

Abstract
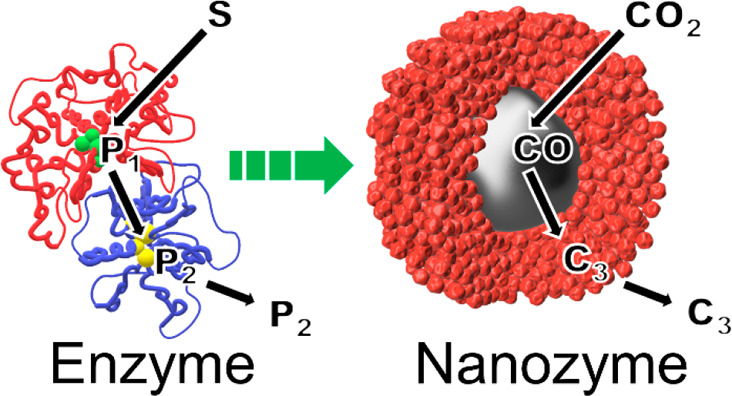
Enzymes can perform complex multistep cascade reactions by linking multiple distinct catalytic sites via substrate channeling. We mimic this feature in a generalized approach with an electrocatalytic nanoparticle for the carbon dioxide reduction reaction comprising a Ag core surrounded by a porous Cu shell, providing different active sites in nanoconfined volumes. The architecture of the nanozyme provides the basis for a cascade reaction, which promotes C–C coupling reactions. The first step occurs on the Ag core, and the subsequent steps on the porous copper shell, where a sufficiently high CO concentration due to the nanoconfinement facilitates C–C bond formation. The architecture yields the formation of n-propanol and propionaldehyde at potentials as low as −0.6 V vs RHE.
The outstanding turnover rates and selectivity of enzymes, as nature’s catalysts, have long provided inspiration to researchers.1,2 Integral to their performance is a key structural feature: a catalytically active site located at the end of a substrate channel. This structural feature allows control over the local solution environment in which the reaction occurs and spatially constrains the reactants for optimal reactivity. Incorporating more than one type of active site in the substrate channel enables multiple steps in a cascade reaction to proceed in one enzyme complex.3,4 This substrate channeling of intermediate products to new active centers prevents their diffusion to the bulk solution. This enables locally high concentrations of an intermediate product, facilitating the formation of more complex molecules from simple reactants.5,6 Mimicking these features in an attempt to obtain the superior catalytic behavior of enzymes in artificial systems is highly desirable in heterogeneous catalysis.7−9
We recently articulated a nanozyme concept where nanoparticles mimic the three-dimensional architecture of enzymes with an active site inside a substrate channel. We showed that the control over the solution environment inside this substrate channel resulted in substantial enhancement in the electrocatalytic activity of a PtNi nanoparticle for the oxygen reduction reaction.10 Herein, we extend the nanozyme concept to cascade reactions for which multiple active sites with different catalytic properties are located in close proximity within a nanoconfined substrate channel. The electrocatalytic CO2 reduction reaction (CO2RR) was used as a model reaction, where the multistep conversion of CO2 into a range of higher organic molecules is achieved via sequential proton-coupled electron transfer reactions.11,12
The CO2RR to higher products can be divided into two major processes: (i) electroreduction of CO2 to CO and (ii) the bimolecular electroreduction of CO to multiple higher organic molecules. The latter reaction depends on multiple parameters including the nature of the catalyst, local pH, electrolyte, temperature, and CO2 and CO concentrations.13 Whereas Ag shows high catalytic activity for converting CO2 to CO with high selectivity at a potential as low as −0.60 V vs RHE, it is inactive for the follow-up reactions.14−16 Cu is able to catalyze the second step of the cascade reaction, the reduction of CO to C≥2 products, at cathodic potentials as low as −0.25 V vs RHE.17−22 However, at −0.60 V vs RHE Cu exhibits a negligible activity for the formation of CO from CO2.23−25 In order to couple the properties of Ag and Cu and decrease the required overpotential for the formation of C≥2 products, the two-step conversion of CO2 on bimetallic catalysts was recently investigated.26−28 Herein, in contrast to conventional bimetallic systems where active sites are exposed to the bulk of the electrolyte, we suggest enzyme-inspired substrate channeling to connect two different catalytic sites in a nanozyme approach toward CO2RR. The proposed nanozyme architecture is shown to support a cascade reaction mechanism, which ultimately shifts the product distribution to C≥2 products such as n-propanol and propionaldehyde at low overpotentials.
A nanozyme with Ag and Cu active sites for CO2RR needs to fulfill two structural requirements to enable substrate channeling for an enzyme-like cascade reaction: (i) accessibility for reactants to the first active site and (ii) channeling of the intermediates to the second active site within a nanoconfined volume for further reaction prior to their diffusion into the bulk solution. Exploiting the aforementioned properties of Ag and Cu for a cascade reaction, the proposed nanozyme in Scheme 1 comprises a Ag core acting as primary active site at the bottom of a mesoporous Cu shell. The Cu shell serves as both the secondary active site and the substrate channel. In this scenario, CO2 is reduced to CO on the Ag, not the Cu. The CO produced is then spatially confined in the Cu channels, where a bimolecular C–C coupling reaction between two CO occurs next, leading toward higher order organic molecules.
Scheme 1. Cascade Reaction Mechanism on a Ag–Cu Nanozyme for CO2RR.
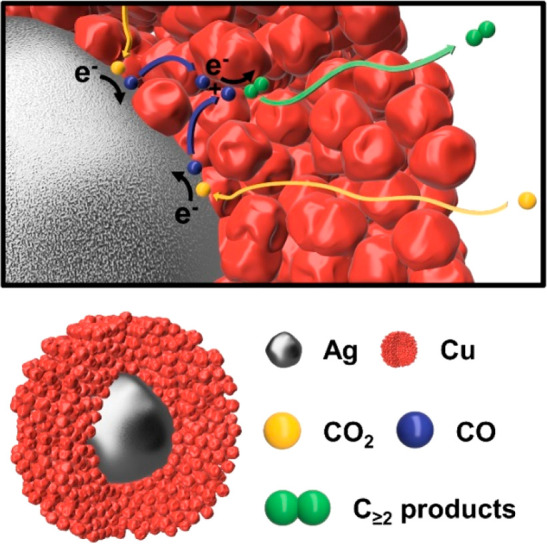
In the nanozyme, we propose the CO2RR is following the cascade reaction mechanism: (i) diffusion of CO2 through the porous Cu shell to the Ag core, (ii) adsorption of CO2 on the Ag surface, (iii) proton-coupled reduction of CO2 to CO, (iv) desorption of CO and diffusion through the porous Cu shell, (v) adsorption of CO on the Cu channel walls, (vi) proton-coupled bimolecular reduction of multiple CO molecules to C≥2 products, and (vii) desorption and diffusion of C≥2 products to the bulk of the solution.
A two-step seeded growth synthesis procedure was employed. First the Ag core29 was synthesized. It was then encapsulated within a porous Cu shell30 (see methods in the SI). Transmission electron microscopy (TEM) reveals that Ag-core–porous-Cu-shell particles, which constitute the nanozyme, were obtained (Figure 1a). The nanozymes exhibit an average diameter of 145 ± 15 nm with an average Ag-core diameter of 75 ± 11 nm and an average porous-Cu-shell thickness of 35 ± 9 nm (Figure S1). Energy dispersive X-ray spectroscopy (EDX) allows for distinction between both metals and indicates that no alloying between Ag and Cu was observable (Figure 1b). The porosity of the Cu shell is visible in dark-field scanning transmission electron microscopy (DF-STEM) and scanning electron microscopy (SEM) images (Figure 1a,c respectively). Selected area electron diffraction (SAED) confirms the presence of face-centered-cubic Ag and cubic Cu2O (Figure 1d and S2). Hence, it is assumed that the active Cu sites of the nanozyme during CO2RR are derived from Cu oxide.31,32 Altogether, electron microscopy provides conclusive evidence for the synthesis of the nanozyme as proposed in Scheme 1.
Figure 1.
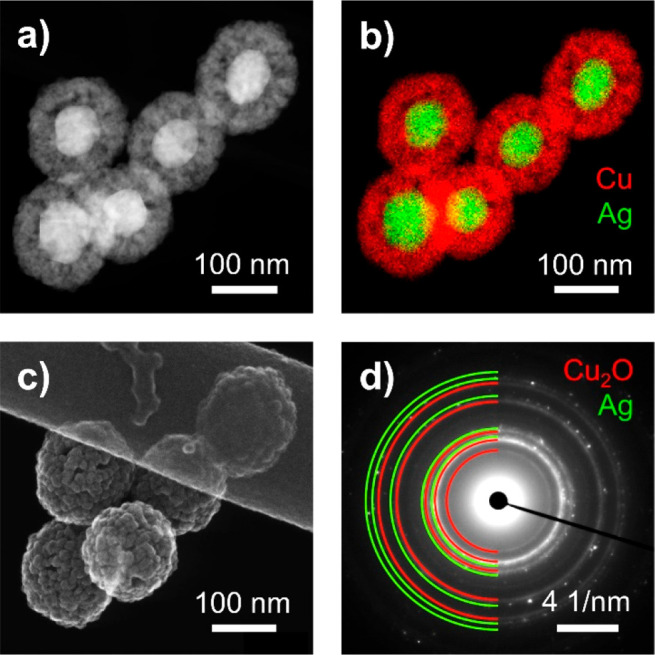
(a) DF-STEM image, (b) EDX elemental mapping of Ag (green) and Cu (red), (c) SEM image, and (d) SAED of the synthesized nanozyme particles.
Electrochemical addressability of the core of the nanozyme was demonstrated by anodically stripping the Ag in sodium thiocyanate solution (Figure S3). Partial dissolution of Ag verified the successful wetting of the channels with electrolyte as well as electric contact down to the Ag core of the nanozyme. Traces of Ag on the surface and throughout the particles in postexperiment TEM images attest to the ability of Ag ions to diffuse through the substrate channels to the exterior of the nanozymes (Figures S3 and S4). Once it was demonstrated that the Ag cores are electrochemically accessible, electrochemical reduction of CO2 was performed next. Different potentials were applied in order to investigate the influence of the structural features of the nanozyme on the potential-dependent product distribution of the CO2RR.
Electrochemical analysis was performed in CO2-saturated 0.1 M KHCO3 as electrolyte (pH = 6.8). Chronoamperograms were recorded for a duration of 120 min each at five different potentials between −0.55 and −0.80 V vs RHE (Figure S5), the potential range in which a substantial increase in CO formation on the Ag core is expected. Gaseous products were analyzed and quantified by online gas chromatography and liquid products by 1H NMR from aliquots of the electrolyte. Along with formate and hydrogen, five major products were detected: carbon monoxide (CO), ethylene (C2H4), ethanol (C2H6O), and n-propanol/propionaldehyde (C3H8O). Methane (CH4) was only present in trace amounts (Figure S6). Comparison of the conversion rate profiles for the different products depending on the electrolysis potential (Figure 2) reveals two major changes in product distribution with increasing potential.
Figure 2.
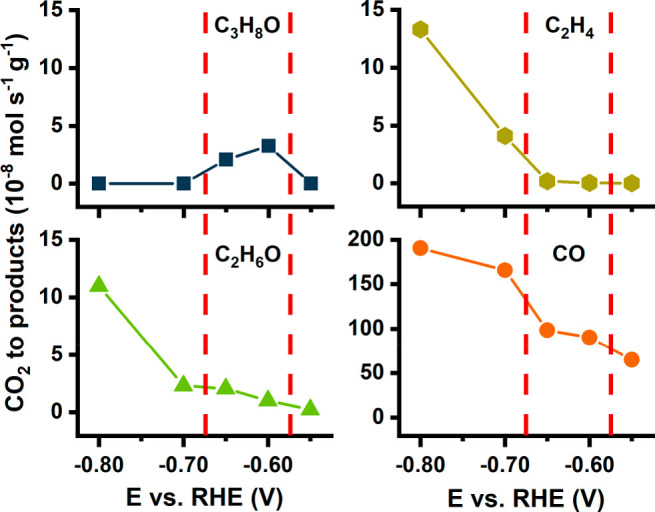
Product formation rates of CO, C2H4, C2H6O, and C3H8O as moles per second and gram of nanozyme particles. Vertical lines separate three distinct potential windows.
At the lowest applied cathodic potential of −0.55 V vs RHE, the only detectable product is CO. At −0.60 and −0.65 V vs RHE an increase in CO formation was accompanied by the conversion of CO2 into C2 products, mainly C2H6O. The formation of C3H8O is also observed, with the formation rate of the C3H8O being higher than that observed for C2H6O. Subsequent 1H NMR analysis revealed that the C3H8O signal is a superposition of n-propanol and propionaldehyde in the ratio of 1 to 1.78 (Figure S7). At more cathodic potentials of −0.70 and −0.80 V vs RHE, a substantial difference in the product distribution is observed with the disappearance of C3 products and a significant increase in the CO, C2H4, and C2H6O production rates.
Changes in the product distribution allow for the designation of three distinct potential windows, each characterized by specific CO2 conversion scenarios (Figure 3a). The formation of CO and the absence of any measurable C≥2 products characterize the first potential window at −0.55 V vs RHE, where the proposed cascade mechanism is not in effect. However, it is possible to reduce CO to C≥2 products on Cu at this potential.17−22 Therefore, it can be hypothesized that slow conversion of CO2 to CO on the Ag core and Cu channels at this potential does not provide a sufficient local CO concentration to promote measurable product formation in highly concentration-dependent bimolecular C–C coupling reactions on Cu inside the substrate channels.
Figure 3.
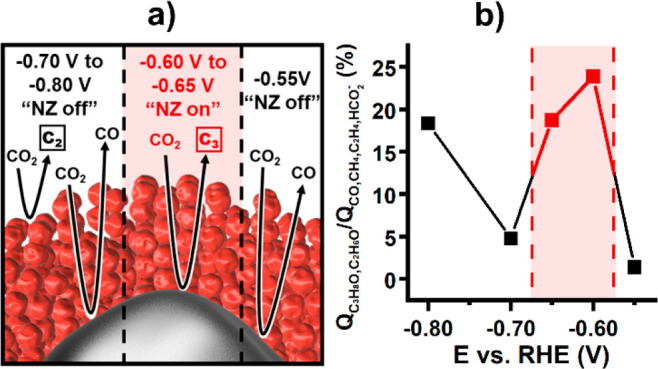
(a) Schematic representation of processes occurring at the specific potential windows with active (red) or inactive (black) nanozyme cascade mechanism. (b) Preference for the formation of C2 and C3 products over other carbon-containing products as charge ratio. Nanozyme activity window is marked in red.
The formation of C3H8O as well as C2H6O at the potentials of −0.60 and −0.65 V vs RHE marks the second window. No formation of C3 products is observed at the same potentials for a control nanoparticle composed of porous Cu without any Ag (Figures S8 and S9). The formation of C3H8O on the nanozyme combined with its absence on the pure porous Cu control nanoparticle verifies the effect of the suggested cascade reaction mechanism within the nanoconfined space with both catalytic sites located in close proximity. It demonstrates the viability of the proposed nanozyme concept for the CO2RR, since a preference for C3 over C2 products is unreported in the literature at −0.60 V vs RHE. In accordance with the cascade mechanism depicted in Scheme 1, we hypothesize that the formation of C3H8O at this potential window is a result of the nanozyme architecture, which combines (i) an increased local concentration of CO inside the substrate channel, which induces a subsequent spillover of CO from Ag onto the surface of Cu,26−28 and (ii) a prolonged residence time of the CO intermediate within the channel system before it diffuses into the bulk of the solution.22,33,34 Both features increase the likelihood of C2–C1 coupling and hence promote the formation rate of this bimolecular reaction.
The disappearance of C3 species at the more cathodic potentials of −0.70 and −0.80 V vs RHE implies that the previously described cascade mechanism is inactive in this potential window. Since very similar behavior in terms of increased C2 product formation is observed for both porous Cu nanoparticles and nanozymes, active CO2RR directly on the Cu active sites is suggested (Figure S9). Compared with the control porous Cu nanoparticles, the nanozyme exhibits, however, a significantly higher formation rate for CO. This finding suggests that the Ag core of the nanozymes remains accessible and active for the formation of CO, yet the cascade is suppressed within the channels. The observation can be rationalized by considering that a significant increase in the hydrogen evolution reaction (HER) rate occurs at these higher overpotentials (Figure S6). At these conditions, HER is known to act as a strong competitor for the reduction of CO2 and CO on Cu by blocking active sites as well as increasing the local pH value.26,35−37 These effects apply particularly to the spatially confined mesoporous architecture of the substrate channels, where hindered mass transport prevents fast equilibration with the bulk solution.10 The changes in the reaction environment in the nanozyme substrate channel appear to be detrimental for the formation rate of C3 species, which are the product of multiple proton-coupled electron transfer reactions and are therefore substantially influenced by the proton concentration as well as the adsorption of multiple intermediate products. The interplay of these effects in the channels creates a narrow potential window, within which the cascade mechanism on nanozymes can be successfully exploited to enhance the electrochemical production of C≥2 products from CO2 (Figure 3b).
In conclusion, we have taken inspiration from architectural features that allow enzymes to perform cascade reactions and transposed them to an electrocatalytic nanoparticle system for the CO2RR reaction. It was demonstrated that the local confinement of different active sites within the nanozyme substrate channels leads to a shift of the potential-dependent product distribution toward C≥2 products at lower overpotentials. The concept is relevant well beyond the CO2RR. The presented results encourage the extension of the nanozyme paradigm to other catalytic multistep reactions to fully exploit the capabilities of different catalytically active sites.
Acknowledgments
W.S. is grateful for financial support by the Deutsche Forschungsgemeinschaft (EXC-2033-390677874) and the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement no. 833408). This work was supported by the DAAD in the framework of the PPP project 57446293 as well as the Ruhr University Research school PLUS through a research stay of P.W. at UNSW, funded by Germany’s Excellence Initiative (DFG GSC 98/3). This research was financially supported by the Australian Research Council of Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036), the ARC Australian Laureate Fellowship (FL150100060), the Discovery Project (DP190102659), and the Mark Wainwright Analytical Centre (MWAC) at UNSW. This work used the facilities supported by Microscopy Australia at the Electron Microscope Unit at UNSW. The authors are grateful to Dr. Douglas Lawes for discussion and assistance with 1H NMR experiments. P.B.O’M. acknowledges the Australian Government Research Training Program Scholarship for financial support. P.W. is grateful to the Association of the Chemical Industry e.V. (VCI) for funding of the Ph.D. fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b07310.
Materials and methods section, TEM and size distribution of nanozymes and copper control, indexed SAED data, additional information on the anodic Ag stripping, chronoamperograms and SEM of nanozyme CO2RR, product formation rates of nanozyme and copper control particles during CO2RR, high-resolution 1H NMR of n-propanol/propionaldehyde peak (PDF)
Author Contributions
# P.B.O’M., P.W., and T.M.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang Y.; Ren J.; Qu X. Nanozymes: Classification, Catalytic Mechanisms, Activity Regulation, and Applications. Chem. Rev. 2019, 119, 4357–4412. 10.1021/acs.chemrev.8b00672. [DOI] [PubMed] [Google Scholar]
- Haseloff J.; Gerlach W. L. Simple RNA Enzymes with New and Highly Specific Endoribonuclease Activities. Nature 1988, 334, 585–591. 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- Wheeldon I.; Minteer S. D.; Banta S.; Barton S. C.; Atanassov P.; Sigman M. Substrate Channelling as an Approach to Cascade Reactions. Nat. Chem. 2016, 8, 299–309. 10.1038/nchem.2459. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Moore B. S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem., Int. Ed. 2019, 58, 6846–6879. 10.1002/anie.201807844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetlove L. J.; Fernie A. R. The Role of Dynamic Enzyme Assemblies and Substrate Channelling in Metabolic Regulation. Nat. Commun. 2018, 9, 2136. 10.1038/s41467-018-04543-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Holden H. M.; Raushel F. M. Channeling of Substrates and Intermediates in Enzyme-Catalyzed Reactions. Annu. Rev. Biochem. 2001, 70, 149–180. 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- Gutzler R.; Stepanow S.; Grumelli D.; Lingenfelder M.; Kern K. Mimicking Enzymatic Active Sites on Surfaces for Energy Conversion Chemistry. Acc. Chem. Res. 2015, 48, 2132–2139. 10.1021/acs.accounts.5b00172. [DOI] [PubMed] [Google Scholar]
- Wu J.; Wang X.; Wang Q.; Lou Z.; Li S.; Zhu Y.; Qin L.; Wei H. Nanomaterials with Enzyme-Like Characteristics (Nanozymes): Next-Generation Artificial Enzymes (II). Chem. Soc. Rev. 2019, 48, 1004–1076. 10.1039/C8CS00457A. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Deng K.; He L.; Liu Y.; Li G.; Zhao H.; Tang Z. Core-shell Palladium Nanoparticle@Metal-Organic Frameworks as Multifunctional Catalysts for Cascade Reactions. J. Am. Chem. Soc. 2014, 136, 1738–1741. 10.1021/ja411468e. [DOI] [PubMed] [Google Scholar]
- Benedetti T. M.; Andronescu C.; Cheong S.; Wilde P.; Wordsworth J.; Kientz M.; Tilley R. D.; Schuhmann W.; Gooding J. J. Electrocatalytic Nanoparticles That Mimic the Three-Dimensional Geometric Architecture of Enzymes: Nanozymes. J. Am. Chem. Soc. 2018, 140, 13449–13455. 10.1021/jacs.8b08664. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc., Faraday Trans. 1 1989, 85, 2309. 10.1039/f19898502309. [DOI] [Google Scholar]
- Gattrell M.; Gupta N.; Co A. A Review of the Aqueous Electrochemical Reduction of CO2 to Hydrocarbons at Copper. J. Electroanal. Chem. 2006, 594, 1–19. 10.1016/j.jelechem.2006.05.013. [DOI] [Google Scholar]
- Ross M. B.; De Luna P.; Li Y.; Dinh C.-T.; Kim D.; Yang P.; Sargent E. H. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2019, 488, 294. 10.1038/s41929-019-0306-7. [DOI] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Ma M.; Trześniewski B. J.; Xie J.; Smith W. A. Selective and Efficient Reduction of Carbon Dioxide to Carbon Monoxide on Oxide-Derived Nanostructured Silver Electrocatalysts. Angew. Chem., Int. Ed. 2016, 55, 9748–9752. 10.1002/anie.201604654. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Rosen J.; Zhou Y.; Hutchings G. S.; Kimmel Y. C.; Chen J. G.; Jiao F. A Selective and Efficient Electrocatalyst for Carbon Dioxide Reduction. Nat. Commun. 2014, 5, 3242. 10.1038/ncomms4242. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508, 504–507. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jiang K.; Fan S.; Kanan M. W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. 10.1021/acscentsci.6b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y.; Takahashi R.; Yoshinami Y.; Murata A. Electrochemical Reduction of CO at a Copper Electrode. J. Phys. Chem. B 1997, 101, 7075–7081. 10.1021/jp970284i. [DOI] [Google Scholar]
- Pang Y.; Li J.; Wang Z.; Tan C.-S.; Hsieh P.-L.; Zhuang T.-T.; Liang Z.-Q.; Zou C.; Wang X.; Luna P. de; Edwards J. P.; Xu Y.; Li F.; Dinh C.-T.; Zhong M.; Lou Y.; Wu D.; Chen L.-J.; Sargent E. H. Efficient Electrocatalytic Conversion of Carbon Monoxide to Propanol Using Fragmented Copper. Nat. Catal. 2019, 2, 251–258. 10.1038/s41929-019-0225-7. [DOI] [Google Scholar]
- Jouny M.; Luc W.; Jiao F. High-rate electroreduction of carbon monoxide to multi-carbon products. Nat. Catal. 2018, 1, 748–755. 10.1038/s41929-018-0133-2. [DOI] [Google Scholar]
- Zhuang T.-T.; Pang Y.; Liang Z.-Q.; Wang Z.; Li Y.; Tan C.-S.; Li J.; Dinh C. T.; de Luna P.; Hsieh P.-L.; Burdyny T.; Li H.-H.; Liu M.; Wang Y.; Li F.; Proppe A.; Johnston A.; Nam D.-H.; Wu Z.-Y.; Zheng Y.-R.; Ip A. H.; Tan H.; Chen L.-J.; Yu S.-H.; Kelley S. O.; Sinton D.; Sargent E. H. Copper Nanocavities Confine Intermediates for Efficient Electrosynthesis of C3 Alcohol Fuels from Carbon Monoxide. Nat. Catal. 2018, 1, 946–951. 10.1038/s41929-018-0168-4. [DOI] [Google Scholar]
- Zhang H.; Li J.; Cheng M.-J.; Lu Q. CO Electroreduction: Current Development and Understanding of Cu-Based Catalysts. ACS Catal. 2019, 9, 49–65. 10.1021/acscatal.8b03780. [DOI] [Google Scholar]
- Hatsukade T.; Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. Insights into the Electrocatalytic Reduction of CO2 on Metallic Silver Surfaces. Phys. Chem. Chem. Phys. 2014, 16, 13814–13819. 10.1039/C4CP00692E. [DOI] [PubMed] [Google Scholar]
- Kuhl K. P.; Hatsukade T.; Cave E. R.; Abram D. N.; Kibsgaard J.; Jaramillo T. F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- Morales-Guio C. G.; Cave E. R.; Nitopi S. A.; Feaster J. T.; Wang L.; Kuhl K. P.; Jackson A.; Johnson N. C.; Abram D. N.; Hatsukade T.; Hahn C.; Jaramillo T. F. Improved CO2 Reduction Activity Towards C2+ Alcohols on a Tandem Gold on Copper Electrocatalyst. Nat. Catal. 2018, 1, 764–771. 10.1038/s41929-018-0139-9. [DOI] [Google Scholar]
- Huang J.; Mensi M.; Oveisi E.; Mantella V.; Buonsanti R. Structural Sensitivities in Bimetallic Catalysts for Electrochemical CO2 Reduction Revealed by Ag-Cu Nanodimers. J. Am. Chem. Soc. 2019, 141, 2490–2499. 10.1021/jacs.8b12381. [DOI] [PubMed] [Google Scholar]
- Lee S.; Park G.; Lee J. Importance of Ag–Cu Biphasic Boundaries for Selective Electrochemical Reduction of CO2 to Ethanol. ACS Catal. 2017, 7, 8594–8604. 10.1021/acscatal.7b02822. [DOI] [Google Scholar]
- Chen B.; Jiao X.; Chen D. Size-Controlled and Size-Designed Synthesis of Nano/Submicrometer Ag Particles. Cryst. Growth Des. 2010, 10, 3378–3386. 10.1021/cg901497p. [DOI] [Google Scholar]
- Zhang L.; Jing H.; Boisvert G.; He J. Z.; Wang H. Geometry Control and Optical Tunability of Metal-Cuprous Oxide Core-Shell Nanoparticles. ACS Nano 2012, 6, 3514–3527. 10.1021/nn300546w. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Kanan M. W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. 10.1021/ja3010978. [DOI] [PubMed] [Google Scholar]
- Mandal L.; Yang K. R.; Motapothula M. R.; Ren D.; Lobaccaro P.; Patra A.; Sherburne M.; Batista V. S.; Yeo B. S.; Ager J. W.; Martin J.; Venkatesan T. Investigating the Role of Copper Oxide in Electrochemical CO2 Reduction in Real Time. ACS Appl. Mater. Interfaces 2018, 10, 8574–8584. 10.1021/acsami.7b15418. [DOI] [PubMed] [Google Scholar]
- Yang K. D.; Ko W. R.; Lee J. H.; Kim S. J.; Lee H.; Lee M. H.; Nam K. T. Morphology-Directed Selective Production of Ethylene or Ethane from CO2 on a Cu Mesopore Electrode. Angew. Chem., Int. Ed. 2017, 56, 796–800. 10.1002/anie.201610432. [DOI] [PubMed] [Google Scholar]
- Merino-Garcia I.; Albo J.; Irabien A. Tailoring Gas-Phase CO2 Electroreduction Selectivity to Hydrocarbons at Cu Nanoparticles. Nanotechnology 2018, 29, 14001. 10.1088/1361-6528/aa994e. [DOI] [PubMed] [Google Scholar]
- Varela A. S.; Kroschel M.; Reier T.; Strasser P. Controlling the Selectivity of CO2 Electroreduction on Copper: The Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8–13. 10.1016/j.cattod.2015.06.009. [DOI] [Google Scholar]
- Seifitokaldani A.; Gabardo C. M.; Burdyny T.; Dinh C.-T.; Edwards J. P.; Kibria M. G.; Bushuyev O. S.; Kelley S. O.; Sinton D.; Sargent E. H. Hydronium-Induced Switching between CO2 Electroreduction Pathways. J. Am. Chem. Soc. 2018, 140, 3833–3837. 10.1021/jacs.7b13542. [DOI] [PubMed] [Google Scholar]
- Clark E. L.; Bell A. T. Direct Observation of the Local Reaction Environment during the Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2018, 140, 7012–7020. 10.1021/jacs.8b04058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.