

This cross-sectional study used screening data from the Anti-Amyloid Treatment in Asymptomatic Alzheimer Disease Study (A4 Study) to assess whether the presence of the apolipoprotein E ε2 allele is protective against β-amyloid accumulation in the presence of ε4 in individuals without cognitive impairment.
Key Points
Question
Does the ε2 allele remain protective against β-amyloid (Aβ) accumulation in the presence of the ε4 allele?
Findings
In this cross-sectional study of 4432 older participants without cognitive impairment, apolipoprotein E ε2 (APOE ε2) was associated with a reduction in both the overall and age-dependent level of Aβ in the presence of ε4, with Aβ levels in the APOE ε24 group (n = 115) increasing at significantly less than half the rate with respect to increasing age compared with the APOE ε34 group (n = 1295).
Meaning
The protective outcome of carrying an ε2 allele in the presence of an ε4 allele against Aβ accumulation may inform future development of disease-modifying Alzheimer disease therapies.
Abstract
Importance
Although the most common recent approach in Alzheimer disease drug discovery is to directly target the β-amyloid (Aβ) pathway, the high prevalence of apolipoprotein E ε4 (APOE ε4) in Alzheimer disease and the ease of identifying ε4 carriers make the APOE genotype and its corresponding protein (apoE) an appealing therapeutic target to slow Aβ accumulation.
Objective
To determine whether the ε2 allele is protective against Aβ accumulation in the presence of the ε4 allele and evaluate how age and the APOE genotype are associated with emerging Aβ accumulation and cognitive dysfunction.
Design, Setting, and Participants
This cross-sectional study used screening data from the Anti-Amyloid Treatment in Asymptomatic Alzheimer Disease Study (A4 Study) collected from April 2014 to December 2017 and analyzed from November 2019 to July 2020. Of the 6943 participants who were a part of the multicenter clinical trial screening visit, 4432 were adults without cognitive impairment aged 65 to 85 years who completed a fluorine 18–labeled (18F)-florbetapir positron emission tomography scan, had APOE genotype information, and had a Clinical Dementia Rating of 0. Participants who were taking a prescription Alzheimer medication or had a current serious or unstable illness that could interfere with the study were excluded.
Main Outcomes and Measures
Aβ pathology, measured by 18F-florbetapir positron emission tomography and cognition, measured by the Preclinical Alzheimer Cognitive Composite.
Results
A total of 4432 participants were included (mean [SD] age, 71.3 [4.7] years; 2634 women [59.4%]), with a mean (SD) of 16.6 (2.8) years of education and 1512 (34.1%) with a positive Aβ level. APOE ε2 was associated with a reduction in both the overall (standardized uptake value ratio [SUVR], ε24, 1.11 [95% CI, 1.08-1.14]; ε34, 1.18 [95% CI, 1.17-1.19]) and the age-dependent level of Aβ in the presence of ε4, with Aβ levels in the APOE ε24 group (n = 115; ε24, 0.005 SUVR increase per year of age) increasing at less than half the rate with respect to increasing age compared with the APOE ε34 group (n = 1295; 0.012 SUVR increase per year of age; P = .04). The association between Aβ and decreasing Preclinical Alzheimer Cognitive Composite scores did not differ by APOE genotype, and the reduced performance on the Preclinical Alzheimer Cognitive Composite in APOE ε4 carriers compared with noncarriers was completely mediated by Aβ (unadjusted difference in composite scores between ε4 carriers and noncarriers = –0.084, P = .005; after adjusting for 18F-florbetapir = –0.006, P = .85; after adjusting for 18F-florbetapir and cardiovascular scores = –0.009, P = .78).
Conclusions and Relevance
These findings suggest that the protective outcome of carrying an ε2 allele in the presence of an ε4 allele against Aβ accumulation is important for potential treatments that attempt to biochemically mimic the function of the ε2 allele in order to facilitate Aβ clearance in ε4 carriers. Such a treatment strategy is appealing, as ε4 carriers make up approximately two-thirds of patients with Alzheimer disease dementia. This strategy could represent an early treatment option, as many ε4 carriers begin to accumulate Aβ in early middle age.
Introduction
Age and the apolipoprotein E (APOE) genotype are among the strongest risk factors for amyloid-β (Aβ) accumulation. Rates of early Aβ accumulation are highest in APOE ε4 allele carriers and lowest in ε2 allele carriers compared with ε3-only carriers.1 Increasing evidence suggests that the APOE genotype and its corresponding protein (apoE) affect the pathogenesis of Alzheimer disease (AD) through multiple biological pathways, including the differential regulation of Aβ aggregation and clearance.2,3 Two-thirds of patients with AD dementia are APOE ε4 allele carriers.4 Although the most common recent approach in AD drug discovery is to directly target the Aβ pathway, the high prevalence of APOE ε4 in AD and the ease of identifying ε4 carriers at any age make APOE pathways an appealing therapeutic target to slow Aβ accumulation.2
Two single-nucleotide variations in APOE and 3 apoE isoforms (apoE2, apoE3, and apoE4) are thought to have a substantial effect on the structure and function of apoE, including Aβ binding.5 From a treatment standpoint, it is unclear whether strategies that increase the “good” forms of apoE (apoE2, apoE3) or decrease the “bad” form (apoE4) would be most successful.3 By mimicking the biochemical properties associated with the apoE2 isoform, it may be possible to increase the Aβ clearance that is reduced with apoE4. However, a central question is whether apoE2 remains protective in the presence of apoE4. This question has been difficult to answer, in large part because the simultaneous carriage of both the ε2 and ε4 alleles is rare—approximately 2% of the population has the ε24 genotype.6 Even in previous meta-analyses, small sample sizes of the ε24 group precluded precise estimates of the effect of ε2 in the presence of ε4 on Aβ pathology.7 Consequently, questions about the potential for therapies targeting apoE, such as synthetic peptides, to reduce Aβ pathology in the presence of apoE4 remain unanswered.
Because genetic risk factors, such as carriage of APOE ε4, can be determined at birth, targeting apoE4 is of particular interest as an early treatment option. Predicting when individuals may become at increased risk for abnormal rates of Aβ accumulation will help to inform the design of primary prevention. In 4432 participants without cognitive impairment who were screened for participation in the Anti-Amyloid Treatment in Asymptomatic Alzheimer Disease (A4 Study) trial,8 we evaluated how the principal risk factors for AD (age and APOE genotype) were associated with early buildup of Aβ, measured by fluroine 18–labeled (18F)-florbetapir positron emission tomography (PET).
Methods
In this cross-sectional study, data were collected from April 2014 to December 2017 and analyzed from November 2019 to July 2020. Participants screened for inclusion in the A4 Study8 were included in this study if they completed an 18F-florbetapir PET scan, had APOE genotype information, completed a battery of neuropsychological testing, scored between 25 and 30 on the Mini-Mental State Examination (MMSE), had a Clinical Dementia Rating of 0, and were aged between 65 and 85 years. Exclusion criteria for the A4 study have been described previously.9 Briefly, participants were excluded from the A4 Study if they were taking a prescription Alzheimer medication or had a current serious or unstable illness that could interfere with the study. Note that participants without evidence of brain Aβ at screening were not randomized to treatment in the A4 Study but were included in the current study regardless of their PET scan result. This study was approved by the institutional review boards of all participating institutions, and written informed consent was obtained from all participants. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
18F-Florbetapir PET Imaging
β-Amyloid PET imaging in the A4 Study was done using 18F-florbetapir data, which was acquired 50 to 70 minutes postinjection. Images were realigned and averaged and then spatially aligned to a standard space template. 18F-florbetapir, sampled in a global neocortical region for Aβ, was expressed as a standardized uptake value ratio (SUVR) with a cerebellar reference region.10 β-Amyloid positivity was defined as participants with an 18F-florbetapir PET SUVR greater than or equal to 1.10.11,12
Cognitive Testing
The A4 Study participants completed a neuropsychological test battery including the Preclinical Alzheimer Cognitive Composite (PACC),13,14 comprising the MMSE, the Logical Memory Delayed Recall, the Free and Cued Selective Reminding Test, and the Digit Symbol Substitution Test. To calculate the PACC, individual components were z-transformed and summed. The resulting sum was then centered on the mean and SD of the Aβ-negative group.
Cardiovascular Risk Factors
Cardiovascular risk scores were calculated based on body mass index, systolic blood pressure, smoking status, and information gathered during an initial health assessment and physical and neurologic examination. During the initial assessment, participants were asked about the chronicity and severity of underlying health conditions. Chronicity (1 = single occurrence; 2 = intermittent; and 3 = persistent) and severity (1 = mild; 2 = moderate; and 3 = severe) scores were used to calculate the cardiovascular risk score, described further in the Statistical Analysis section. During the physical and neurologic examination, participants were classified as normal or abnormal with regard to cardiac health. Cardiac values (0 = normal; 1 = abnormal) were incorporated into the cardiovascular risk score.
Statistical Analysis
18F-Florbetapir SUVR values (both continuous and dichotomized, separately) were regressed on APOE genotype (all 6 genotypes), adjusting for age and sex. The interaction between APOE genotype and age was also assessed. Models with continuous outcomes (18F-florbetapir PET SUVRs and cognitive scores) were modeled using ordinary least-squares regression. Monotone cubic splines were used to evaluate potential nonlinearity in the associations among 18F-florbetapir PET SUVR, age, and cognition.15 Models with Aβ positivity as the outcome were fit with logistic regression. We also evaluated the interaction between 18F-florbetapir SUVR and APOE genotype to predict PACC scores, as well as the individual components of the PACC in an exploratory analysis. Statistical significance of the associations between the outcome and predictors was tested using likelihood ratio tests and the Akaike information criterion (AIC). A lower value of the AIC indicates a better-fitting model. Multiple-comparison P value adjustment of the PACC components was done using a Holm correction.16 All models predicting 18F-florbetapir PET included age and sex. All models predicting cognition included age, sex, and years of education.
Cardiovascular risk scores were also evaluated for their association with the outcomes and the association between APOE genotype and the outcomes. Cardiovascular risk scores were calculated as the sum of z-transformed body mass index, z-transformed natural log systolic blood pressure, z-transformed product of chronicity and severity of cardiovascular symptoms from the initial health assessment, smoking status (0 = nonsmoker; 1 = smoker), and cardiac symptoms from the physical and neurologic examination (0 = normal; 1 = abnormal). The resulting sum was then z-transformed, providing a summary of cardiovascular risk with higher scores indicating more risk.
Associations between demographics and APOE genotype were assessed using a Kruskal-Wallis test for continuous variables and a χ2 test for categorical variables. Changes in the AIC (ΔAIC) less than –2 and 2-sided P < .05 were considered significant. The PACC P values were obtained using the Holm-Bonferroni method. All analyses were done in R software, version 3.6.0 (R Foundation).
Results
Of the 6943 participants who were part of the multicenter clinical trial screening visit, 4432 adults without cognitive impairment were included (2634 women [59.4%] and 1798 men [40.6%]; mean [SD] age, 71.3 [4.7] years). Individuals had mean (SD) of 16.6 (2.8) years of education, and 1512 had a positive Aβ level (34.1%). Cohort characteristics by APOE genotype are summarized in Table 1.
Table 1. Characteristics of the Study Cohort by APOE Genotype.
| Characteristic | APOE | P value | |||||
|---|---|---|---|---|---|---|---|
| ε22 (n = 25) | ε23 (n = 449) | ε33 (n = 2409) | ε24 (n = 115) | ε34 (n = 1295) | ε44 (n = 139) | ||
| Age, mean (SD), y | 71.1 (4.1) | 71.9 (4.9) | 71.5 (4.8) | 71.5 (5.0) | 70.7 (4.3) | 69.8 (3.8) | <.001 |
| Women, No. (%) | 16 (64.0) | 245 (54.6) | 1455 (60.4) | 66 (57.4) | 771 (59.5) | 81 (58.3) | .32 |
| Race No. (%) | |||||||
| White | 25 (100) | 395 (88.0) | 2188 (90.8) | 103 (89.6) | 1203 (92.9) | 128 (92.1) | .003 |
| Black | 0 | 31 (6.9) | 71 (2.9) | 9 (7.8) | 41 (3.2) | 7 (5.0) | |
| Asian | 0 | 15 (3.3) | 117 (4.9) | 2 (1.7) | 33 (2.6) | 2 (1.4) | |
| American Indian | 0 | 1 (0.2) | 3 (0.1) | 0 | 4 (0.3) | 1 (0.7) | |
| Native Hawaiian | 0 | 0 | 1 (0.04) | 0 | 1 (0.08) | 0 | |
| Multiple | 0 | 4 (0.9) | 14 (0.6) | 0 | 9 (0.7) | 0 | |
| Unknown | 0 | 3 (0.7) | 15 (0.6) | 1 (0.9) | 4 (0.3) | 1 (0.7) | |
| Ethnicity, No. (%) | |||||||
| Hispanic or Latino | 0 | 8 (1.8) | 89 (3.7) | 6 (5.2) | 29 (2.2) | 6 (4.3) | .21 |
| Non-Hispanic or non-Latino | 25 (100) | 438 (97.6) | 2300 (95.5) | 108 (93.9) | 1255 (96.9) | 133 (95.7) | |
| Unknown | 0 | 3 (0.7) | 20 (0.8) | 1 (0.9) | 11 (0.8) | 0 | |
| Education, mean (SD), y | 17.2 (3.5) | 16.5 (3.1) | 16.5 (2.8) | 16.6 (3.2) | 16.7 (2.8) | 16.5 (2.7) | .70 |
| Cardiovascular Risk Score, mean (SD) | –0.06 (1.01) | –0.09 (0.98) | 0.00 (0.99) | 0.16 (0.86) | –0.03 (1.01) | –0.14 (0.96) | .08 |
| Aβ positive, No. (%) | 4 (16.0) | 82 (18.3) | 562 (23.3) | 47 (40.9) | 702 (54.2) | 115 (82.7) | <.001 |
| Aβ SUVR, estimated mean (95% CI) | 1.02 (0.95-1.09) | 1.02 (1.01-1.04) | 1.05 (1.04-1.06) | 1.11 (1.08-1.14) | 1.18 (1.17-1.19) | 1.31 (1.28-1.34) | <.001 |
Abbreviations: Aβ, β-amyloid; APOE, apolipoprotein E; SUVR, standardized uptake value ratio.
APOE and 18F-Florbetapir SUVR
APOE genotype was significantly associated with 18F-florbetapir SUVR (χ2 = 708.93; P < .001). Every APOE allele combination was significantly different from all other combinations with the exception of ε22 vs ε23 (1.02 vs 1.02; P = .91) and ε22 vs ε33 (1.02 vs 1.05; P = .43); note the small sample size of the ε22 group (n = 25). A sample size of 272 for the ε22 group was required to detect the observed difference from the ε33 group with 80% power. Mean 18F-florbetapir SUVR estimates and CIs are summarized in Table 1 and shown in Figure 1. Notably, the ε23 group had a significantly lower mean 18F-florbetapir SUVR compared with the ε33 group (1.02 vs 1.05; P = .01), and the ε24 group had a significantly lower mean 18F-florbetapir SUVR compared with the ε34 group (1.11 vs 1.18; P < .001). Adjusting for cardiovascular risk score did not affect the APOE genotype estimates and was not associated with 18F-florbetapir SUVR (β = 0.0001; P = .98).
Figure 1. Apolipoprotein E (APOE), Age, and Fluorine 18–Labeled (18F)-Florbetapir Standardized Uptake Value Ratio (SUVR).
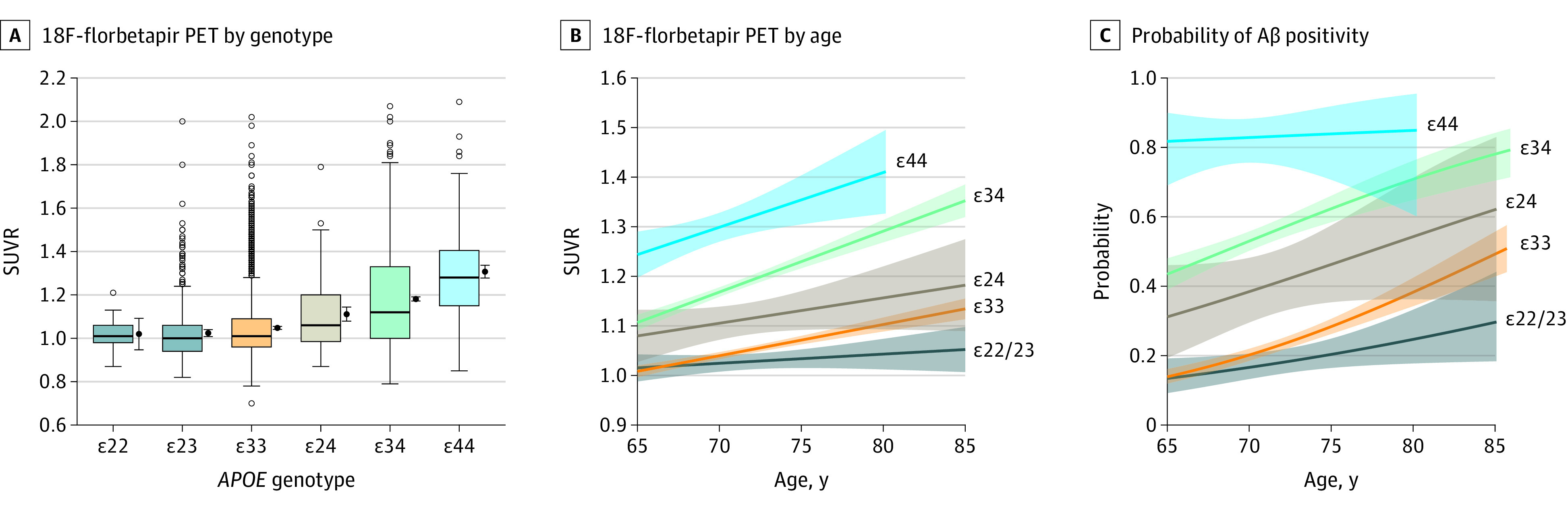
Boxplots of continuous 18F-florbetapir SUVRs are shown for each APOE group (A). The horizontal lines inside the boxes indicate the median; the upper and lower bounds of the boxes indicate the third and first quartiles, respectively; whiskers indicate the most extreme point no more than 1.5 times the interquartile range; and circles indicate outliers. To the right of each boxplot is the mean and 95% CI. The 18F-florbetapir SUVR is plotted by age for each APOE group, separately (ε22 and ε23 participants were combined because of the small sample size over age for the ε22 group) (B). The estimated probability of Aβ positivity is plotted by age for each APOE group, separately (C). Aβ indicates β-amyloid; PET, positron emission tomography. The shaded areas in B and C indicate 95% CIs.
APOE, Age, and 18F-Florbetapir SUVR
There was a significant interaction between APOE genotype and age to predict 18F-florbetapir SUVR (P < .001; ΔAIC = –26.4). The increase in 18F-florbetapir in the ε33 group was 0.006 SUVR per year (for every 1-year increase in age). Comparing each APOE group to the ε33 group, the ε22/ε23 group (combined because of sparse data over age in the ε22 group) increased significantly more slowly (0.002 SUVR per year; P = .01); the ε24 group increased similarly to the ε33 group (0.005 SUVR per year; P = .73); the ε34 group had approximately twice the rate of the ε33 group (0.012 SUVR per year; P < .001); and the ε44 group also had approximately twice the rate, although not significantly different from the ε33 group (0.011 SUVR per year; P = .23), shown in Figure 1. The ε24 group also increased at less than half the rate of the ε34 group (rate difference: 0.005 in the ε24 group vs 0.012 in the ε34 group; P = .04). There was no significant interaction between the APOE genotype and age to predict the odds of Aβ positivity (χ2 = 3.94; P = .41) (Figure 1).
Aβ, APOE, and the PACC
The association between 18F-florbetapir SUVR and decreasing PACC scores did not differ by APOE genotype (ΔAIC = 23.4; P = .97). Cardiovascular risk was associated with worse PACC scores (β = –0.06; P < .001) but did not affect the interaction between age and APOE genotype to predict PACC scores (ΔAIC = 22.9; P = .96). There was also no difference when comparing ε4 carriers to ε4 noncarriers (ΔAIC = 4.4; P = .67 (Figure 2).
Figure 2. Preclinical Alzheimer Cognitive Composite.

Boxplots of the Preclinical Alzheimer Cognitive Composite (PACC) are shown by apolipoprotein E (APOE) genotype (A). The horizontal lines inside the boxes indicate the median; the upper and lower bounds of the boxes indicate the third and first quartiles, respectively; whiskers indicate the most extreme point no more than 1.5 times the interquartile range; and circles indicate outliers. To the right of each boxplot is the mean and 95% CI. PACC scores are plotted against fluroine 18–labeled (18F)-florbetapir standardized uptake value ratios (SUVRs) for APOE ε4 carriers and noncarriers (B). The shaded areas indicate 95% CIs.
When adjusting for age, sex, and education but not 18F-florbetapir SUVR, the APOE ε34 group performed 0.08 points worse on the PACC compared with the APOE ε33 group (β = –0.08; P = .01). There were no other significant differences on the PACC compared with the ε33 group (APOE ε22: β = –0.25, P = .19; APOE ε23: β = –0.01, P = .89; APOE ε24: β = –0.13, P = .14; APOE ε44: β = –0.11; P = .20) (Figure 2). Mean PACC scores and 95% CIs for ε22, ε23, ε33, ε24, ε34, and ε44 were –0.29 (–0.67 to 0.10); –0.04 (–0.13 to 0.04); –0.04 (–0.08 to 0.00); –0.17 (–0.34 to 0.01); –0.12 (–0.17 to –0.07); and –0.14 (–0.30 to 0.01), respectively (Table 2).
Table 2. Association of Cognition With APOE genotype.
| Measure | APOE, mean (95% CI) | |||||
|---|---|---|---|---|---|---|
| ε22 (n = 25) | ε23 (n = 449) | ε33 (n = 2409) | ε24 (n = 115) | ε34 (n = 1295) | ε44 (n = 139) | |
| PACC | –0.29 (–0.67 to 0.10) | –0.04 (–0.13 to 0.04) | –0.04 (–0.08 to 0.00) | –0.17 (–0.34 to 0.01) | –0.12 (–0.17 to –0.07) | –0.14 (–0.30 to 0.01) |
| FCSRT96 | 74.0 (71.8 to 76.3) | 77.0 (76.4 to 77.5) | 76.5 (76.3 to 76.7) | 76.7 (75.7 to 77.7) | 76.0 (75.7 to 76.3) | 75.3 (74.3 to 76.2) |
| Logical Memory Delayed Recall | 12.1 (10.8 to 13.4) | 11.9 (11.6 to 12.2) | 11.8 (11.6 to 11.9) | 11.8 (11.2 to 12.3) | 11.5 (11.4 to 11.7) | 11.9 (11.4 to 12.4) |
| Digit Symbol Substitution | 45.0 (41.5 to 48.4) | 43.1 (42.3 to 43.9) | 44.1 (43.8 to 44.4) | 41.0 (39.5 to 42.6) | 43.8 (43.3 to 44.2) | 42.9 (41.5 to 44.3) |
| MMSE | 28.3 (27.8 to 28.8) | 28.8 (28.7 to 28.9) | 28.8 (28.8 to 28.9) | 28.8 (28.6 to 29.0) | 28.8 (28.7 to 28.8) | 28.8 (28.6 to 29.0) |
Abbreviations: APOE, apolipoprotein E; FCSRT96, Free and Cued Selective Reminding Test; MMSE, Mini-Mental State Examination; PACC, Preclinical Alzheimer Cognitive Composite.
When adjusting for cardiovascular risk, all APOE estimates remained similar, including the effect of APOE ε34 (β = –0.086; P = .007). When also adjusting for 18F-florbetapir SUVR, the effect of the APOE ε34 group was removed (β = –0.012; P = .71). For the ε4 carriers vs noncarriers, the unadjusted difference was –0.084 (P = .005); after adjusting for 18F-florbetapir, the difference was –0.006 (P = .85), and after adjusting for 18F-florbetapir and cardiovascular scores, the difference was –0.009 (P = .78).
Aβ, APOE, and the PACC Components
The association between 18F-florbetapir SUVR and decreasing PACC component scores did not differ by APOE genotype (P > .87; ΔAIC>20.9 for all). When adjusting for demographics but not 18F-florbetapir SUVR or cardiovascular risk, Digit Symbol Substitution Test scores in the APOE ε24 group were reduced (β = –3.06; P = .003). Mean cognitive scores and 95% CIs for ε22, ε23, ε33, ε24, ε34, and ε44 were 45.0 (41.5 to 48.4); 43.1 (42.3 to 43.9); 44.1 (43.8 to 44.4); 41.0 (39.5 to 42.6); 43.8 (43.3 to 44.2); and 42.9 (41.5 to 44.3), respectively (Table 2). This outcome remained after adjusting for 18F-florbetapir SUVR (β = –2.94; P = .005) and also after adjusting for cardiovascular risk (β = –2.97; P = .005).
Discussion
The main findings of this study were (1) APOE ε2 was associated with a reduction in both the overall and the age-dependent level of Aβ in the presence of ε4, (2) large differences in Aβ between APOE groups were already apparent at age 65 years, (3) the association between Aβ and decreasing PACC scores did not differ by APOE genotype, and (4) the associated reduction in performance of the PACC in APOE ε4 carriers compared with noncarriers was completely mediated by Aβ.
There was a large protective outcome of APOE ε2 in the presence of APOE ε4 (Table 1 and Figure 1). β-Amyloid levels in the APOE ε24 group increased at less than half the rate with respect to increasing age compared with the APOE ε34 group. A 2015 meta-analysis did not find a protective effect of carrying the ε2 allele in the presence of ε4 with respect to Aβ positivity.7 However, this study had one-third of the number of ε24 participants (n = 41) compared with the A4 Study, most of whom were younger than 70 years. In Figure 1, separation between the ε24 and ε34 groups becomes clear as the groups approach 70 years of age. The reduced levels of Aβ in ε24 compared with ε34 participants shown here may be one of the primary drivers behind the protective outcome of the ε2 allele against AD dementia, shown previously in a large case-control study.17 The ε24 group demonstrated an associated reduced risk of AD dementia (odds ratio, 2.68 [95% CI, 1.65-4.36]) compared with the ε34 group (odds ratio, 6.13 [95% CI, 5.08-7.41]) when comparing both groups with ε33 participants. However, the presence of the ε2 allele does not completely protect against Aβ positivity, as 16% of ε2 homozygotes in the A4 Study had positive Aβ levels, nor does it completely protect against AD dementia, as 5 of 24 ε2 homozygotes had a neuropathologically confirmed AD dementia diagnosis.17 Although the APOE genotype is one of the strongest risk factors for AD, it does not determine Aβ accumulation or cognitive decline.
One of the largest studies to date evaluating age and APOE (including 2914 participants without cognitive impairment) found Aβ positivity in 13.2% of APOE ε4–negative participants and 37.8% of APOE ε4–positive participants at 65 years and 27.7% and 67.8%, respectively, at 80 years.7 However, a limitation of previous studies is the focus on Aβ positivity (using dichotomous data with conservative thresholds) to define preclinical AD. Using continuous 18F-florbetapir data allows for the estimation of the first increases in Aβ at subthreshold levels. In APOE ε4 carriers, the mean 18F-florbetapir levels were already greatly increased at the minimum age (65 years) compared with APOE ε4 noncarriers. The APOE ε34 and APOE ε44 groups had mean SUVRs of approximately 1.10 and 1.25 at age 65, whereas the ε33 and ε23 groups had mean SUVRs near 1.0. APOE ε4 carrier longitudinal rates of global 18F-florbetapir change have been estimated to be 0.0044 SUVR per year in Aβ-negative individuals and 0.0126 SUVR per year in Aβ-positive individuals,1 suggesting that it would take decades for the APOE ε4 carrier groups to reach the Aβ levels observed at age 65 years in this study. This coincides with the estimated prevalence of Aβ positivity in ε44 individuals between 25% and 30% at age 45 years and 10% in ε34 individuals at age 50 years.7 With Aβ positivity already observable in some individuals in their 40s, the gradual accumulation likely begins much earlier. Indeed, reductions of cerebrospinal fluid Aβ have been observed in ε44 carriers in their 20s.18 A protein-modifying treatment mimicking the protective effect of the ε2 allele against Aβ accumulation may be most effective before significant Aβ deposition. The age at which such a treatment should be initiated would vary greatly by APOE genotype and individual, but if done safely, it could be used to slow Aβ accumulation in early middle age for those at highest risk.
The association between Aβ and performance on the PACC did not differ by APOE genotype. In a recent meta-analysis of 3 large preclinical AD studies,19 2 of the 3 studies did not find an interaction between Aβ and APOE genotype to predict cognitive decline.20,21 The current study, with a sample size 4 times the size of the previous 3 studies combined, shows the same mean PACC score for a given level of Aβ regardless of APOE genotype. When adjusting for Aβ levels, the significant reduction of PACC scores by 0.08 points in ε4 carriers without Aβ adjustment was completely removed. This reduction is quite modest and refers to a reduction of 0.08 SDs within the Aβ-negative group. However, considering that these participants lack cognitive impairment, this outcome may indicate a nonignorable initial level of cognitive dysfunction. This suggests that the negative association of APOE ε4 positivity on cognition is likely mediated entirely by Aβ accumulation at the preclinical stage of AD. Although many Aβ-independent mechanisms of APOE ε4 have been described,3,22 these findings suggest that such Aβ-independent associations of APOE ε4 positivity do not markedly contribute to cognitive decline in the early stages of AD.
Reduced Digit Symbol Substitution test scores in APOE ε2 carriers were unexpected. Although the APOE ε2 allele shows clear protective outcomes against Aβ accumulation, it is also associated with increased risk of atherosclerosis,23 which in turn is linked to increased risk of cognitive decline and vascular dementia.24,25 The associated reduction in executive function and processing speed in the APOE ε24 group observed in this cohort may reflect a non-Alzheimer path to cognitive dysfunction, especially as the adjustment for Aβ burden showed no mediating outcome. Additionally, increased cardiovascular risk scores were associated with decreased cognition in the A4 Study and were highest in the APOE ε24 group, although adjusting for cardiovascular risk factors did not mediate the reduced Digit Symbol Substitution scores observed in the APOE ε24 group. These analyses were exploratory and need to be replicated with longitudinal cognitive trajectories. Still, although the ε2 allele appears to confer protection against Aβ accumulation, safely developing a treatment that mimics its protective outcome will require care not to increase cardiovascular risk or another non-Aβ path to cognitive dysfunction.
Limitations
This study has several limitations. This study was limited to participants without cognitive impairment older than 65 years of age, thereby limiting analyses to the outcome of emerging Aβ pathology in the absence of significant cognitive dysfunction. Importantly, this is a cross-sectional study, and these results do not apply to changes within individuals. Longitudinal follow-up of early middle-aged individuals, with low and intermediate levels of amyloid, will be required to further clarify when APOE groups initially diverge. Importantly, participation in an AD prevention trial is voluntary, which may introduce bias and reduce generalizability. A4 Study participants are highly educated relative to the general population. The vast majority of A4 Study participants are White and are not representative of the population at risk for AD. Exclusion criteria limited participation to those without health conditions that could interfere with the study, which may introduce bias. Although the sample size of our primary group of interest, ε24 carriers, was relatively large compared with previous studies, further subdivision by factors known to be associated with APOE genotype, particularly race/ethnicity,26 were precluded by small sample sizes. Finally, use of the PACC, a cognitive composite, may be limited in its sensitivity to specific cognitive differences depending on the distribution across cognitive domains.
Conclusions
This study’s findings suggest that the protective outcome of carrying an ε2 allele in the presence of an ε4 allele offers potential for a treatment that attempts to mimic this protective outcome in order to facilitate Aβ clearance in ε4 carriers. Such a treatment strategy is appealing, as ε4 carriers make up 67% of patients with AD dementia, and it could represent an early treatment option, as many ε4 carriers begin to accumulate Aβ in early middle age. If the goal is to interfere early in the disease process before activation of downstream pathways, AD prevention trials may consider targeting much younger people before the accumulation of high or even intermediate levels of Aβ develop.
References
- 1.Lim YY, Mormino EC; Alzheimer’s Disease Neuroimaging Initiative . APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology. 2017;89(10):1028-1034. doi: 10.1212/WNL.0000000000004336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suidan GL, Ramaswamy G. Targeting apolipoprotein E for Alzheimer’s disease: an industry perspective. Int J Mol Sci. 2019;20(9):E2161. doi: 10.3390/ijms20092161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106-118. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattsson N, Groot C, Jansen WJ, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 2018;14(7):913-924. doi: 10.1016/j.jalz.2018.02.009 [DOI] [PubMed] [Google Scholar]
- 5.Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740-754. doi: 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240(4852):622-630. doi: 10.1126/science.3283935 [DOI] [PubMed] [Google Scholar]
- 7.Jansen WJ, Ossenkoppele R, Knol DL, et al. ; Amyloid Biomarker Study Group . Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924-1938. doi: 10.1001/jama.2015.4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sperling RA, Donohue MC, Raman R, et al. ; A4 Study Team . Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol. 2020;77(6):735-745. doi: 10.1001/jamaneurol.2020.0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson KA, Schultz AP, Raman R, et al. Tau PET in A4: preliminary report. Alzheimers Dement. 2018;14(7):P1583-P1584. doi: 10.1016/j.jalz.2018.07.144 [DOI] [Google Scholar]
- 11.Joshi AD, Pontecorvo MJ, Clark CM, et al. ; Florbetapir F 18 Study Investigators . Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer’s disease and cognitively normal subjects. J Nucl Med. 2012;53(3):378-384. doi: 10.2967/jnumed.111.090340 [DOI] [PubMed] [Google Scholar]
- 12.Clark CM, Pontecorvo MJ, Beach TG, et al. ; AV-45-A16 Study Group . Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 2012;11(8):669-678. doi: 10.1016/S1474-4422(12)70142-4 [DOI] [PubMed] [Google Scholar]
- 13.Donohue MC, Sperling RA, Salmon DP, et al. ; Australian Imaging, Biomarkers, and Lifestyle Flagship Study of Ageing; Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Cooperative Study . The Preclinical Alzheimer Cognitive Composite: measuring amyloid-related decline. JAMA Neurol. 2014;71(8):961-970. doi: 10.1001/jamaneurol.2014.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donohue MC, Sperling RA, Petersen R, Sun C-K, Weiner MW, Aisen PS; Alzheimer’s Disease Neuroimaging Initiative . Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA. 2017;317(22):2305-2316. doi: 10.1001/jama.2017.6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramsay JO. Monotone regression splines in action. Statist Sci. 1988;3(4):425-441. doi: 10.1214/ss/1177012761 [DOI] [Google Scholar]
- 16.Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6(2):65-70. [Google Scholar]
- 17.Reiman EM, Arboleda-Velasquez JF, Quiroz YT, et al. ; Alzheimer’s Disease Genetics Consortium . Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667. doi: 10.1038/s41467-019-14279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lautner R, Insel PS, Skillbäck T, et al. Preclinical effects of APOE ε4 on cerebrospinal fluid Aβ42 concentrations. Alzheimers Res Ther. 2017;9(1):87. doi: 10.1186/s13195-017-0313-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Insel PS, Weiner M, Mackin RS, et al. Determining clinically meaningful decline in preclinical Alzheimer disease. Neurology. 2019;93(4):e322-e333. doi: 10.1212/WNL.0000000000007831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim YY, Villemagne VL, Pietrzak RH, et al. ; Australian Imaging, Biomarkers and Lifestyle (AIBL) Research Group . APOE ε4 moderates amyloid-related memory decline in preclinical Alzheimer’s disease. Neurobiol Aging. 2015;36(3):1239-1244. doi: 10.1016/j.neurobiolaging.2014.12.008 [DOI] [PubMed] [Google Scholar]
- 21.Mormino E, Betensky RA, Hedden T, et al. ; Alzheimer's Disease Neuroimaging Initiative; Australian Imaging Biomarkers and Lifestyle Flagship Study of Ageing; Harvard Aging Brain Study. Amyloid and APOE4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82(20):1760-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501-518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med (Berl). 2016;94(7):739-746. doi: 10.1007/s00109-016-1427-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinkers DJ, Stek ML, van der Mast RC, et al. Generalized atherosclerosis, cognitive decline, and depressive symptoms in old age. Neurology. 2005;65(1):107-112. doi: 10.1212/01.wnl.0000167544.54228.95 [DOI] [PubMed] [Google Scholar]
- 25.Hofman A, Ott A, Breteler MMB, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 1997;349(9046):151-154. doi: 10.1016/S0140-6736(96)09328-2 [DOI] [PubMed] [Google Scholar]
- 26.Evans DA, Bennett DA, Wilson RS, et al. Incidence of Alzheimer disease in a biracial urban community: relation to apolipoprotein E allele status. Arch Neurol. 2003;60(2):185-189. doi: 10.1001/archneur.60.2.185 [DOI] [PubMed] [Google Scholar]