USA). Unless otherwise stated data are the mean ± S.E.M (n = 5 or higher). P-values lower than 0.05 were considered statistically significant.
3. Results and discussion
3.1. Ligand binding experiments
We studied the binding of the agonists adenosine, NECA and CGS-21680, the partial agonists PSB-0777 and LUF-5834, and the selective antagonist SCH-58261 by homogeneous time-resolved fluorescence (HTRF) experiments, schematized in Fig. 1A, in living HEK-293T cells expressing either wild type (wt) A2AR or a truncated receptor (A2AΔ40R, lacking the last 40 amino acids of the C-terminal end) tagged with HaloTag on the N-terminus (see 2.3–2.5). First, a saturation curve was performed using a fluorescence-conjugated antagonist (Fig. 1B). The obtained KD values were 1.7 nM and 1.2 nM for full-length A2AR and A2AΔ40R, respectively (Table 1). Subsequently, competition assays were performed using 20 nM of the fluorescence-conjugated antagonist and increasing amounts of the ligands (Fig. 1C-1D). The Ki value of each compound was calculated from IC50 and KD values using the Cheng-Prusoff expression [25] (Table 1). The obtained Ki values are in the range of those obtained in radioligand binding assays for isolated membranes from tissues or transfected cells. According to this assay, the Ki values of agonists are in the 13–219 nM range, LUF-5834 being the compound that binds A2AR the strongest and PSB-0777 the weakest. Clearly, removal of the C-terminal domain results in weaker agonist binding to the orthosteric site (a ratio of Ki values in the 1.4–4.4 range, which correspond to binding free energy differences between 0.2 and 0.9 Kcal/mol). On the other hand, removal of the C-terminal domain has a significant impact on the binding of the antagonist SCH-58261 (0.7 nM vs 113.9 nM for full-length A2AR and A2AΔ40R, respectively; binding free energy difference of 3.0 Kcal/mol).
Fig. 1.

Competition curves in HTRF-based assays. Panel A: Scheme of the homogeneous binding technology performed in living HEK-293T cells. Panel B. Saturation isotherm of binding of fluorophore-conjugated red A2AR ligand to HEK-293T cells transiently transfected with HALO-A2AR in the absence (red) or presence of 10 µM SCH-58261 (black); specific binding is depicted in green. Panels C-D: Non-radiolabeled HTRF-based competition curves of specific binding of 20 nM fluorophore-conjugated red A2AR ligand in the presence of increasing concentrations of different agonists and of the selective antagonist SCH-58261, in cells expressing A2AR (C) or A2AΔ40R (D). Data represent the mean ± SEM of a representative experiment (n = 4). HTRF ratio = (665 nm acceptor signal/620 nm donor signal) × 10,000. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
IC50 and Ki values obtained from competition binding assays in HEK-293T cells expressing wild type A2AR or A2AΔ40R. Values reported are the mean of three experiments conducted in triplicates fitted to a monophasic competition model.
| A2AR |
A2AΔ40R |
|||||
|---|---|---|---|---|---|---|
| IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | KiΔCT/ Kiwt | ΔΔGa (kcal/mol) | |
| adenosine | 1658 | 183 | 4804 | 531 | 2.9 | 0.6 |
| NECA | 1118 | 124 | 2190 | 242 | 2.0 | 0.4 |
| CGS-21680 | 746 | 82.5 | 1690 | 187 | 2.3 | 0.5 |
| PSB-0777 | 1984 | 219 | 8680 | 960 | 4.4 | 0.9 |
| LUF-5834 | 115 | 13 | 166.0 | 18 | 1.4 | 0.2 |
| SCH-58261 | 6.3 | 0.7 | 1030 | 114 | 162.3 | 3.0 |
| A2AR |
A2AΔ40R |
||
|---|---|---|---|
| Bmax | KD (nM) | Bmax | KD (nM) |
| 6959 | 1.677 | 9214 | 1.22 |
aExperimental binding free energy differences between wild type (A2AR) and truncated (A2AΔ40R) receptors, calculated as ΔΔG = -RT ln (KiΔCT/Kiwt) where R is 1.987 cal mol−1 K−1 and T is the temperature of 298°K (-RT = −0.592 kcal mol−1)
3.2. Molecular models of agonist binding
The binding of these compounds to A2AR has been extensively studied by site-directed mutagenesis, computer simulations, and supported by the recent crystallographic data of adenosine, NECA and CGS in complex with A2AR (reviewed in [36]). PSB-0777 and LUF-5834 were docked (see 2.12) using, as template, ligands solved in crystal structures with homologous chemical scaffolds (see Supplementary Fig. S2). All molecules but LUF-5834 are structurally related through a common adenosine core. Fig. 2 summarizes the binding modes of all compounds, emphasizing common/different areas of the receptor occupied by each ligand. In order to understand the molecular mechanism of receptor activation (see 3.5), we studied by molecular dynamics (MD) simulations (see 2.12) the interactions of crucial ligand moieties to key receptor amino acids in all these regions. Detailed analysis of the simulations shows that these binding modes were stable during the unbiased 1 μs MD simulations (three replicas) as shown by the relatively low movement in root mean-square deviation (rmsd) plots of the receptor and ligand heavy atoms, as well as the conservation of the secondary structure elements of A2AR (Supplementary Figs. S3-S5).
Fig. 2.

Ligand-receptor complexes. Cross-section through the A2AR, highlighting the agonists adenosine (white sticks), NECA (cyan), CGS-21680 (pink), PSB-0777 (green) and LUF-5834 (blue) occupying the binding site. Color rectangles highlight regions of the orthosteric binding cavity that are occupied by functional groups of the studied agonists (see 3.2). The PIF motif (in yellow) located below the orthosteric binding cavity, and the side chain of the highly conserved R3.50 (in green) of the DRY motif near the G protein binding site are highlighted. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As shown in Fig. 2, three regions (blue, green and yellow rectangles) are invariably occupied by functional groups of all the studied compounds, illustrated by the binding of adenosine in the central panel. The heterocyclic site (blue rectangle) corresponds to the central region and accommodates the adenine moiety of adenosine, NECA, CGS-21680, and PSB-0777, and the aminopyridine group of LUF-5834 through aromatic/hydrophobic interactions with F168, M5.38, M7.35, I7.39 (numbering in Ballesteros and Weinstein scheme [37], except for residues outside helices in which sequence numbers are used) and polar interactions of the exocyclic nitrogen with N6.55 (Supplementary Fig. S3). The second region (green rectangle) accommodates the 5′-hydroxymethyl (adenosine and PSB-0777), N-ethylcarboxamido (NECA and CGS-21680) and hydroxyphenyl group of LUF-5834 (Fig. 2). The 5′-hydroxymethyl group attached to the ribose ring of adenosine and PSB-0777 and the hydroxyphenyl group of LUF-5834 hydrogen bond H6.52 during all the simulation time (Supplementary Fig. S3). NECA and CGS-21680 replace the 5′-hydroxymethyl group by the longer N-ethylcarboxamido group that can directly interact with both H6.52 and T3.36 (Supplementary Fig. S3). Several X-ray structures of A2AR contain crystallized water molecules in the environment of T3.36 and N5.42 [4] that are important for the activity of GPCRs [38], [39]. Supplementary Fig. S6 shows that during the MD simulations adenosine, PSB-0777 and LUF-5834 maintain this water bridge, whereas NECA and CGS-21680 cannot because the N-ethylcarboxamido group interacts with T3.36. Finally, the third region (yellow rectangle) is occupied by the ribose moiety of adenosine derivatives and by the carbonitrile substituent of LUF-5834. Most important interactions in this site imply the formation of hydrogen bonds from the hydroxyl groups at positions C2 and C3 of the ribose moiety of adenosine derivatives with the S7.42 and H7.43 residues.
Three additional regions are occupied by the ligands (orange, cyan and black in Fig. 2). The ethyl group of the N-ethylcarboxamido moiety of NECA and CGS-21680 extends deep into the binding pocket (orange), forming hydrophobic interactions with C5.46 (Supplementary Fig. S3). These interactions between aliphatic chains and sulfur-containing amino acids have been shown to be of high energy [40]. The long substituents of CGS-21680 and PSB-0777 expand toward the extracellular loops through a cavity within TMs 1, 2 and 7 (black rectangle), which has been shown to influence G protein and β-arrestin signaling in other GPCRs [41], [42]. The negatively charged SO3− group of PSB-0777 interacts with His264 in ECL3, whereas the longer chain of CGS-21680 permits the negatively charged COO− to form an ionic interaction with K153 in ECL2 (Supplementary Fig. S7). Finally, the imidazole substituent of LUF-5834 interacts with residues Y1.35, A2.61, I2.64, S2.65, and I7.39 in the sideward region of the binding cavity (cyan rectangle). This binding orientation of LUF-5834 has been previously proposed using mutagenesis studies [43]. This region is generally occupied by inverse agonists and, thus, we believe contributes to the low activity of LUF-5834 regarding the rest of the agonists.
3.3. Agonist-induced signaling responses
Next, we measured four functional read-outs that correspond to different steps of the signaling pathways in cells expressing full-length A2AR or A2AΔ40R: cAMP production, β-arrestin recruitment, ERK1/2 phosphorylation, and dynamic mass redistribution (DMR) assays (Fig. 3 and Table 2). Immediately following receptor activation, cAMP levels increase as the result of adenylyl cyclase activation by Gs. Next, receptor phosphorylation by G protein kinases triggers β-arrestin recruitment. Later, ERK signaling is regulated by G protein or/and β-arrestin. DMR accounts for events that occur much later in the signaling pathway such as protein trafficking, rearrangement of cytoskeleton and adhesion or morphological changes. The amount of transfected cDNA for A2AR and A2AΔ40R was adjusted to obtain similar receptor expression levels (Supplementary Fig. S1B). β-arrestin recruitment and ERK1/2 phosphorylation were analyzed by time response curves (Supplementary Figs. S1C-S1D). In our cAMP assay conditions, adenosine, NECA, and CGS-21680 behaved as full agonists, whereas PSB-0777 and LUF-5834 were partial agonists on full-length A2AR. The effect was specific for all agonists as shown by the blockade of cAMP production using the selective receptor antagonist SCH-58261 (Supplementary Fig. S8). β-arrestin recruitment assays showed that PSB-0777 and NECA are more efficient in recruitment than adenosine and CGS-21680. Remarkably, LUF-5834, which is a partial agonist in the cAMP assay, is as efficient as CGS-21680 and adenosine in β-arrestin recruitment. pERK1/2 dose–response curves follow similar patterns as cAMP curves, with the exceptions of PSB-0777 that behaves as a full agonist, and LUF-5834 that is a very weak partial agonist. DMR responses were all very similar with PSB-0777 providing a slightly larger signal at higher concentrations.
Fig. 3.
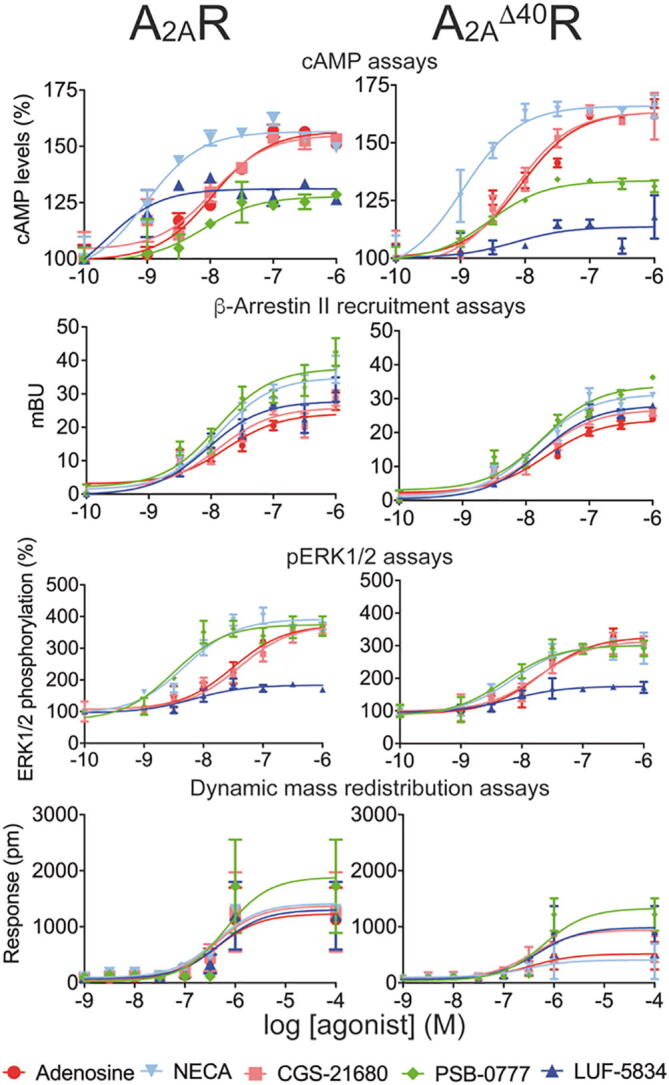
Signaling in cells expressing either A2AR or A2AΔ40R. Dose response curves on 0.5 µM forskolin-induced cAMP levels, on ß-arrestin recruitment, on ERK1/2 phosphorylation, and on dynamic mass redistribution (DMR). Data (n = 12, each in triplicates) for cAMP are given in percentage (100% represents the forskolin effect). Data (n = 10, each in triplicates) for BRET assays, used to determine β-arrestin recruitment, are given in milliBRET Units (mBU). Data (n = 6, each in triplicates) for ERK1/2 phosphorylation are expressed as % with respect to basal levels. DMR tracings are representing the picometer (pm)-shifts of reflected light wavelengths over time upon ligand treatment.
Table 2.
pEC50 and Emax values obtained in HEK-293T cells expressing wild type A2AR or truncated A2AΔ40R for cAMP production, β-arrestin II recruitment, ERK1/2 phosphorylation and dynamic mass redistribution (DMR) response (Fig. 3).
| cAMP assays |
β -arrestin assays |
pERK1/2 assays |
DMR assays |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2AR |
A2AΔ40R |
A2AR |
A2AΔ40R |
A2AR |
A2AΔ40R |
A2AR |
A2AΔ40R |
|||||||||
| pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| Adenosine | 8.0 | 156.6 | 8.1 | 163.5 | 7.8 | 24.1 | 7.7 | 23.7 | 7.5 | 373.0 | 7.7 | 326.9 | 6.4 | 1231 | 6.4 | 517.8 |
| NECA | 9.0 | 156.6 | 9.0 | 166.0 | 7.9 | 34.9 | 7.8 | 31.3 | 8.3 | 392.1 | 8.2 | 302.6 | 6.4 | 1416 | 6.5 | 407.9 |
| CGS-21680 | 8.0 | 155.1 | 8.2 | 163.4 | 7.9 | 25.9 | 7.8 | 26.8 | 7.4 | 373.5 | 7.7 | 315.2 | 6.4 | 1374 | 6.4 | 941.5 |
| PSB-0777 | 8.1 | 127.7 | 8.6 | 133.5 | 7.9 | 37.6 | 7.7 | 33.8 | 8.5 | 374.7 | 8.3 | 300.7 | 6.2 | 1887 | 6.2 | 1332 |
| LUF-5834 | 9.7 | 131.1 | 8.2 | 113.6 | 8.0 | 27.8 | 7.8 | 28.0 | 8.2 | 183.9 | 8.2 | 175.8 | 6.3 | 1306 | 6.3 | 988.6 |
In agreement with previous reports [10], removal the last 40 amino acids of the C-terminus in the A2AΔ40R construct caused a right-shift of the cAMP dose–response curves (a ratio of EC50 values in the 2.2–9.8 range, similar to the range of Ki ratios) with almost no change in Emax for all agonists, with the exception of LUF-5834 whose effect became almost negligible in the truncated form of the receptor (Fig. 3 and Table 2). C-terminal truncation provides a small decrease in β-arrestin recruitment with no changes in Emax rank order relative to full-length A2AR. pERK1/2 dose–response curves of A2AΔ40R were very similar to full-length A2AR except that pEC50 increased for the truncated receptor, which goes against the general trend, and the potency of NECA and PSB-0777 became close to that of adenosine and CGS-21680. Finally, DMR read-outs of A2AΔ40R were almost negligible for NECA and adenosine while we could observe a higher signal for PSB-0777 compared to that of the other compounds in A2AR and A2AΔ40R.
3.4. Addressing biased agonism
Bias factors were calculated taking as reference the effect of adenosine, the endogenous compound, and the Gs-mediated signal, i.e. cAMP response elicited by adenosine (see 2.8). Bias factors for CGS-21680, PSB-0777, LUF-5834 and NECA agonists in cAMP, β-arrestin recruitment, ERK1/2 phosphorylation and DMR signaling responses are summarized in the radar plot of Fig. 4. Adenosine and CGS-21680 are balanced agonists with similar bias factors for the four pathways, whereas NECA has small factors for DMR and β-arrestin recruitment and LUF-5834 has small factors for all responses other than cAMP. By contrast, PSB-0777 has a higher bias for the pERK1/2 response. Truncation of the C-terminus makes LUF-5834 and PSB-0777 more balanced. In fact, LUF-5834 acquires the largest bias factors for β-arrestin recruitment, ERK1/2 phosphorylation and DMR. By contrast, NECA keeps the small factors for β-arrestin recruitment and DMR. Overall the results are consistent with the C-terminus being dispensable for both G-protein and arrestin recruitment and also for MAPK activation (ERK1/2 phosphorylation and DMR), with only minor differences in signaling compared to full-length A2AR.
Fig. 4.

Radar plot representation of bias factors. Plots show the bias factors of the different compounds in the different functional outcomes in cells expressing A2AR or A2AΔ40R. Adenosine and the Gs-cAMP signaling pathway were used as reference for calculations.
3.5. Molecular mechanisms of agonist-induced receptor activation
The first step to understand the mechanisms of agonist-induced receptor activation, was to compare by MD simulations the trajectories of amino acids at positions 3.40, 5.50, and 6.44 (Fig. 5), which have been named as the “P-I-F” motif [44], the “transmission switch” [45], the “triad core” [46] or the connector region [6], in the presence of the agonists adenosine, NECA, CGS-21680, PSB-0777 and LUF-5834 and the antagonist SCH-58261. These residues, located below the ligand binding cavity and above the G protein or arrestin binding cavity (see Fig. 2), adopt different positions upon binding of agonists or antagonists [47], [48], and we have been using them to predict the effect of the ligand on the conformational state of the receptor [49], [50], [51]. Clearly, the agonist-bound, active-like complexes are characterized relative to the antagonist-bound, inactive-like, complex by the proposed inward movement of TM5 at the highly conserved P5.50, rotation of TM3 due to a steric clash with the bulky I3.40, and an outward movement of F6.44 in TM6 (Fig. 5). Unfortunately, these movements are similar for full and partial agonists and cannot be used to explain the different agonist pharmacological profiles.
Fig. 5.

Receptor side-chain movements in response to agonists. Plot of the centroids (calculated from 100 snapshots) of the Cβ atoms of a selected group of 34 amino acids (Table S1) located above (in the ligand binding cavity) and below (in the G protein or β-arrestin binding cavity) the “transmission switch” amino acids obtained during 1 μs of MD simulations of A2AR in the presence of the agonists adenosine, NECA, CGS-21680, PSB-0777 and LUF-5834 and a selective antagonist SCH-58261. The distances between the centroids of the agonist-bound conformations and the centroid of the antagonist-bound conformation were statistically correlated with Emax values measured in cAMP production and β-arrestin recruitment (Table S1). (A) Movement of the salt bridge between E169ECL2 and H264ECL3 that is proposed to govern the residence time of ligands. (B) Movement of E131.39, and the nearby H2787.43, that correlates with β-arrestin recruitment. (C) The proposed mechanism of receptor activation at the “transmission switch” amino acids (inward movement of TM 5, an anticlockwise rotation of TM 3, and an outward movement of TM 6, see arrows) is observed but similar for full and partial agonists. (D) Movement of T883.36 and W2466.48 that correlate with cAMP production (Supplementary Fig. S9).
Thus, in order to understand the structural arrangements of the binding cavity, triggered by these chemically different agonists, facilitating G protein binding or β-arrestin recruitment with different efficacy, we studied the trajectories of a selected group of 34 amino acids either located above (in the ligand binding cavity) and below (in the G protein or arrestin binding cavity) the “transmission switch” amino acids in the presence of agonists and the antagonist (see Supplementary Table S1). In the bivariate correlation analysis, dependent variables are Emax values measured in cAMP production and β-arrestin recruitment, whereas independent variables are the movement of the chosen amino acid in the agonist-bound, active-like complexes relative to the antagonist-bound, inactive-like complex. This is measured as the distance between the centroid (calculated from 100 snapshots obtained during 1 μs of unbiased MD simulations) of the Cβ positions of the chosen amino acids of A2AR bound to agonists and the centroid of A2AR bound to the antagonist. No deviations from normality (Shapiro-Wilk test) are observed in all variables, thus, correlation analyses were performed using a Pearson test for continuous, normally distributed variables.
Although ligand efficacy is a function of multiple factors, we found a clear statistically significant correlation between Emax in cAMP assays and the movement of the side chains of T3.36 (p = 0.033) and W6.48 (p = 0.015) above the “transmission switch” and Y5.58 (p = 0.039) below the “transmission switch” (Supplementary Table S1). Y5.58 is a key amino acid in the process of receptor activation as it stabilizes the extended conformation of R3.50 in the active state [52]. Positions 3.36 and 6.48 have been described as conformational toggle or trigger switches involved in the initial agonist-induced receptor activation in other GPCRs such as cannabinoid CB1R [53], [54], serotonin 5HT4R [55], melanocorin MC4R [56], mGlu2R [57], or A2AR [58]. With the aim of understanding at the molecular level the different activation trends among agonists, we explored the amount of time, in the MD simulations, the side chains of T3.36 and W6.48 spend in the gauche+ (g+, χ1 = -60°), gauche- (g-, χ1 = 60°) or trans (t, χ1 = 180°) conformations (see Supplementary Fig. S9). Clearly, the antagonist-bound structure favors the g+/g+ conformations of T3.36 and W6.48, whereas full agonists adenosine, NECA, and CGS-21680 favor the g-/g+ conformations. Notably, partial agonists PSB-0777 and LUF-5834 cannot achieve these g-/g+ conformations as frequently as full agonists. In this respect, it should be noted that Thr residues in this g- conformation are capable of hydrogen bonding the backbone carbonyl in the previous turn of the helix, which is known to trigger a local opening of the helix [59] that might be necessary for receptor activation. Importantly, the T3.36A mutation impedes signaling [43], [60] and the W6.48A mutation has significant deleterious effects on receptor function [43].
In the case of Emax in β-arrestin recruitment, we found a statistically significant correlation only with the movement of the side chain of E1.39 (p = 0.014) above the “transmission switch” (Supplementary Table S1). Because E1.39 and H7.43 might form an ionic interaction we calculated the distance between these side chains in the presence of the different agonists. However, we have not observed a statistically significant correlation between this distance and Emax in β-arrestin recruitment.
4. Conclusion
The agonist-dependent selectivity for intracellular pathways of A2AR has been studied using, in synergy, molecular modeling tools and pharmacological assays. This combination of expertise has permitted to understand the structural arrangements of the binding cavity, triggered by chemically different agonists, facilitating G protein binding with different efficacy. The mechanism of agonist-induced β-arrestin recruitment seems more difficult to rationalize. First, different ligands stimulation might induce distinct patterns of receptor phosphorylation, which direct specific β-arrestin conformations and functional outcomes [61], [62], [63]. Second, there is increasing evidence that biased signaling could also be a consequence of binding kinetics [64], [65], [66]. In particular, a relation between residence time of ligands with biased signaling in both A1R and A2AR [67], [68] that is governed by a salt bridge between E169ECL2 and H264ECL3 in A2AR (Fig. 5A) [69], [70] has been shown. Finally, the last 40 amino acids of the C-terminal end of A2AR receptor are dispensable for both G-protein and arrestin recruitment and also for MAPK activation (ERK1/2 phosphorylation and DMR).
5. Notes
Authors declare no conflict of interest.
Funding
This work was partially supported by grants from the Spanish Ministry of Economy and Competitiveness (BFU2015-64405-R, SAF2017-84117-R, RTI2018-094204-B-I00 and PID2019-109240RB-I00; they may include FEDER funds), the Alzheimer’s Association (AARFD-17-503612) and by a grant from Fundació la Marató de TV3 (201413-30).
CRediT authorship contribution statement
Gemma Navarro: Conceptualization, Validation, Supervision, Project administration, Resources, Writing - original draft, Funding acquisition. Angel Gonzalez: Validation, Supervision, Project administration, Resources. Stefano Campanacci: Investigation. Rafael Rivas-Santisteban: Investigation. Irene Reyes-Resina: Investigation. Nil Casajuana-Martin: Investigation. Arnau Cordomí: Investigation, Validation, Supervision. Leonardo Pardo: Conceptualization, Validation, Project administration, Resources, Writing - original draft, Funding acquisition. Rafael Franco: Conceptualization, Validation, Project administration, Resources, Writing - original draft, Funding acquisition.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.09.028.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Mickael M.E., Rajput A., Steyn J., Wiemerslage L., Burglin T. An optimised phylogenetic method sheds more light on the main branching events of rhodopsin-like superfamily. Comp Biochem Physiol Part D Genomics Proteomics. 2016;20:85–94. doi: 10.1016/j.cbd.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Fredriksson R., Lagerstrom M.C., Lundin L.G., Schioth H.B. The g-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm B.B., IJzerman A.P., Jacobson K.A., Linden J., Muller C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaakola V.P., Griffith M.T., Hanson M.A., Cherezov V., Chien E.Y., Lane J.R. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandy-Szekeres G., Munk C., Tsonkov T.M., Mordalski S., Harpsoe K., Hauser A.S. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018;46:D440–D446. doi: 10.1093/nar/gkx1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weis W.I., Kobilka B.K. The molecular basis of G protein-coupled receptor activation. Annu Rev Biochem. 2018;87:897–919. doi: 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez A., Cordomí A., Caltabiano G., Campillo M., Pardo L. Impact of helix irregularities on sequence alignment and homology modelling of G protein-coupled receptors. ChemBioChem. 2012;13:1393–1399. doi: 10.1002/cbic.201200189. [DOI] [PubMed] [Google Scholar]
- 8.Ciruela F., Canela L., Burgueno J., Soriguera A., Cabello N., Canela E.I. Heptaspanning membrane receptors and cytoskeletal/scaffolding proteins: focus on adenosine, dopamine, and metabotropic glutamate receptor function. J Mol Neurosci. 2005;26:277–292. doi: 10.1385/JMN:26:2-3:277. [DOI] [PubMed] [Google Scholar]
- 9.Keuerleber S., Gsandtner I., Freissmuth M. From cradle to twilight: the carboxyl terminus directs the fate of the A(2A)-adenosine receptor. Biochim Biophys Acta. 2011;1808:1350–1357. doi: 10.1016/j.bbamem.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Klinger M., Kuhn M., Just H., Stefan E., Palmer T., Freissmuth M. Removal of the carboxy terminus of the A2A-adenosine receptor blunts constitutive activity: differential effect on cAMP accumulation and MAP kinase stimulation. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:287–298. doi: 10.1007/s00210-002-0617-z. [DOI] [PubMed] [Google Scholar]
- 11.Navarro G., Ferre S., Cordomi A., Moreno E., Mallol J., Casado V. Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers. J Biol Chem. 2010;285:27346–27359. doi: 10.1074/jbc.M110.115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kofalvi A., Moreno E., Cordomi A., Cai N.S., Fernandez-Duenas V., Ferreira S.G. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biol. 2020;18:9. doi: 10.1186/s12915-020-0739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro G., Cordomi A., Brugarolas M., Moreno E., Aguinaga D., Perez-Benito L. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018;16:24. doi: 10.1186/s12915-018-0491-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lera Ruiz M., Lim Y.H., Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. 2014;57:3623–3650. doi: 10.1021/jm4011669. [DOI] [PubMed] [Google Scholar]
- 15.Gutierrez-de-Teran H., Sallander J., Sotelo E. Structure-based rational design of adenosine receptor ligands. Curr Top Med Chem. 2017;17:40–58. doi: 10.2174/1568026616666160719164207. [DOI] [PubMed] [Google Scholar]
- 16.Domenici M.R., Ferrante A., Martire A., Chiodi V., Pepponi R., Tebano M.T. Adenosine A2A receptor as potential therapeutic target in neuropsychiatric disorders. Pharmacol Res. 2019;147 doi: 10.1016/j.phrs.2019.104338. [DOI] [PubMed] [Google Scholar]
- 17.Dungo R., Deeks E.D. Istradefylline: first global approval. Drugs. 2013;73:875–882. doi: 10.1007/s40265-013-0066-7. [DOI] [PubMed] [Google Scholar]
- 18.Wisler J.W., Rockman H.A., Lefkowitz R.J. Biased G protein-coupled receptor signaling: changing the paradigm of drug discovery. Circulation. 2018;137:2315–2317. doi: 10.1161/CIRCULATIONAHA.117.028194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valant C., May L.T., Aurelio L., Chuo C.H., White P.J., Baltos J.A. Separation of on-target efficacy from adverse effects through rational design of a bitopic adenosine receptor agonist. PNAS. 2014;111:4614–4619. doi: 10.1073/pnas.1320962111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith J.S., Lefkowitz R.J., Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243–260. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burgueno J., Blake D.J., Benson M.A., Tinsley C.L., Esapa C.T., Canela E.I. The adenosine A2A receptor interacts with the actin-binding protein alpha-actinin. J Biol Chem. 2003;278:37545–37552. doi: 10.1074/jbc.M302809200. [DOI] [PubMed] [Google Scholar]
- 22.Medrano M., Aguinaga D., Reyes-Resina I., Canela E.I., Mallol J., Navarro G. Orexin A/hypocretin modulates leptin receptor-mediated signaling by allosteric modulations mediated by the ghrelin GHS-R1A receptor in hypothalamic neurons. Mol Neurobiol. 2018;55:4718–4730. doi: 10.1007/s12035-017-0670-8. [DOI] [PubMed] [Google Scholar]
- 23.Zhou X.E., He Y., de Waal P.W., Gao X., Kang Y., Van Eps N. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell. 2017;170(457–469) doi: 10.1016/j.cell.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Pinilla E., Rabal O., Reyes-Resina I., Zamarbide M., Navarro G., Sanchez-Arias J.A. Two affinity sites of the cannabinoid subtype 2 receptor identified by a novel homogeneous binding assay. J Pharmacol Exp Ther. 2016;358:580–587. doi: 10.1124/jpet.116.234948. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y.C., Prusoff W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopal S., Ahn S., Rominger D.H., Gowen-MacDonald W., Lam C.M., Dewire S.M. Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol. 2011;80:367–377. doi: 10.1124/mol.111.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Black J.W., Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- 28.Kenakin T., Christopoulos A. Measurements of ligand bias and functional affinity. Nat Rev Drug Discov. 2013;12:483. doi: 10.1038/nrd3954-c2. [DOI] [PubMed] [Google Scholar]
- 29.Webb B., Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinform. 2014;47 doi: 10.1002/0471250953.bi0506s47. 5 6 1-32. [DOI] [PubMed] [Google Scholar]
- 30.Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez-Espigares I., Torrens-Fontanals M., Tiemann J.K.S., Aranda-Garcia D., Ramirez-Anguita J.M., Stepniewski T.M. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat Methods. 2020;17:777–787. doi: 10.1038/s41592-020-0884-y. [DOI] [PubMed] [Google Scholar]
- 32.Massink A., Gutierrez-de-Teran H., Lenselink E.B., Ortiz Zacarias N.V., Xia L., Heitman L.H. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol Pharmacol. 2015;87:305–313. doi: 10.1124/mol.114.095737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu W., Chun E., Thompson A.A., Chubukov P., Xu F., Katritch V. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez-de-Teran H., Massink A., Rodriguez D., Liu W., Han G.W., Joseph J.S. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor. Structure. 2013;21:2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordomi A., Caltabiano G., Pardo L. Membrane protein simulations using AMBER force field and berger lipid parameters. J Chem Theory Comput. 2012;8:948–958. doi: 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- 36.Jespers W., Schiedel A.C., Heitman L.H., Cooke R.M., Kleene L., van Westen G.J.P. Structural mapping of adenosine receptor mutations: ligand binding and signaling mechanisms. Trends Pharmacol Sci. 2018;39:75–89. doi: 10.1016/j.tips.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Ballesteros J.A., Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 38.Pardo L., Deupi X., Dolker N., Lopez-Rodriguez M.L., Campillo M. The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. ChemBioChem. 2007;8:19–24. doi: 10.1002/cbic.200600429. [DOI] [PubMed] [Google Scholar]
- 39.Venkatakrishnan A.J., Ma A.K., Fonseca R., Latorraca N.R., Kelly B., Betz R.M. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc Natl Acad Sci USA. 2019 doi: 10.1073/pnas.1809251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez-Tamayo J.C., Cordomi A., Olivella M., Mayol E., Fourmy D., Pardo L. Analysis of the interactions of sulfur-containing amino acids in membrane proteins. Protein Sci. 2016;25:1517–1524. doi: 10.1002/pro.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J.J., Horst R., Katritch V., Stevens R.C., Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCorvy J.D., Butler K.V., Kelly B., Rechsteiner K., Karpiak J., Betz R.M. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol. 2018;14:126–134. doi: 10.1038/nchembio.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lane J.R., Klein Herenbrink C., van Westen G.J., Spoorendonk J.A., Hoffmann C., IJzerman A.P. A novel nonribose agonist, LUF5834, engages residues that are distinct from those of adenosine-like ligands to activate the adenosine A(2a) receptor. Mol Pharmacol. 2012;81:475–487. doi: 10.1124/mol.111.075937. [DOI] [PubMed] [Google Scholar]
- 44.Wacker D., Wang C., Katritch V., Han G.W., Huang X.P., Vardy E. Structural Features for Functional Selectivity at Serotonin Receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 46.Huang W., Manglik A., Venkatakrishnan A.J., Laeremans T., Feinberg E.N., Sanborn A.L. Structural insights into micro-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sansuk K., Deupi X., Torrecillas I.R., Jongejan A., Nijmeijer S., Bakker R.A. A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol Pharmacol. 2011;79:262–269. doi: 10.1124/mol.110.066068. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen S.G., Choi H.J., Fung J.J., Pardon E., Casarosa P., Chae P.S. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Troupiotis-Tsailaki A., Zachmann J., Gonzalez-Gil I., Gonzalez A., Ortega-Gutierrez S., Lopez-Rodriguez M.L. Ligand chain length drives activation of lipid G protein-coupled receptors. Sci Rep. 2017;7:2020. doi: 10.1038/s41598-017-02104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Izquierdo C., Gomez-Tamayo J.C., Nebel J.C., Pardo L., Gonzalez A. Identifying human diamine sensors for death related putrescine and cadaverine molecules. PLoS Comput Biol. 2018;14 doi: 10.1371/journal.pcbi.1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Llinas Del Torrent C., Casajuana-Martin N., Pardo L., Tresadern G., Perez-Benito L. Mechanisms underlying allosteric molecular switches of metabotropic glutamate receptor 5. J Chem Inf Model. 2019;59:2456–2466. doi: 10.1021/acs.jcim.8b00924. [DOI] [PubMed] [Google Scholar]
- 52.Park J.H., Scheerer P., Hofmann K.P., Choe H.W., Ernst O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 53.McAllister S.D., Hurst D.P., Barnett-Norris J., Lynch D., Reggio P.H., Abood M.E. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 54.Krishna Kumar K., Shalev-Benami M., Robertson M.J., Hu H., Banister S.D., Hollingsworth S.A. Structure of a signaling cannabinoid receptor 1-G protein complex. Cell. 2019;176(448–458) doi: 10.1016/j.cell.2018.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pellissier L.P., Sallander J., Campillo M., Gaven F., Queffeulou E., Pillot M. Conformational toggle switches implicated in basal constitutive and agonist-induced activated states of 5-hydroxytryptamine-4 receptors. Mol Pharmacol. 2009;75:982–990. doi: 10.1124/mol.108.053686. [DOI] [PubMed] [Google Scholar]
- 56.Ersoy B.A., Pardo L., Zhang S., Thompson D.A., Millhauser G., Govaerts C. Mechanism of N-terminal modulation of activity at the melanocortin-4 receptor GPCR. Nat Chem Biol. 2012;8:725–730. doi: 10.1038/nchembio.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez-Benito L., Doornbos M.L.J., Cordomi A., Peeters L., Lavreysen H., Pardo L. Molecular switches of allosteric modulation of the metabotropic glutamate 2 receptor. Structure. 2017;25(1153–1162) doi: 10.1016/j.str.2017.05.021. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez D., Pineiro A., Gutierrez-de-Teran H. Molecular dynamics simulations reveal insights into key structural elements of adenosine receptors. Biochemistry. 2011;50:4194–4208. doi: 10.1021/bi200100t. [DOI] [PubMed] [Google Scholar]
- 59.Deupi X., Olivella M., Sanz A., Dolker N., Campillo M., Pardo L. Influence of the g- conformation of Ser and Thr on the structure of transmembrane helices. J Struct Biol. 2010;169:116–123. doi: 10.1016/j.jsb.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 60.Bertheleme N., Singh S., Dowell S.J., Hubbard J., Byrne B. Loss of constitutive activity is correlated with increased thermostability of the human adenosine A2A receptor. Br J Pharmacol. 2013;169:988–998. doi: 10.1111/bph.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nobles K.N., Xiao K., Ahn S., Shukla A.K., Lam C.M., Rajagopal S. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang F., Yu X., Liu C., Qu C.X., Gong Z., Liu H.D. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun. 2015;6:8202. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Z., Yang F., Zhang D., Liu Z., Lin A., Liu C. Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Mol Pharmacol. 2017;92:201–210. doi: 10.1124/mol.116.107839. [DOI] [PubMed] [Google Scholar]
- 64.Klein Herenbrink C., Sykes D.A., Donthamsetti P., Canals M., Coudrat T., Shonberg J. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grundmann M., Kostenis E. Temporal bias: time-encoded dynamic GPCR signaling. Trends Pharmacol Sci. 2017;38:1110–1124. doi: 10.1016/j.tips.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 66.Jensen D.D., Lieu T., Halls M.L., Veldhuis N.A., Imlach W.L., Mai Q.N. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo D., Mulder-Krieger T., Ijzerman A.P., Heitman L.H. Functional efficacy of adenosine A(2)A receptor agonists is positively correlated to their receptor residence time. Br J Pharmacol. 2012;166:1846–1859. doi: 10.1111/j.1476-5381.2012.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yun Y., Chen J., Liu R., Chen W., Liu C., Wang R. Long residence time adenosine A1 receptor agonists produce sustained wash-resistant antilipolytic effect in rat adipocytes. Biochem Pharmacol. 2019;164:45–52. doi: 10.1016/j.bcp.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 69.Segala E., Guo D., Cheng R.K., Bortolato A., Deflorian F., Dore A.S. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J Med Chem. 2016;59:6470–6479. doi: 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- 70.Guo D., Pan A.C., Dror R.O., Mocking T., Liu R., Heitman L.H. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol Pharmacol. 2016;89:485–491. doi: 10.1124/mol.115.102657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.