

Abstract
Several thiophene featuring compounds are known for their promising antiproliferative activity. Prompted by the urgent need to identify new potent anticancer agents, 16 compounds of benzamides, benzylamines, and urea analogues incorporating a cyclohepta[b]thiophene scaffold were synthesized and biologically evaluated with a cell proliferation assay using the A549 nonsmall cell lung cancer cell line. Compound 17 demonstrated both potent and broad-spectrum anticancer activity with submicromolar 50% growth inhibition (GI50) values. It also showed superior antiproliferative activity (vs nocodazole) in OVACAR-4, OVACAR-5, CAKI-1, and T47D cell lines with GI50 values of 2.01 (vs 22.28), 2.27 (vs 20.75), 0.69 (vs 1.11), and 0.362 (vs 81.283) μM, respectively. Additionally, compound 17 displayed minimal cytotoxicity based on 50% lethal concentration (LC50) values toward all tested cell lines. Further cell-based mechanistic studies of compound 17 revealed its ability to induce cell cycle arrest of A549 cells as evidenced by dose dependent G2/M accumulation. Furthermore, induction of early apoptosis along with activation of caspase 3, 8, and 9 were confirmed in A549 cells treated with compound 17. Targeting tubulin polymerization may explain the mechanism of the antiproliferative activity of compound 17 based on cell cycle analysis, detected apoptosis, and in vitro inhibition of tubulin polymerization. In vitro data were further supported by in vivo antitumor efficacy studies of compound 17 in a CT26 murine model for which the results showed a reduction in the tumor growth compared to untreated mice. Overall, compound 17 has the potential to function as a promising candidate for further development of potent anticancer chemotherapeutics.
Keywords: cyclohepta[b]thiophene, benzyl urea, antiproliferative activity, cell cycle arrest, early apoptosis, caspase, in vivo antitumor efficacy
Over the past decade, cancer has been one of the leading causes of human deaths worldwide.1 In 2018, an estimate of 9.6 million deaths due to cancer was reported by the World Health Organization.2 Chemotherapeutic agents are one of the available treatments targeting different types of cancer; however, there are many limitations associated with these agents including drug resistance, systemic cytotoxicity, and narrow therapeutic index.3−6 Therefore, developing new chemotherapeutic agents with validated mechanism(s) are required to overcome these limitations. Gene mutations can cause cancer through aberrant cell division including inhibiting normal controls on the system, such as cell cycle arrest. The cell cycle is a highly regulated multistage process that results in cell proliferation. Dysregulation of this process can result in malignant transformation and chemotherapy resistance.7−9 Mitosis is a part of the cell cycle where duplicated DNA separates by the action of mitotic spindles. Microtubules are the major structural element of the mitotic spindle that are generated by tubulin polymerization. Thus, targeting microtubule formation or function has demonstrated crucial effects on cell cycle and cell proliferation.10−14
Several drugs that interfere with mitosis by inhibiting tubulin polymerization have been reported as anticancer therapeutics.15,16 For example, taxanes and vinca alkaloids are broad-spectrum anticancer drugs that disrupt and interfere with microtubule dynamics.17,18 However, a number of limitations are associated with these antimitotic drugs including complex synthetic routes, poor bioavailability, drug resistance (in cancer cells), and systemic toxicity.19−22 Compounds incorporating thiophene are of great interest as anticancer agents.23−26 Several drug candidates bearing thiophene (for example I–III, Figure 1) have been reported to act as anticancer agents by targeting certain oncogenic kinases such as FLT3 tyrosine kinase, checkpoint kinase, PI3K, IKK-2, or Akt.27−32 Nocodazole, a thiophene bearing drug (Figure 1), suppresses mitosis by binding with β-tubulin causing G2/M cell cycle arrest.33,34 ELR510444 (IV, Figure 1) is a phase II drug candidate that bears thiophene and was reported to inhibit the proliferation of MDA-MB-435 and MDA-MB-231 cells with IC50 values of 9.0 nM and 30.9 nM, respectively, by disruption of microtubules.35 Moreover, benzothiophene derivatives (V and VI, Figure 1) were reported as combretastatin A-4 analogues that exert their anticancer effects by binding to the colchicine binding site and inhibiting tubulin polymerization.36−38 In view of the above-mentioned findings and in order to broaden the chemical diversity of anticancer agents, a series of benzamides, benzylamines, and urea derivatives possessing the cyclohepta[b]thiophene scaffold were synthesized and tested for their in vitro and in vivo antitumor activity; then further mechanistic studies of the most promising derivative were performed.
Figure 1.

Representative examples of reported anticancer thiophene derivatives.
2. Results and Discussion
2.1. Chemistry
The target compounds were synthesized as depicted in Scheme 1. The synthesis of 2-amino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (1, Scheme 1) was accomplished by a one-pot multicomponent reaction of sulfur, malonitrile, cyclohexanone, and diethylamine in ethanol for 24 h.39 Benzamides (2–5, Scheme 1) were synthesized by treatment of compound 1 with the corresponding acid chloride in pyridine at 80 °C. Benzylation of compound 1 with the appropriate benzyl bromide in DMF afforded the corresponding benzyl amine derivatives (6–11, Scheme 1). 1H NMR spectra of benzyl amines 6–11 showed the coupling of a NH proton with the neighbor benzyl CH2 protons and appeared as a triplet and doublet, respectively. Coupling the amine 1 with the suitable aryl isocyanate in pyridine–DMF (1:1) produced the desired ureidothiophenes at a 58–73% yield.
Scheme 1. Synthesis of Compounds 2–17.

Conditions: (a) acid chlorides, pyridine, 80 °C, 12–18 h, 30-55% yield; (b) benzyl bromides, DMF, 75 °C, 12–16 h, 67–81% yield; (c) aryl isocyanates, DMF–pyridine (1:1), room temperature, 6–10 h, 58–73% yield.
2.2. In Vitro Biological Screening
2.2.1. Cell Viability Testing by MTS Assay
All of the synthesized compounds were evaluated for their anticancer activity against the human nonsmall cell lung cancer cell line A549 using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS assay) in preliminary testing. The GI50 values (compound’s concentration required to inhibit the growth of 50% of the cells) were determined after testing five concentrations (100, 50, 25, 10, 1, 0.1 μM) and the results are shown in Table 1 and Figure 2.
Table 1. GI50 Values (μM) of the Final Compounds 2–17 against A549 Cell Line.
| compound no. | R | GI50(μM)a |
|---|---|---|
| 2 | 2-I | >50 |
| 3 | 2-Br | >50 |
| 4 | 3-Br | >50 |
| 5 | 4-Br | 30.89 ± 2.45 |
| 6 | 2-I | >50 |
| 7 | 2-Br | >50 |
| 8 | 3-Br | >50 |
| 9 | 4-Br | >50 |
| 10 | 2-F | >50 |
| 11 | 2-C10H7 | 3.6 ± 3.73 |
| 12 | C6H5 | >50 |
| 13 | 2-BrC6H4 | >50 |
| 14 | 4-CH3C6H4 | >50 |
| 15 | 4-OCH3C6H4 | >50 |
| 16 | 4-n-C4H9C6H4 | >50 |
| 17 | C6H5CH2 | 1.6 ± 1.37 |
Bold values used to point out promising activity.
Figure 2.

Dose response curves of the tested compounds (2–16): (A) benzamides; (B) benzylamines; (C) urea derivatives.
2.2.2. Cell Viability Testing by NCI
2.2.2.1. Single Dose Testing
Compounds 6, 13, and 17 were selected by the National Cancer Institute (NCI) to be tested at 10 μM concentration against a diverse panel of 60 human cancer cell lines representing nine cancer diseases of different origins. The percentage growth inhibition (GI%) values for 60 cancer cell lines treated with the investigated compounds are listed in Table 2. Also, the mean growth inhibition percentage (GI%) of the tested compounds are shown in Figure 3. Close examination of the data in Table 2 showed that the benzyl urea derivative, compound 17, displayed the highest growth inhibitory activity toward most of the tested cell lines. Compound 17 also exhibited potent antiproliferative activities (GI > 80%) toward 20 cancer cell lines and cytotoxic activities against seven cancer cell lines. Furthermore, compound 6 displayed GI% values greater than 25% against UACC-62, IGORV1, CAKI-1, and T-47D cell lines. Compound 13 had no effect on 47 of the 60 cell lines tested and was only marginally effective (10.41−24.42% GI) against the remaining 13 cell lines.
Table 2. Growth Inhibition Percentage (GI%) of Cancer Cell Lines at 10 μM Concentration of Compounds 6, 13, and 17a.
| growth
inhibition % |
growth
inhibition % |
||||||
|---|---|---|---|---|---|---|---|
| panel/cell line | 6 | 13 | 17 | panel/cell line | 6 | 13 | 17 |
| Leukemia | Melanoma | ||||||
| CCRF-CEM | − | − | 85.27 | M14 | − | − | 89.11 |
| HL-60(TB) | − | 24.42 | L | MDA-MB-435 | − | − | L |
| K-562 | − | − | 87.26 | SK-MEL-2 | − | − | 60.92 |
| MOLT-4 | 15.69 | − | 82.03 | SK-MEL-28 | − | − | 32.65 |
| RPMI-8226 | − | − | 85.15 | SK-MEL-5 | − | − | 82.37 |
| SR | − | 21.48 | 70.54 | UACC-257 | − | − | 38.44 |
| Nonsmall Cell Lung Cancer | Ovarian Cancer | ||||||
| A549/ATCC | 14.29 | 11.46 | 74.5 | IGORV1 | 28.09 | 11.17 | 72.78 |
| EKVX | 21.15 | − | 60.65 | OVCAR-3 | − | − | L |
| HOP-62 | − | − | 65.8 | OVCAR-4 | 10.04 | − | 57.05 |
| HOP-92 | 11.24 | 10.41 | 55.24 | OVCAR-5 | − | − | 56.46 |
| NCI-H226 | 12.54 | − | 77.52 | OVCAR-8 | 11.23 | − | 78.15 |
| NCI-H23 | 12.51 | − | 62.47 | NCI/ADR-RES | 10.34 | − | 82.82 |
| NCI-H322M | 10.07 | 12.70 | 55.14 | SK-OV-3 | − | − | 75.02 |
| NCI-H460 | − | − | 99.55 | ||||
| NCI-H522 | 13.32 | 19.20 | 87.93 | ||||
| Colon Cancer | Renal Cancer | ||||||
| COLO 205 | − | − | 97.85 | 786-0 | − | − | 73.74 |
| HCC-2998 | − | − | 75.53 | ACHN | − | − | 56.60 |
| HCT-116 | − | − | 91.1 | CAKI-1 | 25.18 | − | 70.17 |
| HCT-15 | − | − | 84.82 | RXF 393 | − | − | L |
| HT29 | − | − | 90.32 | SN12C | − | − | 69.72 |
| KM 12 | − | − | 87.4 | TK-10 | − | 10.41 | 58.93 |
| SW-620 | − | − | 78.55 | UO-31 | 20.28 | 14.43 | 53.35 |
| CNS Cancer | Breast Cancer | ||||||
| SF-268 | − | − | 59.03 | MCF-7 | 19.97 | − | 80.89 |
| SF-295 | − | − | 80.11 | MDA-MB-231/ATCC | 14.63 | − | 89.32 |
| SF-539 | − | − | L | HS 578T | − | 11.06 | 90.11 |
| SNB-19 | − | − | 71.77 | BT-549 | − | − | 73.82 |
| SNB-75 | 15.19 | 14.53 | L | T-47D | 26.67 | − | 62.19 |
| U251 | − | − | 75.19 | MDA-MB-468 | − | − | L |
| Melanoma | Prostate Cancer | ||||||
| LOX IMVI | − | − | 73.56 | PC-3 | − | − | 71.28 |
| MALME-3M | 12.48 | 11.90 | 55.32 | DU-145 | − | − | 96.46 |
| UACC-62 | 30.5 | 12.26 | 81.14 | ||||
Notation: (−) GI < 10%; (L) compound proved lethal to the cancer cell line.
Figure 3.
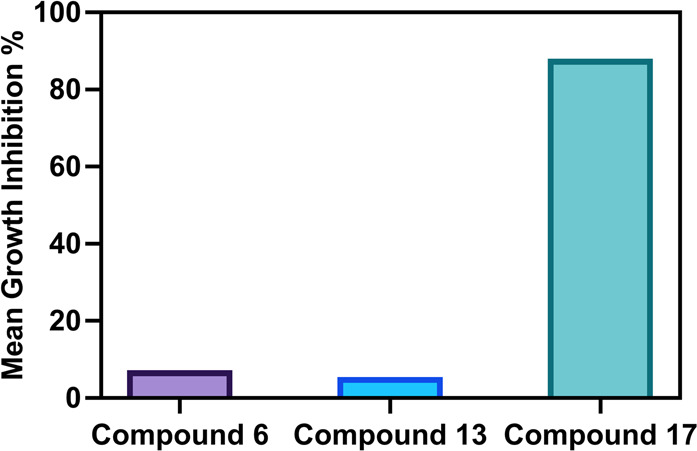
Mean growth inhibition percentage (GI%) of the investigated compounds against NCI-60 cancer cell lines.
2.2.2.2. Five-Dose Testing
Compound 17 emerged as the most effective broad-spectrum anticancer agent out of the synthesized compounds and was then selected for further investigation in a five-dose testing assay (100, 10, 1, 0.1, and 0.01 μM concentrations). To measure potency, efficacy, and lethality of the tested compound against 60 cell lines; the GI50, TGI (compound’s concentration causing total growth inhibition), and LC50 (compound’s concentration producing 50% lethality) were determined. The GI50 values of compound 17 were compared with those of the antimitotic drug nocodazole (Table 3). Nocodazole is a thiophene bearing antimitotic drug that has a similar mechanism of action to that of compound 17. Compound 17 displayed potent anticancer activity with GI50 values in the one digit micromolar range (1.06–6.51 μM) against the following 14 cancer cell lines of various origins NCI-H226, NCI-23, HCC-2998, SF-268, SK-MEL-2, SK-MEL-28, UACC-257, OVCAR-4, OVCAR-5, OVCAR-8, 786-0, TK-10, MDA-MB-231/ATCC, and HS-578T. Interestingly, compound 17 demonstrated pronounced anticancer activity in the submicromolar range (GI50 = 0.28–0.62 μM) toward all leukemia and prostate cancer cell lines. For example, it showed GI50 values of 0.279 μM, 0.363 μM, and 0.613 μM toward HL-60(TB), SR, and PC-3, respectively. In addition, the GI50 values toward most of the nonsmall cell lung cancer, colon, CNS, melanoma, ovarian, renal, and breast cancer cell lines were lower than one micromolar (0.22–0.88 μM). For instance, the GI50 values of compound 17 for MDA-MB-435, A498, and NCI-H460 were 0.22, 0.269, and 0.415 μM, respectively.
Table 3. GI50 Values (μM) of Compound 17 and Nocodazole from NCI-60 Cancer Cell Lines Screening.
| GI50 values (μM) |
GI50 values (μM) |
||||
|---|---|---|---|---|---|
| cell lines | compound 17 | nocodazole | cell lines | compound 17 | nocodazolea |
| Leukemia | Melanoma | ||||
| CCRF-CEM | 0.364 | 0.014 | M14 | 0.44 | 0.013 |
| HL-60(TB) | 0.279 | 0.011 | MDA-MB-435 | 0.22 | 0.011 |
| K-562 | 0.387 | 0.011 | SK-MEL-2 | 6.51 | 0.019 |
| MOLT-4 | 0.531 | 0.020 | SK-MEL-28 | 4.13 | 0.095 |
| RPMI-8226 | 0.435 | 0.013 | SK-MEL-5 | 0.62 | 0.018 |
| SR | 0.363 | 0.015 | UACC-257 | 1.17 | 0.148 |
| UACC-62 | 0.575 | 0.018 | |||
| Nonsmall Cell Lung Cancer | Ovarian Cancer | ||||
| A549/ATCC | 0.645 | 0.018 | IGROV1 | 0.793 | 0.024 |
| EKVX | 0.524 | 0.039 | OVCAR-3 | 0.348 | 0.017 |
| HOP-62 | 0.815 | 0.028 | OVCAR-4 | 2.01 | 22.28 |
| HOP-92 | 0.888 | 0.292 | OVCAR-5 | 2.27 | 20.75 |
| NCI-H226 | 1.56 | 0.062 | OVCAR-8 | 2.04 | 0.029 |
| NCI-H23 | 1.06 | 0.016 | NCI/ADR-RES | 0.447 | 0.014 |
| NCI-H322M | 0.434 | 0.023 | SK-OV-3 | 0.799 | 0.017 |
| NCI-H460 | 0.415 | 0.017 | |||
| NCI-H522 | 0.722 | 0.012 | |||
| Colon Cancer | Renal Cancer | ||||
| COLO | 0.557 | 0.019 | 786-0 | 1.03 | 0.078 |
| HCC-2998 | 2.00 | 0.038 | A498 | 0.269 | 0.019 |
| HCT-116 | 0.519 | 0.013 | ACHN | 0.627 | 0.225 |
| HCT-15 | 0.434 | 0.019 | CAKI-1 | 0.69 | 1.109 |
| HT29 | 0.396 | 0.013 | RXF 393 | 0.472 | 0.070 |
| KM12 | 0.469 | 0.013 | SN 12C | 0.743 | 0.073 |
| SW-620 | 0.430 | 0.013 | TK-10 | 5.93 | 2.951 |
| UO-31 | 0.673 | 0.058 | |||
| CNS Cancer | Breast Cancer | ||||
| SF-268 | 1.89 | 0.089 | MCF-7 | 0.376 | 0.050 |
| SF-295 | 0.535 | 0.012 | MDA-MB-468 | 0.340 | NT |
| SF-539 | 0.37 | 0.017 | HS 578T | 1.19 | 0.016 |
| SNB-19 | 0.61 | 0.086 | T-47D | 0.362 | 81.28 |
| SNB-75 | 0.27 | 0.015 | MDA-MB-231/ATCC | 1.23 | 0.171 |
| U251 | 0.626 | 0.040 | |||
| Melanoma | Prostate Cancer | ||||
| LOX IMVI | 0.567 | 0.021 | PC-3 | 0.613 | 0.020 |
| MALME-3M | 0.623 | 0.027 | DU-145 | 0.620 | 0.014 |
NT: not tested.
Upon comparing the potency of compound 17 with that of the antimitotic drug nocodazole, both compounds displayed antiproliferative activity in HOP-92 and ACHN cell lines with submicromolar GI50 values of 0.888 vs 0.292 μM and 0.627 vs 0.225 μM, respectively. Remarkably, compound 17 demonstrated potent antiproliferative activity against OVACAR-4, OVACAR-5, CAKI-1, and T47D cell lines (GI50 = 2.01, 2.27, 0.69, and 0.362 μM, respectively) compared to nocodazole which showed higher GI50 values (22.28, 20.75, 1.11, and 81.283 μM) against the same cell lines. The GI50 values of compound 17 versus nocodazole were 5.93 vs 2.95 μM, respectively, when used to treat the TK-10 renal cancer cell line.
Regarding the efficacy parameters (TGI and LC50 values shown in Table 4), it was revealed that compound 17 could have low cellular toxicity since LC50 values were greater than 70 μM against the 60 cell lines. In terms of TGI values, compound 17 exhibited TGI values in the low micromolar range (0.5–13.4 μM) against 11 cancer cell lines. For example, TGI values for MDA-MB-435, SF-539, and MDA-MB-231/ATCC cell lines were 0.564 μM, 4.19 μM, and 13.4 μM, respectively.
Table 4. TGI and LC50 Values (μM) of Compound 17 and Nocodazole from NCI-60 Cell Lines Screening.
| compound 17 |
nocodazole |
compound
17 |
nocodazole |
||||||
|---|---|---|---|---|---|---|---|---|---|
| cell lines | TGI | LC50 | TGI | LC50 | cell lines | TGI | LC50 | TGI | LC50 |
| Leukemia | Melanoma | ||||||||
| CCRF-CEM | 100 | >100 | 19.9 | 100 | M14 | 100 | >100 | 5.01 | >100 |
| HL-60(TB) | 0.828 | >100 | 0.05 | 79.43 | MDA-MB-435 | 0.564 | 70.0 | 0.1 | 1.58 |
| K-562 | 100 | >100 | 39.8 | 79.43 | SK-MEL-2 | 100 | >100 | 12.59 | >100 |
| MOLT-4 | 100 | >100 | 12.58 | 79.43 | SK-MEL-28 | 100 | >100 | 63.09 | >100 |
| RPMI-8226 | 100 | >100 | 6.31 | >100 | SK-MEL-5 | 7.52 | >100 | 6.31 | 50.11 |
| SR | 100 | >100 | 6.31 | 79.43 | UACC-257 | 100 | >100 | 100 | >100 |
| UACC-62 | 100 | >100 | 63.09 | >100 | |||||
| Non-Small Cell Lung Cancer | Ovarian Cancer | ||||||||
| A549/ATCC | 100 | >100 | 50.12 | 100 | IGROV1 | 100 | >100 | 39.81 | >100 |
| EKVX | 100 | >100 | 31.62 | >100 | OVCAR-3 | 100 | 86.7 | 100 | 79.43 |
| HOP-62 | 41.2 | >100 | 100 | >100 | OVCAR-4 | 1.34 | >100 | 63.09 | >100 |
| HOP-92 | 60.5 | >100 | 7.94 | >100 | OVCAR-5 | 100 | >100 | 100 | >100 |
| NCI-H226 | 100 | >100 | 1.26 | >100 | OVCAR-8 | 100 | >100 | 15.8 | >100 |
| NCI-H23 | 100 | >100 | 0.63 | 63.09 | NCI/ADR-RES | 54.8 | >100 | 0.16 | >100 |
| NCI-H322M | 100 | >100 | 79.43 | >100 | SK-OV-3 | 100 | >100 | 10.00 | >100 |
| NCI-H460 | 100 | >100 | 10 | 79.43 | |||||
| NCI-H522 | 51.3 | >100 | 0.039 | >100 | |||||
| Colon Cancer | Renal Cancer | ||||||||
| COLO | 3.07 | 76.30 | 1.99 | 25.11 | 786–0 | 100 | >100 | 31.62 | 79.43 |
| HCC-2998 | 100 | >100 | 12.56 | >100 | A498 | 1.83 | >100 | 1.58 | 79.43 |
| HCT-116 | 100 | >100 | 15.84 | >100 | ACHN | 100 | >100 | 79.43 | >100 |
| HCT-15 | 100 | >100 | 2.51 | >100 | CAKI-1 | 100 | >100 | 63.09 | >100 |
| HT29 | 100 | >100 | 1.00 | 25.11 | RXF 393 | 7.58 | >100 | 6.31 | 79.43 |
| KM12 | 100 | >100 | 39.81 | >100 | SN 12C | 100 | >100 | 31.62 | >100 |
| SW-620 | 100 | >100 | 100 | >100 | TK-10 | 100 | >100 | 63.09 | >100 |
| UO-31 | 100 | >100 | 39.81 | 79.43 | |||||
| CNS Cancer | Breast Cancer | ||||||||
| SF-268 | 100 | >100 | 63.09 | >100 | MCF-7 | 100 | >100 | 50.12 | >100 |
| SF-295 | 51.4 | >100 | 0.03 | 63.09 | MDA-MB-468 | 5.29 | >100 | 79.43 | >100 |
| SF-539 | 4.19 | >100 | 0.501 | >100 | HS 578T | 100 | >100 | 79.43 | >100 |
| SNB-19 | 100 | >100 | 39.81 | >100 | T-47D | 100 | >100 | 100 | >100 |
| SNB-75 | 28.2 | >100 | 63.09 | >100 | MDA-MB-231/ATCC | 13.4 | >100 | 100 | >100 |
| U251 | 50.07 | >100 | 1.99 | 50.12 | |||||
| Melanoma | Prostate Cancer | ||||||||
| LOX IMVI | 100 | >100 | 39.81 | >100 | PC-3 | 100 | >100 | 15.84 | >100 |
| MALME-3M | 100 | >100 | 39.81 | >100 | DU-145 | 3.34 | >100 | 1.58 | 79.43 |
2.2.3. Cell Cycle Analysis of A549 using Flow Cytometry
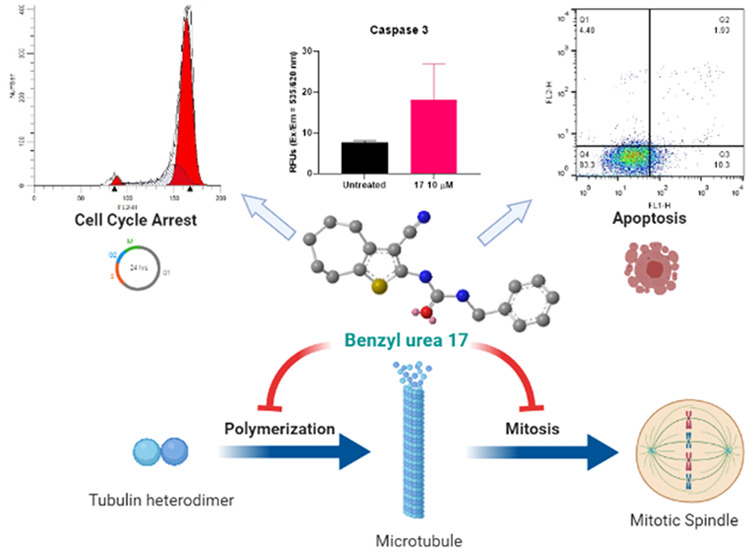
To investigate whether compound 17 can affect the cell cycle dynamics, cell cycle analysis of A549 cells treated with compound 17 (1, 10 μM) was carried out. The cell cycle analysis was performed 24 h after treating cells to detect cell populations in the different cell cycle phases prior to a significant number of cells undergoing apoptosis (Figure 4 and Table 5). The results indicated that compound 17 induced cell cycle arrest through the accumulation of cells in the G2/M phase in a dose-dependent manner.
Figure 4.

Analysis of cell cycle of A549 cells treated with compound 17. (A) Effect of compound 17 (1 and 10 μM) on different cell cycle phases; (B) The percentage of cells in G2/M phase; (C) Cell cycle profiles of A549 cells treated with compound 17. Statistical analysis was performed using one-way ANOVA, ***P < 0.001.
Table 5. Analysis of Cell Cycle Phases of A549 Cells Treated with Compound 17.
| groups | G0/G1 | G2/M | S phase |
|---|---|---|---|
| Untreated | 43.26 ± 1.27 | 15.59 ± 2.00 | 41.15 ± 0.73 |
| Compound 17 (1 μM) | 43.56 ± 0.73 | 19.31 ± 1.01 | 37.12 ± 1.67 |
| Compound 17 (10 μM) | 2.38 ± 0.27 | 74.41 ± 4.28 | 23.21 ± 4.02 |
2.2.4. Apoptosis Detection by Annexin V/Propidium Iodide Staining
Early apoptosis of A549 cells treated with compound 17 was further investigated as a possible anticancer mode of action by using annexin V and propidium iodide staining. On the basis of the results shown in Figure 5, compound 17 induced apoptosis in A549 cells. Cells treated with 10 μM of compound 17 showed a higher percentage of cells in Q3 (early apoptosis) compared to untreated cells (10.04% vs 0.45%). In addition, 2.1% of cells were detected in the late apoptosis stage (Q2) for the drug treated group versus 0.32% cells in the untreated group.
Figure 5.

Apoptosis detection in A549 cells treated with compound 17. (A) Percentages of early apoptotic cells in different treated groups; (B) percentages of cells in Q1 (dead cells), Q2 (late apoptotic cells), Q3 (early apoptotic cells), and Q4 (live cells); (C) profile of cell count on different phases Q1–Q4 for control and upon treatment with compound 17 (10 μM) and paclitaxel (5 nM). Statistical analysis was performed using one-way ANOVA, ***P < 0.001.
2.2.5. Caspase 3, 8, and 9 Activity Assays
Programmed cell death occurs in response to an extrinsic pathway (external stimuli) or an intrinsic pathway (mitochondrial response) involving caspase 9 or caspase 8 and followed by caspase 3 activation.40 To confirm programmed cell death in cells treated with our compound 17, caspase 3, 8, and 9 activities were determined in A549 cells treated with 10 μM of compound 17. Higher caspase activities were detected in treated cells than untreated cells (Figure 6).
Figure 6.

Detection of (A) caspase 3, (B) caspase 8, and (C) caspase 9 levels in A549 cells treated with compound 17 (10 μM) for 48 h. Statistical analysis was performed using two-tailed t test, **P < 0.01.
2.2.6. Tubulin Polymerization Assay
Since antimitotic drugs that interfere with microtubule assembly have been proven to cause cell cycle arrest after G2/M accumulation and further apoptosis, the effect of compound 17 on tubulin polymerization was evaluated.41 As shown in Figure 7, compound 17 inhibited tubulin polymerization at 3 μM. CaCl2 was used as a microtubule destabilizing agent, whereas paclitaxel (at 3 μM) was used as microtubule stabilizer (CaCl2 and paclitaxel were recommended as controls by the manufacturer (Cytoskeleton Inc., Denver, CO) to be used as assay controls). Also, tubulin alone was used as a control.
Figure 7.

Effect of compound 17 on tubulin polymerization. Purified tubulin in reaction buffer was incubated at 37 °C in the absence or presence of 3 μM of compound 17, 3 μM paclitaxel, or 75 μM of CaCl2. Tubulin polymerization rate was then measured every minute and over 60 min at excitation of 360 nm and emission of 450 nm using a fluorimeter (SpectraMax M5Microplate Reader).
2.2.7. Cell Viability Testing of Compound 17 against CT-26 Murine Colon Carcinoma
The CT-26 colon carcinoma is widely implemented in biomedical research for evaluation of anticancer drug candidates in vivo since it is highly tumorigenic and exhibits a low tendency to metastasize.43 Prior to in vivo testing, the antitumor activity of compound 17 against the CT-26 murine colon carcinoma cell line was evaluated using the MTS assay (Figure 8). The investigated compound displayed promising in vitro antiproliferative activity with GI50 = 11.26 μM after being incubated with cells for 24 h. Therefore, the study was extended to in vivo testing against the CT26 murine model.
Figure 8.
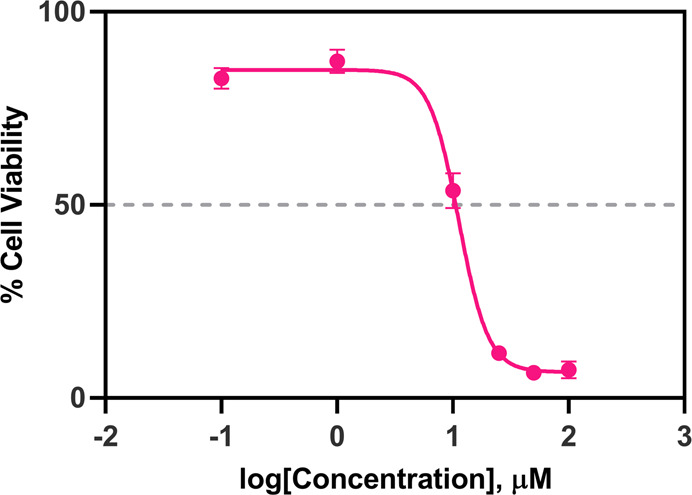
Dose response curves of CT26 cells treated with compound 17. IC50 = 11.26 μM.
2.2.8. In Vivo Antitumor Efficacy Study
The syngeneic CT-26 colon carcinoma model was used to investigate the antitumor efficacy of compound 17. Treatment of CT-26 tumors with compound 17 displayed a trend toward reduction in tumor growth when compared to the untreated group (Figure 9A) albeit not significant (P > 0.05). The soluble drug treatment group displayed stable body weight except for one mouse that was euthanized at day 17 due to weight loss of more than 20% of the initial body weight (Figure 9B). Increasing the dose could potentially improve the antitumor efficacy.23
Figure 9.

Antitumor efficacy study of compound 17: (A) tumor progression curve; (B) mice body weight change over time during treatments (n = 4 per group, P > 0.05).
3. Experimental Section
All solvents and reagents were obtained from Sigma-Aldrich (St. Louis, MI) and used without further purification. Compound 1 was synthesized using a previously reported synthesis protocol.39 Chemical reactions were monitored using silica G TLC plates, w/UV 254, polyester backed, 200 μm, 4 cm × 8 cm, and UV was applied to visualize the spots. Also, n-hexane/ethyl acetate mixture (5:1) was used as eluent. 1H NMR and 13C NMR spectra were recorded on Bruker spectrometers (Bruker Bioscience, Billerica, MA) at 400 MHz for 1H and 100 MHz for 13C using CDCl3 and DMSO-d6 as solvents. Chemical shifts (δ) are reported in parts per million (ppm). High resolution mass spectra (HRMS) were obtained in positive ion mode using electrospray ionization (ESI) with a double-focusing magnetic sector mass spectrometer.
3.1. Chemistry
3.1.1. Synthesis of Benzamides 2–5
2-Amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (1) was heated with equimolar amounts of the corresponding benzoyl chloride in pyridine (10 mL) at 80 °C for 12–18 h. The reaction mixture was allowed to cool and then poured into 1 M HCl (100 mL). The aqueous layer was extracted with CHCl3 (3 × 50 mL), dried over anhydrous Na2SO4, and evaporated to dryness. The crude mixture was purified by flash column chromatography using hexane and ethyl acetate (9:1).
3.1.1.1. N-(3-Cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)-2-iodobenzamide (2)
1H NMR (DMSO-d6, δ ppm): 1.59–1.61 (m, 4H, 2CH2), 1.80–1.82 (m, 2H, CH2), 2.67–2.73 (m, 4H, 2CH2), 7.81–7.93 (m, 3H, Ar–H), 8.10–8.12 (m, 1H, Ar–H), 11.81 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.40, 29.12, 29.91, 29.92, 32.90, 69.56, 99.85, 116.41, 123.29, 129.14, 129.76, 131.71, 133.04, 136.55, 137.50, 145.06, 164.90.
3.1.1.2. 2-Bromo-N-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)benzamide (3)
1H NMR (DMSO-d6, δ ppm): 1.57–1.61 (m, 4H, 2CH2), 1.78–1.80 (m, 2H, CH2), 2.65–2.73 (m, 4H, 2CH2), 7.41–7.55 (m, 4H, Ar–H), 12.23 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.40, 29.14, 29.86, 30.21, 32.93, 98.39, 116.00, 120.79, 121.48, 129.21, 130.83, 132.12, 134.09, 135.27, 137.59, 138.47, 145.21. HRMS (ESI): calculated for C17H15BrN2OS (M + H)+, 375.0167; found, 375.0154.
3.1.1.3. 3-Bromo-N-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)benzamide (4)
1H NMR (DMSO-d6, δ ppm): 1.59–1.61 (m, 4H, 2CH2), 1.80–1.82 (m, 2H, CH2), 2.49–2.61 (m, 4H, 2CH2), 7.51 (t, 1H, Ar–H, J = 8.0 Hz), 7.92 (d, 2H, Ar–H, J = 7.6 Hz), 8.12 (s, 1H, Ar–H), 11.80 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.41, 29.13, 29.93, 31.32, 32.91, 123.08, 123.11, 123.23, 128.98, 129.79, 132.24, 132.27, 132.42, 133.25, 137.10, 138.54, 163.58. HRMS (ESI): calculated for C17H15BrN2OS (M – H)+, 373.0010; found, 372.9998.
3.1.1.4. 4-Bromo-N-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)benzamide (5)
1H NMR (DMSO-d6, δ ppm): 1.60–1.62 (m, 4H, 2CH2), 1.81–1.82 (m, 2H, CH2) 2.68–2.81 (m, 4H, 2CH2), 7.77 (d, 2H, Ar–H, J = 8.40 Hz), 7.87 (d, 2H, Ar–H, J = 8.60 Hz), 11.69 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.41, 29.13, 29.89, 29.92, 32.90, 100.09, 120.40, 127.82, 131.87, 133.06, 134.00, 134.05, 137.78, 165.92. HRMS (ESI): calculated for C17H15BrN2OS (M – H)+, 373.0010; found, 373.0004.
3.1.2. Synthesis of Benzylamines 6–11
A mixture of compound 1 (0.01 mol) and the appropriate benzylamine (0.01 mol) in DMF (15 mL) was stirred at 75 °C for 12–16 h in the presence of triethylamine (1 mL). The cooled mixture was poured into 1 M HCl (100 mL) followed by extraction with CHCl3 (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude mixture was purified by flash column chromatography on silica gel using hexane and ethyl acetate (8:2).
3.1.2.1. 2-((2-Iodobenzyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (6)
1H NMR (DMSO-d6, δ ppm): 1.51–1.56 (m, 4H, 2CH2), 171–1.72 (m, 2H, CH2), 2.47–2.52 (m, 4H, 2CH2), 4.25 (d, 2H, CH2,J = 5.60 Hz), 7.01–7.05 (td, 1H, Ar–H, J = 8.00, 1.80 Hz), 7.32 (dd, 1H, Ar–H, J = 8.00, 1.60 Hz), 7.36–7.38 (td, 1H, Ar–H, J = 7.40, 1.00 Hz), 7.85 (dd, 1H, Ar–H, J = 7.80, 1.00 Hz), 7.93 (t, 1H, NH, J = 5.80 Hz). 13C NMR (DMSO-d6, δ ppm): 28.37, 29.23, 29.89, 30.18, 32.77, 56.65, 87.12, 100.39, 118.09, 122.04, 129.50, 129.97, 130.88, 138.24, 140.67, 140.72, 162.68. HRMS (ESI): calculated for C17H17IN2S (M + H)+, 409.0235; found, 409.0230.
3.1.2.2. 2-((2-Bromobenzyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (7)
13C NMR (DMSO-d6, δ ppm): 23.23, 24.34, 24.36, 24.99, 25.51, 51.65, 83.39, 117.61, 119.03, 124.15, 129.44, 129.94, 130.83, 133.68, 134.07, 138.10, 164.23. HRMS (ESI): calculated for C17H17BrN2S (M + H)+, 361.0374; found, 361.0361.
3.1.2.3. 2-((3-Bromobenzyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (8)
1H NMR (DMSO-d6, δ ppm): 1.52–1.54 (m, 4H, 2CH2), 1.71–1.72 (m, 2H, CH2), 2.48–2.49 (m, 4H, 2CH2), 4.29 (d, 2H, CH2, J = 6.00 Hz), 7.28–7.34 (2H, m, Ar–H), 7.45 (d, 1H, Ar–H, J = 7.60 Hz), 7.53 (s, 1H, Ar–H), 7.93 (t, 1H, NH, J = 6.00 Hz). 13C NMR (DMSO-d6, δ ppm): 28.34, 29.22, 29.87, 30.13, 32.75, 50.82, 87.12, 117.82, 121.95, 123.35, 128.04, 131.50, 131.57, 132.18, 138.04, 142.72, 162.95. HRMS (ESI): calculated for C17H17BrN2S (M + H)+, 361.0374; found, 361.0363.
3.1.2.4. 2-((4-Bromobenzyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (9)
1H NMR (DMSO-d6, δ ppm): 1.52–1.56 (m, 4H, 2CH2), 172–1.73 (m, 2H, CH2), 2.48–2.51 (m, 4H, 2CH2), 4.28 (d, 2H, CH2, J = 6.00 Hz), 7.30 (d, 2H, Ar–H, J = 8.40 Hz), 7.54 (d, 2H, Ar–H, J = 8.40 Hz), 7.96 (t, 1H, NH, J = 6.0 Hz). 13C NMR (DMSO-d6, δ ppm): 28.36, 29.23, 29.87, 30.12, 32.76, 50.70, 86.94, 118.15, 121.76, 121.85, 131.06, 132.83, 138.07, 139.25, 162.83. HRMS (ESI): calculated for C17H17BrN2S (M + H)+, 361.0374; found, 361.0366.
3.1.2.5. 2-((2-Fluorobenzyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (10)
1H NMR (DMSO-d6, δ ppm): 1.52–1.54 (m, 4H, 2CH2), 1.71–1.73 (m, 2H, CH2), 2.49 (m, 4H, 2CH2), 4.34 (d, 2H, CH2, J = 6.00 Hz), 7.15–7.20 (m, 2H, Ar–H), 7.31–7.40 (m, 2H, Ar–H), 7.87 (t, 1H, NH, J = 5.80 Hz). 13C NMR (DMSO-d6, δ ppm): 28.35, 29.23, 29.86, 30.12, 32.75, 45.04, 87.00, 116.74, 116.93, 118.09, 121.87, 125.99, 126.23, 130.83, 130.90, 138.20, 162.73. HRMS (ESI): calculated for C17H17FN2S (M + H)+, 301.1175; found, 301.1171.
3.1.2.6. 2-((Naphthalen-1-ylmethyl)amino)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carbonitrile (11)
1H NMR (DMSO-d6, δ ppm): 1.74–1.79 (m, 4H, 2CH2), 1.90–1.92 (m, 2H, CH2), 2.70–2.76 (m, 4H, 2CH2), 4.87 (s, 2H, CH2), 5.22 (s, 1H, NH) 7.36–7.67 (m, 4H, Ar–H), 7.95–8.08 (m, 3H, Ar–H). 13C NMR (DMSO-d6, δ ppm): 28.33, 29.24, 30.35, 30.38, 32.98, 50.77, 117.60, 123.26, 124.12, 126.41, 127.17, 127.57, 127.84, 130.00, 130.17, 132.38, 133.03, 134.97, 138.43. HRMS (ESI): calculated for C21H20N2S (M + H)+, 333.1425; found, 333.1418.
3.1.3. Synthesis of Urea Derivatives 12–17
A solution of compound 1 (0.01 mol) in pyridine (5 mL) was added to a solution of the corresponding isocyanate (0.015 mol) in DMF (5 mL) under nitrogen and allowed to stir for 6–10 h. The reaction mixture was quenched with 1 M HCl (150 mL), and the aqueous layer was extracted with CHCl3 (3 × 50 mL), dried over anhydrous Na2SO4, and evaporated to dryness. The crude mixture was purified by flash column chromatography using hexane and ethyl acetate (9:1).
3.1.3.1. 1-(3-Cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)-3-phenylurea (12)
1H NMR (DMSO-d6, δ ppm): 1.63–1.67 (m, 4H, 2CH2), 1.79–1.83 (m, 2H, CH2), 2.63–2.68 (m, 4H, 2CH2), 7.07 (t, 1H, Ar–H, J = 7.40 Hz), 7.30 (t, 2H, Ar–H, J = 8.00 Hz), 7.43 (dd, 2H, Ar–H, J = 8.60, 1.00 Hz), 7.74 (s, 1H, NH), 8.70 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.41, 29.06, 30.19, 30.48, 32.99, 93.39, 117.18, 117.63, 120.62, 124.93, 130.19, 131.56, 135.92, 138.86, 150.02, 152.21.
3.1.3.2. 1-(2-Bromophenyl)-3-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)urea (13)
1H NMR (DMSO-d6, δ ppm): 1.57–1.59 (m, 4H, 2CH2), 1.77–1.79 (m, 2H, CH2), 2.60–2.63 (m, 4H, 2CH2), 7.01 (t, 1H, Ar–H, J = 7.80 Hz), 7.34 (t, 1H, Ar–H, J = 78.0 Hz), 7.62 (d, 1H, Ar–H, J = 8.00 Hz), 7.99 (d, 1H, Ar–H, J = 8.40 Hz), 8.84 (s, 1H, NH), 10.75 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.43, 29.21, 29.75, 29.90, 32.86, 94.25, 114.97, 116.71, 124.09, 126.50, 129.67, 131.00, 134.16, 136.40, 137.71, 148.47, 152.60. HRMS (ESI): calculated for C17H16BrN3OS (M + H)+, 390.0276; found, 390.0275.
3.1.3.3. 1-(3-Cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)-3-(p-tolyl)urea (14)
1H NMR (DMSO-d6, δ ppm): 1.26 (s, 3H, CH3), 1.61–1.68 (m, 4H, 2CH2), 1.83–1.87 (m, 2H, CH2), 2.30–2.43 (m, 4H, 2CH2), 7.09–7.15 (m, 2H, Ar–H), 7.30–7.34 (m, 2H, Ar–H), 7.49 (s, 1H, NH), 8.49 (s, 1H, NH). HRMS (ESI): calculated for C18H19N3OS (M + H)+, 326.1327; found, 326.1326.
3.1.3.4. 1-(3-Cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)-3-(4-methoxyphenyl)urea (15)
1H NMR (DMSO-d6, δ ppm): 1.67–1.71 (m, 4H, 2CH2), 1.84–1.85 (m, 2H, CH2), 2.63–2.69 (m, 4H, 2CH2), 3.79 (s, 3H, OCH3), 6.86 (d, 2H, Ar–H, J = 8.80 Hz), 7.34 (d, 2H, Ar–H, J = 8.80 Hz), 7.60 (s, 1H, NH), 8.61 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.40, 29.06, 30.07, 30.17, 33.00, 56.58, 93.69, 103.82, 115.47, 117.47, 123.40, 131.49, 135.98, 149.81, 152.78, 157.69. HRMS (ESI): calculated for C18H19N3O2S (M + H)+, 342.1276; found, 342.1267.
3.1.3.5. 1-(4-Butylphenyl)-3-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)urea (16)
1H NMR (DMSO-d6, δ ppm): 0.90 (t, 3H, CH3, J = 7.20 Hz), 1.30–1.35 (m, 2H, CH2), 1.51–1.57 (m, 2H, CH2), 1.57–1.65 (m, 4H, 2CH2), 1.80–1.85 (m, 2H, CH2), 2.55 (t, 2H, CH2, J = 7.80 Hz), 2.62–2.66 (m, 4H, CH2), 7.11 (d, 2H, Ar–H, J = 8.40 Hz), 7.32 (d, 2H, Ar–H, J = 8.40 Hz), 7.63 (s, 1H, NH), 8.63 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 15.00, 23.35, 28.42, 29.07, 30.07, 30.28, 33.00, 34.73, 36.08, 93.37, 117.62, 121.07, 130.11, 131.48, 135.90, 136.23, 139.89, 150.07, 152.37. HRMS (ESI): calculated for C21H25N3OS (M + H)+, 368.1797; found, 368.1784.
3.1.3.6. 1-Benzyl-3-(3-cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-2-yl)urea (17)
1H NMR (DMSO-d6, δ ppm): 1.61–1.66 (m, 4H, 2CH2), 1.82–1.84 (m, 2H, CH2), 2.53–2.56 (m, 2H, CH2), 2.61–2.63 (m, 2H, CH2), 4.45 (d, 2H, CH2, J = 5.20 Hz), 6.21 (t, 1H, NH, J = 5.60 Hz), 7.23–7.31 (m, 5H, Ar–H), 8.74 (s, 1H, NH). 13C NMR (DMSO-d6, δ ppm): 28.43, 29.14, 30.05, 33.05, 45.34, 92.97, 117.37, 128.42, 128.49, 129.67, 130.71, 135.78, 139.32, 150.50, 155.00. HRMS (ESI): calculated for C18H19N3OS (M + H)+, 326.1327; found, 326.1317.
3.2. Biological Evaluation
3.2.1. In Vitro Screening
A549 and CT-26 cells were purchased from ATCC (Manassas, VA). The cells were cultured in RPMI-1640 supplemented with 1% Pen/Strep (100 U mL–1, Gibco, Carlsbad, CA) and 10% FBS (Atlanta Biologicals, Flowery Branch, GA). All the cells were incubated in a humidified incubator (Sanyo Scientific Autoflow, Hudson, MA) at 37 °C under a 5% CO2 flow. In addition, CellTiter 96 aqueous MTS reagent powder (MTS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was obtained from Promega Corporation, San Luis Obispo, CA. NP-40 Surfact-Amps Detergent Solution and propidium iodide were purchased from Thermo scientific (Waltham, MA). RNAase A solution was obtained from Invitrogen. Apoptosis was detected by eBioscience Annexin V Apoptosis Detection Kit (Thermo Fisher Scientific, Waltham, MA).
3.2.1.1. Cell Viability Testing Using MTS
A549 cells were seeded in 96-well plates at 2 × 103 cells in 100 μL of medium per well. After 24 h, the medium was replaced by 100 μL of fresh medium followed by the addition of 100 μL of each treatment (n = 3) and 100 μL of fresh medium to the untreated control group (n = 3). The treated cells were incubated at 37 °C with 5% CO2 for 48 h. The medium was discarded, and 100 μL of fresh medium and 20 μL of MTS reagent were added to each well and incubated at 37 °C with 5% CO2 for 2 h. A Spectramax plus spectrophotometer, (Molecular Devices, San Jose, CA) at 490 nm was used to record the optical density on each well. Then, the relative cell viability was calculated using the following equation:
| 1 |
3.2.1.2. Cell Viability Testing by NCI
Cell viability testing over a panel of 60 human cancer cell lines was conducted at the National Cancer Institute, Bethesda, MD, following the standard procedure.42
3.2.1.3. Cell Cycle Analysis of the A549 Cell Line with Flow Cytometry
A549 cancer cells were seeded in 6-well plates at a density of 150 × 103 cells/well for 24 h. Then, the medium was removed, and the cells were treated with compound 17 at different concentrations (1, 10, and 50 μM) and paclitaxel (2.5 nM), and 2 mL of fresh medium was added to untreated group (each group was n = 3). After 36 h, the medium was discarded, and each well was washed with 2 mL of PBS. The cells were collected, resuspended in PBS, fixed with 70% ethanol, permeabilized with 1% NP40, stained with propidium iodide 50 μg/mL, and analyzed using FACScan flow cytometer (BD Biosciences, San Jose, CA). ModFit LT and Flowjo software were used to analyze data, generate DNA histograms, and determine the cell counts of each cell cycle phase.
3.2.1.4. Apoptosis Detection by Annexin V/Propidium Iodide Staining
In 6-well plates, A549 cells were seeded at a density of 150 × 103 cells/well for 24 h. Afterward, the old medium was replaced by 2 mL of compound 17 (10 μM), paclitaxel (5 nM), or fresh medium (each group was n = 3) for 48 h. Subsequently, cells were collected, rinsed with PBS, and 1X binding buffer, resuspended in 1X binding buffer, and stained with 5 μL of annexin V for 15 min at room temperature and protected from light. Then, 1 mL of binding buffer was used to wash and resuspend the cells, and 5 μL of propidium iodide was added as a final stain. The FACScan flow cytometer (BD Biosciences, San Jose, CA) was used for sample acquisition and analysis.
3.2.1.5. Caspase 3, 8, and 9 Activity Assay
A549 cells were seeded in 96-well plates at a density of 8 × 103 cells/well in 100 μL for 24 h. After incubation for 24 h, the medium was discarded, and 100 μL of compound 17 (10 μM) or 100 μL of fresh medium was added to the plated cells. After 48 h, the caspase activity was detected following the manual instructions provided by the supplier (Caspase 3, Caspase 8, and Caspase 9 Multiplex Activity Assay Kit Fluorometric, abcam, Burlingame, CA).
3.2.1.6. Tubulin Polymerization Assay
Fluorescence based tubulin polymerization assay (Cytoskeleton Inc., BK011P, Denver, CO) was performed according to the manufacturer’s instructions. Briefly, a stock solution of compound 17 was diluted with ultrapure water to make a final concentration of 3 μM. Similarly, a working solution of paclitaxel was prepared in the same way, and used as a control (stabilizes tubulin polymerization). In addition, approximately 75 μM of CaCl2 was used as another control (destabilizes tubulin polymerization). Before starting the experiment, 96-well plate and fluorimeter (SpectraMax M5Microplate Reader, Molecular Devices, San Jose, CA) were prewarmed to 37 °C. The fluorimeter was also set up for kinetic measurements every minute at excitation 360 nm and emission 450 nm. Next, samples and tubulin were added to a 96-well plate, and the plate was inserted into the plate reader. A recording of the measurements was started immediately. Data were then processed and analyzed.
3.2.2. Cell Viability Testing of Compound 17 against CT-26 Murine Colon Carcinoma
The murine CT-26 colon carcinoma cells were seeded in 96-well plates at a density of 2 × 103 cells/well for 24 h, then the medium was replaced by 100 μL of fresh medium (i.e., after incubation for 24 h). Compound 17 was added in 6 different concentrations (100, 50, 25, 10, 1, 0.1 μM) (n = 3) in 100 μL of fresh medium. The plates were incubated at 37 °C with 5% CO2 for 24 h. Subsequently, the medium was discarded, and 100 μL of fresh medium and 20 μL of MTS reagent were added to each well and incubated at 37 °C with 5% CO2 for 2 h. Spectramax plus Spectrophotometer (Molecular Devices, San Jose, CA) set up at 490 nm was used to record the optical density on each well. Then, the relative cell viability was calculated as described in eq 1.
3.2.3. In Vivo Antitumor Efficacy Study
Six to 8 week old female BALB/cJ mice were challenged with CT26 murine colon carcinoma cells at density 1 × 106 cells/mouse subcutaneously in the right flank. After 1 week, the tumor volumes were measured, and the mice were randomized into two groups. The first group (n = 4) was treated with 250 μg of compound 17 in 10% (v/v) Tween-80 solution per mouse. Mice in the second group (n = 4) were not treated (i.e., naïve). The treatment was administered intraperitoneally for 8 successive days. The mice body weight was also recorded every day, and the tumor volumes were monitored every 2 days starting from day 8 by measuring the tumor diameter (D, mm) and height (H, mm) using a digital caliper and following this equation:
| 2 |
The mice were euthanized once the tumor diameter exceeded 20 mm or the tumor height exceeded 10 mm. This animal study was approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC).
3.2.4. Statistical Analysis
Data obtained from the cell cycle analysis and apoptosis detection experiments were initially analyzed by one-way analysis of variance (ANOVA) using the F-test and followed by a Tukey’s multiple comparison test to compare all pairs of treatments. An unpaired two-tailed t test was used to compare data obtained from caspase 3, 8, and 9 levels experiment. Data of the tumor progression and body weight were analyzed with a linear mixed-effects model using SAS PROC MIXED (SAS Institute Inc., Cary, NC), as previously described.44 In all tests, differences were considered statistically significant when P < 0.05.
Conclusions
In summary, a series of fused thiophene incorporating benzamides, benzyl amines, and aryl ureas were synthesized. The benzyl urea derivative 17 was identified as a potent and broad spectrum antiproliferative agent that displayed GI50 values in submicromolar concentrations against several leukemia, nonsmall cell lung cancer, colon, CNS, melanoma, ovarian, renal, prostate and breast cancer cell lines. In addition, in vivo antitumor efficacy was confirmed using the CT-26 murine tumor model. Tubulin polymerization inhibition is proposed to be the potential mechanism of action of compound 17, and this was consistent with cell cycle arrest resulting from G2/M accumulation. Early apoptosis was detected using annexin V and propidium iodide in A549 cells; also caspase 3, 8, and 9 activity were confirmed in the same cell line treated with compound 17. Therefore, the scaffold reported herein will be a platform for the development of a second generation of potent antiproliferative drug candidates with in vivo antitumor efficacy. We are planning to determine the binding site of compound 17 on tubulin. A structure activity relationship study (SAR) will be performed by synthesizing different substituted benzyl urea analogues in addition to ring size optimization of the cyclohepta[b]thiophene scaffold. Moreover, animal studies with human xenograft tumors and different doses of compound 17 will be explored, and the safety profile of this compound will be investigated as well.
Acknowledgments
We would like to express our sincere gratitude to the National Cancer Institute (NCI, Bethesda, MD) for NCI-60 screening of the new compounds and P30 CA086862 for support. We acknowledge the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center. We acknowledge the Lyle and Sharon Bighley Chair to A.K.S. for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00096.
1HMR; 13CNMR; HRMS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Our world in data. https://ourworldindata.org/causes-of-death (accessed 2020-04-10).
- World Health Organization , Media Centre, Cancer. https://www.who.int/news-room/fact-sheets/detail/cancer (accessed 2020-04-10).
- Vanneman M.; Dranoff G. (2012) Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 12, 237–251. 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin J. J.; Romero M. R.; Blazquez A. G.; Herraez E.; Keck E.; Briz O. (2009) Importance and limitations of chemotherapy among the available treatments for gastrointestinal tumours. Anti-Cancer Agents Med. Chem. 9, 162–84. 10.2174/187152009787313828. [DOI] [PubMed] [Google Scholar]
- Zimmermann S.; Dziadziuszko R.; Peters S. (2014) Indications and limitations of chemotherapy and targeted agents in non-small cell lung cancer brain metastases. Cancer Treat. Rev. 40, 716–722. 10.1016/j.ctrv.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Sui X; Chen R; Wang Z; Huang Z; Kong N; Zhang M; Han W; Lou F; Yang J; Zhang Q; Wang X; He C; Pan H (2013) Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 4, e838. 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall A. S.; Christofk H. R. (2017) Cell cycle: Division enzyme regulates metabolism. Nature 546, 357–358. 10.1038/nature22504. [DOI] [PubMed] [Google Scholar]
- Chow A. Y. (2010) Cell Cycle Control by Oncogenes and Tumor Suppressors: Driving the Transformation of Normal Cells into Cancerous Cells. Nature Education 3, 7. [Google Scholar]
- Yano S.; Takehara K.; Tazawa H.; Kishimoto H.; Urata Y.; Kagawa S.; Fujiwara T.; Hoffman R. M. (2017) Cell-cycle-dependent drug-resistant quiescent cancer cells induce tumor angiogenesis after chemotherapy as visualized by real-time FUCCI imaging. Cell Cycle 16, 406–414. 10.1080/15384101.2016.1220461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J. J.; Diaz J. F.; Miller J. H. (2013) The Binding Sites of Microtubule-Stabilizing Agents. Chem. Biol. 20, 301–315. 10.1016/j.chembiol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. (2004) Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Nogales E. (2001) Structural Insights into Microtubule Function. Annu. Rev. Biophys. Biomol. Struct. 30, 397–420. 10.1146/annurev.biophys.30.1.397. [DOI] [PubMed] [Google Scholar]
- Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Diaz J. F.; Altmann K. H.; Steinmetz M. O. (2013) Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 339, 587–90. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Tian Y.; Liew K. M. (2012) A multiscale model to predict the elastic property of microtubules. J. Comput. Theor. Nanosci. 9, 789–793. 10.1166/jctn.2012.2097. [DOI] [Google Scholar]
- Hait W. N.; Rubin E.; Goodin S. (2005) Tubulin-targeting agents. Cancer Chemother. Biol. Response Modif. 22, 35–59. 10.1016/S0921-4410(04)22003-8. [DOI] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. (2004) Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–65. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Hajek R.; Vorlicek J.; Slavik M. (1996) Paclitaxel (Taxol): a review of its antitumor activity in clinical studies Minireview. Neoplasma 43, 141–54. [PubMed] [Google Scholar]
- Lee C.-T.; Huang Y.-W.; Yang C.-H.; Huang K.-S. (2015) Drug delivery systems and combination therapy by using vinca alkaloids. Curr. Top. Med. Chem. 15, 1491–1500. 10.2174/1568026615666150414120547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaletti G.; Cavalletti E.; Oggioni N.; Sottani C.; Minoia C.; D’Incalci M.; Zucchetti M.; Marmiroli P.; Tredici G. (2001) Distribution of paclitaxel within the nervous system of the rat after repeated intravenous administration. J. Peripher. Nerv. Syst. 6, 61. 10.1046/j.1529-8027.2001.01008-5.x. [DOI] [PubMed] [Google Scholar]
- Windebank A. J. (1999) Chemotherapeutic neuropathy. Curr. Opin. Neurol. 12, 565–71. 10.1097/00019052-199910000-00010. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Sikic B. I. (1999) Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. 17, 1061–70. 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]
- Gao P.; Rush B. D.; Pfund W. P.; Huang T.; Bauer J. M.; Morozowich W.; Kuo M. S.; Hageman M. J. (2003) Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 92, 2386–98. 10.1002/jps.10511. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Cui Y.; Zhao L.; Zhu T.; Lee R. J.; Liao W.; Sun F.; Li Y.; Teng L. (2019) Thiophene Derivatives as New Anticancer Agents and Their Therapeutic Delivery Using Folate Receptor-Targeting Nanocarriers. ACS Omega 4 (5), 8874–8880. 10.1021/acsomega.9b00554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J.; Zheng Y.; Wang Y.; Sheng W.; Li Y.; Liu X.; Si S.; Shao R.; Zhen Y. (2018) A new compound of thiophenylated pyridazinone IMB5043 showing potent antitumor efficacy through ATM-Chk2 pathway. PLoS One 13, 0191984 10.1371/journal.pone.0191984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying X.; Tu W.; Li S.; Wu Q.; Chen X.; Zhou Y.; Hu J.; Yang G.; Jiang S. (2019) Hyperbaric oxygen therapy reduces apoptosis and dendritic/synaptic degeneration via the BDNF/TrkB signaling pathways in SCI rats. Life Sci. 229, 187–199. 10.1016/j.lfs.2019.05.029. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Cui Y.; Zhao L.; Zhu T.; Lee R. J.; Liao W.; Sun F.; Li Y.; Teng L. (2019) Thiophene Derivatives as New Anticancer Agents and Their Therapeutic Delivery Using Folate Receptor-Targeting Nanocarriers. ACS Omega 4, 8874–8880. 10.1021/acsomega.9b00554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch R. J.; Baumann C. A.; Liu J.; Gibbs A. C.; Ott H.; Lattanze J.; Player M. R. (2006) Bioorg. Med. Chem. Lett. 16, 3282–3286. 10.1016/j.bmcl.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Zabludoff S. D.; Deng C.; Grondine M. R.; Sheehy A. M.; Ashwell S.; Caleb B. L.; Green S.; Haye H. R.; Horn C. L.; Janetka J. W.; Liu D.; Mouchet E.; Ready S.; Rosenthal J. L.; Queva C.; Schwartz G. K.; Taylor K. J.; Tse A. N.; Walker G. E.; White A. M. (2008) AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 7, 2955. 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Gui X.; Yang H.; Li T.; Tan X.; Shi P.; Li M.; Du F.; Chen Z. J. (2019) Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. 10.1038/s41586-019-1006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumble M.; Crouthamel M. C.; Zhang S. Y.; Schaber M.; Levy D.; Robell K.; Liu Q.; Figueroa D. J.; Minthorn E. A.; Seefeld M. A.; Rouse M. B.; Rabindran S. K.; Heerding D. A.; Kumar R. (2014) Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS One 9, e100880 10.1371/journal.pone.0100880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls M.; Baxi S. M.; Mehta P. P.; Liu K. K.; Zhu J.; Estrella H.; Li C.; Zientek M.; Zong Q.; Smeal T.; Yin M. J. (2014) Targeting small cell lung cancer harboring PIK3CA mutation with a selective oral PI3K inhibitor PF-4989216. Clin. Cancer Res. 20, 631–43. 10.1158/1078-0432.CCR-13-1663. [DOI] [PubMed] [Google Scholar]
- Wu C. P.; Murakami M.; Hsiao S. H.; Chou A. W.; Li Y. Q.; Huang Y. H.; Hung T. H.; Ambudkar S. V. (2017) Overexpression of ATP-Binding Cassette Subfamily G Member 2 Confers Resistance to Phosphatidylinositol 3-Kinase Inhibitor PF-4989216 in Cancer Cells. Mol. Pharmaceutics 14, 2368–2377. 10.1021/acs.molpharmaceut.7b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K.; Schwarz P. M.; Ludueña R. F. (2002) Interaction of nocodazole with tubulin isotypes. Drug Dev. Res. 55, 91–96. 10.1002/ddr.10023. [DOI] [Google Scholar]
- Feng R.; Li S.; Lu C.; Andreas C.; Stolz D. B.; Mapara M. Y.; Lentzsch S. (2011) Targeting the Microtubular Network as a New Antimyeloma Strategy. Mol. Cancer Ther. 10, 1886–1896. 10.1158/1535-7163.MCT-11-0234. [DOI] [PubMed] [Google Scholar]
- Risinger A. L.; Westbrook C. D.; Encinas A.; Mulbaier M.; Schultes C. M.; Wawro S.; Lewis J. D.; Janssen B.; Giles F. J.; Mooberry S. L. (2011) ELR510444, a novel microtubule disruptor with multiple mechanisms of action. J. Pharmacol. Exp. Ther. 336, 652–60. 10.1124/jpet.110.175331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Brancale A.; Ricci A.; Hamel E.; Bortolozzi R.; Basso G.; Viola G. (2011) Convergent Synthesis and Biological Evaluation of 2-Amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl Thiazoles as Microtubule Targeting Agents. J. Med. Chem. 54, 5144–5153. 10.1021/jm200392p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Carrion M. D.; Cara C. L.; Preti D.; Fruttarolo F.; Pavani M. G.; Tabrizi M. A.; Tolomeo M.; Grimaudo S.; Di Cristina A.; Balzarini J.; Hadfield J. A.; Brancale A.; Hamel E. (2007) Synthesis and Biological Evaluation of 2- and 3-Aminobenzo[b]thiophene Derivatives as Antimitotic Agents and Inhibitors of Tubulin Polymerization. J. Med. Chem. 50, 2273–2277. 10.1021/jm070050f. [DOI] [PubMed] [Google Scholar]
- Simoni D.; Romagnoli R.; Baruchello R.; Rondanin R.; Grisolia G.; Eleopra M.; Rizzi M.; Tolomeo M.; Giannini G.; Alloatti D.; Castorina M.; Marcellini M.; Pisano C. (2008) Novel A-Ring and B-Ring Modified Combretastatin A-4 (CA-4) Analogues Endowed with Interesting Cytotoxic Activity. J. Med. Chem. 51, 6211–6215. 10.1021/jm8005004. [DOI] [PubMed] [Google Scholar]
- Gewald K.; Schinke E.; Bottcher H. (1966) 2-Aminothiopheneaus methlenaktiven nitriilen carbonyl verbindungen and schwefet. Chem. Ber. 99, 94–100. 10.1002/cber.19660990116. [DOI] [Google Scholar]
- Fulda S.; Debatin K. M. (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25 (34), 4798–811. 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Zhang L. H.; Wu L.; Raymon H. K.; Chen R. S.; Corral L.; Shirley M. A.; Narla R. K.; Gamez J.; Muller G. W.; Stirling D. I.; Bartlett J. B.; Schafer P. H.; Payvandi F. (2006) The synthetic compound CC-5079 is a potent inhibitor of tubulin polymerization and tumor necrosis factor-alpha production with antitumor activity. Cancer Res. 66 (2), 951–9. 10.1158/0008-5472.CAN-05-2083. [DOI] [PubMed] [Google Scholar]
- Sato N.; Michaelides M.; Wallack M. (1981) Characterization of tumorigenicity, mortality, metastasis, and splenomegaly of two cultured murine colon lines. Cancer Res. 41, 2267–2272. [PubMed] [Google Scholar]
- DTP Human Tumor Cell Line Screen Process https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm.
- Wafa E. I.; Geary S. M.; Ross K. A.; Goodman J. T.; Narasimhan B.; Salem A. K. (2019) Pentaerythritol-based lipid A bolsters the antitumor efficacy of a polyanhydride particle-based cancer vaccine. Nanomedicine 21, 102055. 10.1016/j.nano.2019.102055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.