

Abstract

Melanoma is a lethal form of skin cancer. Despite recent breakthroughs of BRAF-V600E and PD-1 inhibitors showing remarkable clinical responses, melanoma can eventually survive these targeted therapies and become resistant. To solve the drug resistance issue, we designed and synthesized ligand-drug conjugates that couple cytotoxic drugs, which have a low cancer resistance issue, with the melanocortin 1 receptor (MC1R) agonist melanotan-II (MT-II), which provides specificity to MC1R-overexpressing melanoma. The drug-MT-II conjugates maintain strong binding interactions to MC1R and induce selective drug delivery to A375 melanoma cells through its MT-II moiety in vitro. Furthermore, using camptothecin as the cytotoxic drug, camptothecin-MT-II (compound 1) can effectively inhibit A375 melanoma cell growth with an IC50 of 16 nM. By providing selectivity to melanoma cells through its MT-II moiety, this approach of drug-MT-II conjugates enables us to have many more options for cytotoxic drug selection, which can be the key to solving the cancer resistant problem for melanoma.
Keywords: melanoma, ligand-drug conjugate, MC1R, GPCR internalization, targeted cancer therapy
Melanoma is the most deadly form of skin cancer in the United States, with an estimated 96 480 new cases and 7230 deaths in 2019.1 Once metastasized, the median overall survival for malignant melanoma patients is 5.3 months.2 Despite recent breakthroughs on targeted therapies including BRAF-V600E inhibitors and programmed cell death protein 1 (PD-1) inhibitors, current treatments can only improve survival, and tumor cells eventually become resistant to these treatments. The BRAF-V600E inhibitor vemurafenib was shown to prolong the median overall survival of patients with BRAF V600E mutant melanoma to 15.9 months.3 However, a mice study showed that 20% of melanoma tumors became resistant to vemurafenib treatment after 56 days.4 Similarly, 60% to 70% of metastatic melanoma patients are innately resistant to PD-1 inhibitor treatments.5 These targeted therapies achieve high specificity to cancer and less side effects through interfering with specific targeted molecules required for carcinogenesis and tumor growth, but there are many ways that cancer cells could circumvent such interventions and become resistant to the therapies.6,7 On the other hand, cytotoxic drugs target biological processes that are fundamental for cell proliferation or survival, so that the therapeutic effect cannot be easily bypassed through activation of a compensating signal pathway.8 Nevertheless, current cytotoxic drugs usually have poor specificity to cancer cells and thus are also toxic to healthy noncancer cells, thus causing side effects.
Recently, a new strategy of ligand-targeted cancer therapeutics has been widely explored in different clinical trials, especially for targeting folate receptor (FR) positive cancers and prostate-specific membrane antigen (PSMA) positive prostate cancer.9,10 The design utilizes a ligand that can bind to and activate an overexpressed receptor on cancer cells to provide selectivity. The ligand is conjugated to a cytotoxic drug through a cleavable linker. Once bound to the receptor, the conjugate molecule is internalized into the early endosome through endocytosis,11 where the acidic environment causes the conjugate molecule to dissociate with the receptor.12 The conjugate molecule will then be sorted to other endosomal compartments, where the cleavable linker is degraded, and the cytotoxic drug is released into cancer cells. This design uses specific ligand–receptor interactions between the conjugate molecule and cancer cells to enhance selectivity, and thus would produce much less damage to healthy noncancer cells compared to using the cytotoxic drug itself.13
The melanocortin 1 receptor (MC1R) is a G protein-coupled receptor that is mainly expressed on melanocytes to regulate skin pigmentation. Upon sun exposure, the endogenous agonist α-melanocyte-stimulating hormone (α-MSH) is produced, which activates MC1R on melanocytes to induce melanin production and skin pigmentation.14 MC1R is found to be highly expressed in 80% of malignant melanomas,15 and thus has been demonstrated as a selective target for melanoma imaging.16−21 Even with nonselective peptide ligands that can also bind to other melanocortin receptor subtypes, the in vivo imaging studies demonstrated high melanoma uptake and low normal organ uptake except for the kidney.19,20 Ever since its development, the cyclized peptide melanotan-II (Ac-Nle-cyclo[Asp-His-D-Phe-Arg-Trp-Lys]-NH2, MT-II) has been widely used as melanocortin receptor agonists due to its strong potency and serum stability.22 MT-II also serves as a template for further modifications and conjugations to improve its selectivity,23 oral bioavailability,24 and to expand its applications (such as melanoma imaging and therapy19,20). In this paper, we describe the design, synthesis, and biological evaluation of novel ligand-targeted chemotherapeutic agents targeting MC1R overexpressing melanoma.
Results
Design of Drug-MT-II Conjugates with Self-Cleavable Linker
α-MSH is a linear peptide with a short half-life of around 10 min.25 To maintain a reasonable serum stability of the final conjugate molecule, the cyclized peptide MT-II, with an enhanced half-life of 1.5 h,26 was used for MC1R targeting. MT-II has previously been shown to induce β-arrestin mediated receptor internalization of MC1R.27 The linker was designed to have a disulfide bond, which can be reduced by the excessive amount of glutathione inside the endosome.28 Once reduced, the thiol group in the intermediate 15 can carry out intramolecular nucleophilic attack to release the free drug 11 through two slightly different mechanisms (Scheme 1).29 A 6-aminohexanoic acid was introduced as part of the linker to create space and prevent the cytotoxic drug from interfering with ligand–receptor interactions. The ability of the drug-MT-II conjugate scaffold to selectively target melanoma cells was tested by replacing the therapeutic drug with a fluorescent probe fluorescein 12 in the design of fluorescein-MT-II conjugate (2, Figure 1). The topoisomerase I inhibitor camptothecin (CPT, 11) was selected as the cytotoxic drug in the design of CPT-MT-II conjugate (1, Figure 1).
Scheme 1. Proposed Mechanism of Disulfide-Mediated Release of Camptothecin from MC1R Targeting Prodrugs in the Endosome of Melanoma Cells.
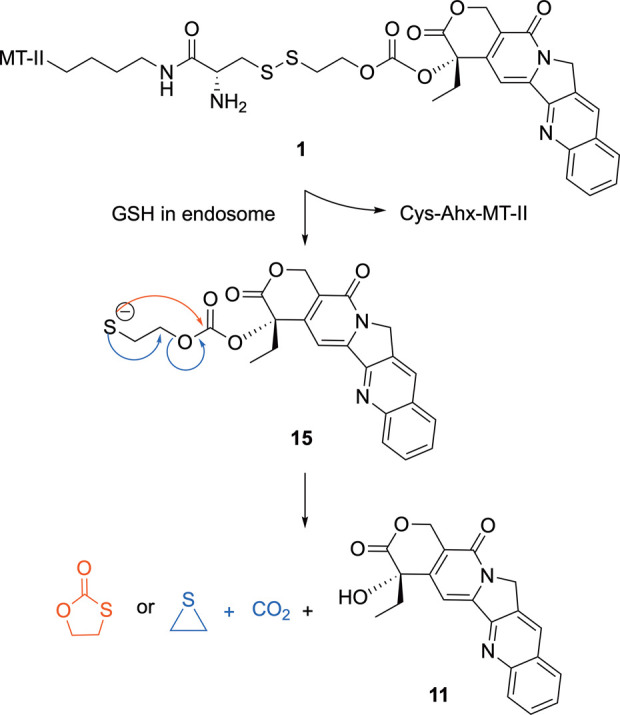
Figure 1.

Structures of CPT-MT-II conjugate 1 and fluorescein-MT-II conjugate 2.
Synthesis of CPT-MT-II (1) and Fluorescein-MT-II (2)
Scheme 2 shows the Fmoc solid-phase peptide synthesis used to prepare the linker-MT-II peptide 3. After the linear peptide Fmoc-Cys(Trt)-6-Ahx-Nle-Asp(Allyl)-His(Trt)-D-Phe-Arg(Pbf)-Trp(Boc)-Lys(Alloc) was synthesized on Rink amide resin, the side chain protecting groups on Asp and Lys residues were removed using the standard Allyl/Alloc deprotection method,30 and side-chain-to-side-chain cyclization was performed. Fmoc deprotection and TFA cleavage were further carried out to produce 3.
Scheme 2. Synthesis of the Linker-MT-II Peptide 3.

Reagents and conditions: (a) 20% piperidine/DMF, 20 min; Fmoc-Lys(Alloc)–OH, HCTU, DIPEA/DMF, 1 h; (b) 20% piperidine/DMF, 20 min; Fmoc-Trp(Boc)–OH, HCTU, DIPEA/DMF, 1 h; (c) 20% piperidine/DMF, 20 min; Fmoc-Arg(Pbf)–OH, HCTU, DIPEA/DMF, 1 h; (d) 20% piperidine/DMF, 20 min; Fmoc-d-Phe-OH, HCTU, DIPEA/DMF, 1 h; (e) 20% piperidine/DMF, 20 min; Fmoc-His(Trt)–OH, HCTU, DIPEA/DMF, 1 h; (f) 20% piperidine/DMF, 20 min; Fmoc-Asp(Allyl)–OH, HCTU, DIPEA/DMF, 1 h; (g) 20% piperidine/DMF, 20 min; Fmoc-Nle–OH, HCTU, DIPEA/DMF, 1 h; (h) 20% piperidine/DMF, 20 min; Fmoc-6-Ahx–OH, HCTU, DIPEA/DMF, 1 h; (i) 20% piperidine/DMF, 20 min; Fmoc-Cys(Trt)–OH, HCTU, DIPEA/DMF, 1 h; (j) PhSiH3, Pd(PPh3)4/DCM, 1 h; (k) HCTU, DIPEA/DMF, 2 h; (l) 20% piperidine/DMF, room temperature, 20 min; (m) TFA/TIS/H2O (95:2.5:2.5), 3 h.
The synthesis of the carbonate linker 10 is shown in Scheme 3. Compound 5 with a sulfenyl chloride functional group was added to 2-mercaptoethanol 4 to form the disulfide bond. A disulfide exchange reaction was performed with pyridine 6 in DCM at reflux to yield alcohol 7. Carbonylation was achieved with triphosgene 8 added to 7, which was followed by ester exchange reaction with hydroxybenzotriazole 9 to yield to carbonate linker 10. The synthesis of 10 was modified based on previously reported procedures.31
Scheme 3. Synthesis of Linker 10.

Scheme 4 shows the final steps to the synthesis of the CPT-MT-II conjugate 1 and fluorescein-MT-II conjugate 2. The hydroxyl group on CPT 11 was connected to the carbonate linker 10 through nucleophilic substitution, yielding CPT-linker compound 13. A thiol–disulfide exchange reaction was then performed between the disulfide groups on 13 and the thiol group on the Cys residue of 3 to yield the CPT-MT-II conjugate 1. Similarly, the hydroxyl group on fluorescein 12 reacted with the carbonate linker 10 to form the fluorescein-linker compound 14, which further reacted with 3 to yield the fluorescein-MT-II conjugate 2.
Scheme 4. Synthesis of the CPT-MT-II Conjugate 1 and Fluorescein-MT-II Conjugate 2.

Binding of CPT-MT-II and Fluorescein-MT-II to Melanocortin Receptors
To evaluate binding affinity of 1 and 2 to MC1R, competitive binding assays were performed on human MC1R overexpressing HEK293 cells with 125I labeled NDP-α-MSH as the competing ligand (Figure 2, Table 1). The Ki values of CPT-MT-II and fluorescein-MT-II binding to MC1R were determined to be 57 ± 7 nM and 172 ± 20 nM, respectively, while the Ki value of MT-II was shown to be 1.5 ± 0.2 nM. Despite having lower binding affinities than MT-II, both 1 and 2 were demonstrated to retain full displacement of 125I-NDP-α-MSH within micromolar range concentration. As MT-II is not selective to MC1R and can also bind to and activate other melanocortin receptor subtypes,24,32 further competitive binding assays were performed with HEK293 cells that overexpress MC3R, MC4R, or MC5R. CPT-MT-II was demonstrated to have weak selectivity to MC1R, with around 3–6-fold higher binding affinity to MC1R as compared to MC3R, MC4R, and MC5R (Table 1, Figure S7).
Figure 2.
Competitive binding assay results of MT-II, CPT-MT-II 1 and fluorescein-MT-II 2 binding to MC1R in competition with 125I labeled NDP-α-MSH.
Table 1. Ki Values of MT-II, CPT-MT-II and Fluorescein-MT-II Binding to Melanocortin Receptorsa.
|
Ki (nM) |
||||
|---|---|---|---|---|
| compounds | MC1R | MC3R | MC4R | MC5R |
| MT-II | 1.5 ± 0.2 | 1.3 ± 0.2 | 1.4 ± 0.2 | 2.9 ± 0.3 |
| CPT-MT-II (1) | 57 ± 7 | 300 ± 34 | 173 ± 48 | 194 ± 10 |
| fluorescein-MT-II (2) | 172 ± 20 | ND | ND | ND |
ND: not determined.
Specific Drug Delivery of Drug-MT-II Conjugates to Melanoma Cell Line A375 over HEK 293
To analyze the ability of drug-MT-II conjugate scaffold to release drug into melanoma cells, A375 malignant melanoma cells, which overexpress MC1R,15,33 were treated with 3 μM of 2 for 0.5, 1.5, or 2.5 h. The media with excess 2 was washed away, and the intracellular fluorescence was monitored by live-cell confocal microscopy under the same microscope settings. No significant fluorescence uptake was observed when A375 cells were treated for 0.5 h (data not shown) or 1.5 h (Figure 3A). After 2.5 h incubation, strong fluorescence was observed to be evenly distributed inside the A375 cells (Figure 3B). A Z-stack image was taken, which further confirmed that the observed fluorescence was not from the cell membrane but came from both nucleus and cytoplasm (Figure S8). To test if the fluorescence uptake was mediated by ligand–receptor interactions between MC1R and the MT-II moiety of 2, A375 cells were simultaneously treated with 3 μM of 2 and 30 μM of MT-II for 2.5 h and examined under confocal microscopy. With an excess amount of MT-II present to occupy the MC1R binding sites, fluorescence uptake by A375 cells was not observed (Figure 3C), suggesting that ligand–receptor interactions between MC1R and fluorescein-MT-II is crucial to deliver fluorescein into A375 cells. To test if drug-MT-II conjugate scaffold also releases the therapeutic drug into healthy noncancer cells, HEK293 kidney epithelial cells were treated with 3 μM of 2 for 2.5 h and monitored under confocal microscope. The results demonstrated fluorescence uptake by HEK293 cells with much weaker fluorescence intensity compared to uptake by A375 cells (Figure 3D), suggesting a selective delivery of fluorescein into melanoma cells using 2. Taken together, these results imply that the drug-MT-II conjugate scaffold can selectively deliver the therapeutic drug into the cytoplasm of melanoma cells through interactions between its MT-II moiety and MC1R on melanoma cells.
Figure 3.

Live-cell confocal microscopic image of A375 (A–C) and HEK293 (D) cells treated with 3 μM of fluorescein-MT-II 2 with or without 30 μM of MT-II for indicated periods of time.
CPT-MT-II Conjugate Effectively Reduces Cell Viability of A375 Cells
To analyze the ability of drug-MT-II conjugates to affect the viability of melanoma cells, A375 malignant melanoma cells were treated with different concentrations of CPT 11, MT-II, or 1 for 24 h, and the cell viability was measured with XTT assay. The results suggest that both CPT and CPT-MT-II can effectively kill A375 melanoma cells, while MT-II alone does not have any impact on melanoma cell viability (Figure 4). CPT-MT-II has an enhanced cytotoxicity with an IC50 value of 16 ± 2 nM, comparing to the IC50 value of CPT (47 ± 2 nM). Camptothecin is known to have poor solubility.34 Thus, the enhanced potency for CPT-MT-II conjugate is possibly due to improved solubility and cell membrane penetration of CPT-MT-II.
Figure 4.
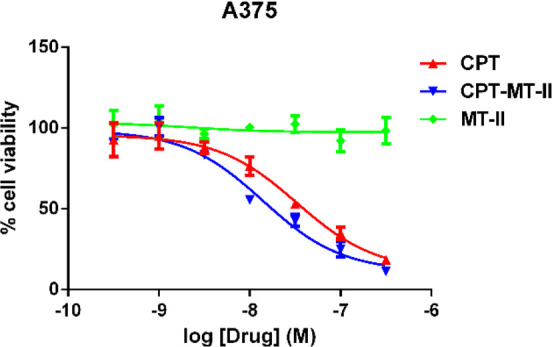
Dose–response XTT cell viability assay of camptothecin (CPT, 11), CPT-MT-II conjugate (1) and MT-II on the cell viability of human A375 melanoma cell line after 24 h incubation.
Discussion
Recent breakthroughs on targeted therapies, including BRAF-V600E inhibitors and PD-1 inhibitors, have successfully prolonged the median overall survival for malignant melanoma patients. Nevertheless, tumor cells eventually relapse and become resistant to these treatments. To completely cure melanoma would require cytotoxic drugs that target biological processes essential for cell proliferation or survival, so that melanoma cells cannot easily bypass the therapeutic target to survive the treatment, or that they are quickly eliminated before they can accumulate enough mutations to become resistant. However, cytotoxic drugs normally have poor selectivity to cancer cells and thus possess high risks of side effects. Here we report the design, synthesis, and biological evaluations of the first ligand-drug conjugate targeting melanoma, which can selectively deliver cytotoxic drugs to melanoma cells through the overexpressed receptor MC1R. Our biological data support the hypothesis that a drug-MT-II conjugate can bind to MC1R, and mediate selective drug delivery to melanoma cells through its interactions with MC1R. Using camptothecin as an example cytotoxic drug, we further confirmed that CPT-MT-II can effectively kill melanoma cells in low nanomolar range in vitro. In our next paper, we will specifically address the PK/PD and safety for the CPT-MT-II in vivo. Taken together, our design of drug-MT-II conjugate motif is a promising approach to selectively deliver cytotoxic drugs that target pathways essential for cell proliferation or survival to MC1R overexpressing melanomas, in order to effectively combat drug resistance issues and extenuate side effects.
The competitive binding assay suggests that both 1 and 2 can bind to MC1R and fully displace the radioligand 125I-NDP-α-MSH, though the binding affinity to MC1R decreased by 40–123 fold. Similar decrease in the binding affinity was also seen in a recent paper when the cytotoxic drug doxorubicin was conjugated to the formyl peptide receptor 1 (FPR1) agonist through a pH-sensitive linker.35 Such decrease in binding affinity could be partly due to the fact that both CPT and fluorescein can interact with serum albumin.34,36 As bovine serum albumin (BSA) was used in the binding assay to reduce nonspecific bindings, interactions between the conjugate drugs and BSA may limit the amounts of conjugate drugs available to the melanocortin receptors on the cell surface, and thus cause the binding affinity to decrease. Another possible reason for the decrease in binding affinity is that intramolecular interactions between CPT/fluorescein and MT-II interfered with binding. Such interactions can be reduced by using a longer and more hydrophilic spacer. Nevertheless, even small increases in the spacer size have been demonstrated to markedly reduce tumor penetration.37
Using confocal microscopy, we demonstrated that the release of fluorescein from 2 into A375 takes at least 90 min. The proposed drug release mechanism for ligand-drug conjugates include conjugate binding to the receptor, endocytosis, release of the cleavable linker, and diffusion of the drug through the membrane of the endosome.9,10 Even though the process of MT-II binding to the MC1R and causing endocytosis could be observed within 10 min,27 36% of surface bound MC1R agonist was shown to internalize 1 h after 125I-NDP-α-MSH incubation.38 The half time of reducing the disulfide bond in ligand-drug conjugates in the presence of glutathione was demonstrated to be 1 h.39 Considering the excessive time required for fluorescein to penetrate the membrane of the lysosome, 90 to 150 min is a reasonable time window for drug-MT-II conjugates to deliver the drug into melanoma cells. A similar long time frame was also reported in the development of a ligand-drug conjugate targeting the formyl peptide receptor (FPR1).35 When doxorubicin was conjugated to the FPR1 agonist through a pH-sensitive linker, doxorubicin was shown to mainly colocalize with FPR1 in the endosome 1 h after incubation.35 Doxorubicin was demonstrated to pass through the endosome membrane and enrich in the nucleus 6 h after incubation.35 In our study, we chose the pH-sensitive dye fluorescein, which has strong fluorescence intensity at neutral to basic pH but only retains a minimal level of fluorescence intensity when the pH is less than 5.5.40 Fluorescein would lose its fluorescence when trafficked in the acidic environment of endosomes and lysosomes. Fluorescein would regain fluorescence when it reaches the cytoplasm, thus mimicking the complete drug delivery process of ligand–drug conjugates.
Because selective delivery to MC1R overexpressing melanomas is provided by the MT-II motif of the drug–MT-II conjugate, our design opens up opportunities for a much wider range of therapeutic drugs selection. From a synthetic perspective, any cytotoxic drug with an −NH2 or −OH group can be used for drug-MT-II conjugates through the same synthetic strategies. To achieve maximal killing of cancer cells, drugs with high potency are favored due to limited number of receptors on the membrane.9,41 Potential personalized therapy may be developed by screening for cytotoxic drugs that have low resistance problems and are most effective at killing a person’s tumor cells without having to consider selectivity, and then provide selectivity to melanoma cells through our drug–MT-II conjugate design. Moreover, a potential combinational therapy with MT-II conjugated to different cytotoxic drugs can be developed based on the drug–MT-II conjugate design, as combinational therapy is widely considered the best option for combating drug resistance in cancer.42 These approaches may greatly reduce the chances that the melanoma tumor become resistant to the therapy.
GPCRs such as protease-activated receptor-1 (PAR-1), angiotensin-II receptor, G protein-coupled receptor 30 (GPR30), lysophosphatidic acid receptor (LPAR) and gastrin releasing peptide receptor (GRP-R) have been demonstrated to overexpress in a variety of cancers and play important roles in carcinogenesis, tumor progression and metastasis.43,44 Distinct from previously developed ligand-drug conjugates that target non-GPCR receptors such as FR, PSMA, and glucose transporter 1 (GLUT1),9,10 we demonstrate through our drug-MT-II conjugates targeting the overexpressed MC1R on melanoma that ligand–drug conjugates can target GPCRs for cancer specific drug delivery. Though FPR1 is also a GPCR, ligand-induced internalization of FPR1 was shown to happen through the clathrin-independent mechanism,35,45 whereas MT-II stimulates MC1R internalization through the β-arrestin-mediated clathrin-dependent mechanism27 that is the most common for GPCR internalization. With our results demonstrating its feasibility, we anticipate more ligand–drug conjugates targeting different GPCRs to be developed to address the problem of cancer resistance in the future.
Materials and Methods
General
Organic solvents and reagents were purchased from Aldrich and used without further purification. ESI-MS was performed with the Bruker amaZon ion trap system. Reverse-phase high-performance liquid chromatography (RP-HPLC) was performed with Agilent 1100 series. Semipreparative RP-HPLC on a C18 bonded silica column (Vydac 218TP152022, 250_22 mm, 15–20 μm, 300 Å) was used for compound purification and to analyze the purity, eluted with a linear gradient of acetonitrile (gradient, 2–100% B in A over 40 min, flow rate 3 mL/min). System 1: solvent A, 0.1% TFA in water; solvent B, 0.08% TFA in acetonitrile. System 2: solvent A, 1% formic acid in water; solvent B, 1% formic acid in methanol) and aqueous 0.1% TFA (v/v). The major peak of all compounds accounted for ≥95% of the combined total peak area monitored by a UV detector at 254 nm. Cells were grown in minimum essential medium (MEM, Gibco) supplemented with 10% FBS, 1% penstrep, and 1 mM sodium pyruvate at 37 °C and 5% CO2.
Synthesis of Cys-6-Ahx-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2 (3)
Nα-Fmoc-amino acids were obtained from GL Biochem and ChemCruz. The side chain protected amino acids were Fmoc–Cys(Trt)–OH, Fmoc–Asp(OAll)–OH, Fmoc–His(trt)–OH, Fmoc–Arg(pbf)–OH, Fmoc–Trp(Boc)–OH, and Fmoc–Lys(Alloc)–OH. Fmoc–Rink amide resin was purchased from Novabiochem. All peptides were synthesized by the N-Fmoc solid-phase peptide strategy using DIEA and HCTU as the coupling reagents. Rink amide resin (0.37 mmol/g) was placed into a 5 mL polypropylene syringe with a frit on the bottom and swollen in DCM (2 mL) and DMF (2 mL) for 1 h. The Fmoc protecting group on the rink linker was removed by 20% piperidine in DMF. After 20 min the solution of piperidine was removed and the resin was washed with DMF (2 mL, 4 times) and DCM (2 mL, 4 times). N-Fmoc amino acid (3 equiv) and HCTU (3 equiv) were dissolved in DMF, and then DIEA (3 equiv) was added. The coupling mixture was transferred into the syringe with the resin and shaken for 1 h. Coupling completion was monitored with a Kaiser test. The coupling mixture was removed, and the resin was washed with DMF (2 mL, 4 times) and DCM (2 mL, 4 times). N-Fmoc groups were removed with 20% piperidine in DMF in 20 min. Each coupling and deprotection step was repeated until a linear peptide was assembled. Allyl and Alloc deprotection was carried out by adding Pd(PPh3)4 (0.35 equiv) and PhSiH3 (20 equiv) in DCM under argon. The deprotecting solution was left to react in the presence of argon and shaken for 30 min. Next, the peptide resin was washed four times with DCM and the process was repeated once. The subsequent cyclization was performed by adding HCTU (3 equiv) and DIEA (3 equiv) in DMF for 2 h. The N-Fmoc group on the Cys residue was removed with 20% piperidine in DMF for 20 min. The final wash of the resin was done with DMF (2 mL, four times) and DCM (2 mL, four times). The product was cleaved from the resin with a mixture of 95% TFA, 2.5% TIPS, and 2.5% water during 3 h. Side chain protecting groups were removed during the cleavage step as well. The cleaved mixture was evaporated on a rotary evaporator, and the crude peptide was dissolved in H2O/methanol and purified by HPLC before being lyophilized. m/z calculated, 1197.62; m/z observed, 1198.78 (M+H)+.
Synthesis of 1H-Benzo[d][1,2,3]triazol-1-yl[2-(pyridin-2-yldisulfanyl)-ethyl] Carbonate (10)31
2-Mercaptoethanol (4) (0.77 g, 9.9 mmol) was dissolved in 5 mL of ACN, and the solution was added dropwise to a solution of chlorocarbonylsulfenyl chloride (5) (1.30 g, 9.9 mmol) in 8 mL of ACN precooled at 0 °C. The solution was stirred at 0 °C for 30 min. A solution of 2-mercaptopyridine (6) (1.00 g, 9.0 mmol) in 20 mL of ACN was added dropwise to the solution, and the mixture was stirred at reflux for 2 h, during which a white precipitate formed. The mixture with white precipitate was then stirred at 0 °C for 1 h and filtered. The filter cake was washed with ACN to provide the compound 7 as a white amorphous solid (1.35 g, 80%). Compound 7 (1.00 g, 4.47 mmol) was dissolved in DCM (5 mL) and Et3N (0.45 g, 4.47 mmol) and added dropwise to a solution of triphosgene (8) (0.44 g, 1.49 mmol) at 0 °C. The solution was stirred at room temperature for 1.5 h, followed by dropwise addition of a solution of hydroxybenzotriazole (9) (0.60 g, 4.47 mmol) in DCM (10 mL) and Et3N (0.45 g, 4.47 mmol). The mixture was then stirred at room temperature for 16 h and then diluted with CHCl3 to 50 mL and washed with H2O (100 mL × 3) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The resulting yellow oil was triturated with hexane and filtered to provide the product 10 as a white solid (1.36 g, 87%):
1H NMR (400 MHz, CDCl3) δ: 3.28 (t, J = 6.4 Hz, 2 H), 4.83 (t, J = 6.4 Hz, 2 H), 7.11 (ddd, J = 1.3, 7.2, 8.1 Hz, 1 H), 7.57 (ddd, J = 1.2, 7.2, 8.2 Hz, 1 H), 7.67 (ddd, J = 0.9, 1.7, 8.0 Hz, 1 H), 7.71 (dt, J = 1.3, 8.1 Hz, 1 H), 7.79 (ddd, J = 1.3, 7.3, 8.4 Hz, 1 H), 8.05 (ddd, J = 0.9, 1.8, 8.0 Hz, 1 H), 8.23 (ddd, J = 0.9, 1.8, 8.0 Hz, 1 H), 8.46 (ddd, J = 0.9, 1.7, 8.1 Hz, 1 H).
Synthesis of CPT-Linker Compound (13)
Camptothecin (11, 25 mg, 0.07 mmol) was dissolved in DCM (5 mL), followed by addition of carbonate reagent 10 (37 mg, 0.10 mmol), DMAP (9.1 mg, 0.07 mmol), and Et3N (15 mg, 0.15 mmol). The mixture was stirred at room temperature overnight, purified by semipreparative RP-HPLC, and lyophilized to provide a yellow solid (37.2 mg, 95%). m/z calculated, 561.10; m/z observed, 562.21 (M+H)+.
1H NMR (400 MHz, CDCl3) δ: 1.00 (t, J = 7.5 Hz, 3 H), 2.32 (m, 2 H), 3.06 (t, J = 6.6 Hz, 3 H), 4.36 (m, 2 H), 5.3 (s, 2 H), 5.53 (s, 2 H), 7.03 (m, 1 H), 7.33 (s, 1 H), 7.65 (m, 3 H), 7.83 (m, 1 H), 7.94 (d, J = 8.2 Hz, 1 H), 8.22 (d, J = 8.6 Hz, 1 H), 8.39 (s, 1 H), 8.41 (d, J = 4.7 Hz, 1 H).
Synthesis of Fluorescein-Linker Compound (14)
Fluorescein (12, 33 mg, 0.10 mmol) was dissolved in THF (5 mL), followed by the addition of carbonate reagent 10 (40 mg, 0.12 mmol), DMAP (13 mg, 0.10 mmol), and Et3N (21 mg, 0.21 mmol). The mixture was stirred at room temperature overnight and purified by semipreparative RP-HPLC, and lyophilized to provide a yellow solid (38.0 mg, 70%). m/z calculated, 545.06; m/z observed, 568.16 (M+Na)+.
1H NMR (400 MHz, CDCl3) δ: 3.15 (t, J = 6.5 Hz, 2 H), 3.78 (s, 1 H), 4.53 (dt, J = 6.5, 1.7 Hz, 2H), 6.56 (dd, J = 8.6, 2.4 Hz, 1 H), 6.63 (dd, J = 8.6, 3.7 Hz, 1 H), 6.74 (dd, J = 7.1, 2.4 Hz, 1 H), 6.82 (dd, J = 17.8, 8.6 Hz, 1 H), 6.86 (dd, J = 8.6, 2.3 Hz, 1 H), 6.91 (dd, J = 8.6, 2.3 Hz, 1 H), 7.15 (m, 3 H), 7.66 (m, 4 H), 8.03 (m, 1 H), 8.49 (m, 1 H).
Synthesis of CPT-MT-II (1)
Linker-MT-II peptide (3, 6.11 mg, 0.0050 mmol) was dissolved in argon-purged, saturated sodium bicarbonate solution (1 mL). A solution of CPT-linker compound 13 (2.86 mg, 0.0050 mmol) in THF (1 mL) was added dropwise to the reaction mixture, and the solution was stirred for 30 min. The product CPT-MT-II (1) was purified by preparative RP-HPLC and lyophilized to provide a yellow solid (4.21 mg, 51%). m/z calculated, 1647.71; m/z observed, 1649.19 (M+H)+.
Synthesis of Fluorescein-MT-II (2)
Linker-MT-II peptide (3, 1.33 mg, 0.0011 mmol) was dissolved in argon-purged, saturated sodium bicarbonate solution (0.5 mL). A solution of fluorescein-linker compound 14 (0.93 mg, 0.0017 mmol) in THF (0.5 mL) was added dropwise to the reaction mixture, and the solution was stirred for 30 min. The product CPT-MT-II (1) was purified by preparative RP-HPLC and lyophilized to provide a yellow solid (1.02 mg, 57%). m/z calculated, 1631.67; m/z observed, 816.91 (M+2H)2+.
Competitive Binding Assays
Competitive binding assays were performed on whole cells using previously described procedures.46−48 Stably transfected HEK 293 cell lines overexpressing human MC1R, MC3R, MC4R, or MC5R were seeded on 96-well plates 48 h before the assay and grown to 100 000 cells per well. For the assay, the medium was removed, and cells were washed twice with a binding buffer containing minimum essential medium with Earle’s salt (MEM, GIBCO), 25 mM HEPES (pH 7.4), 0.2% bovine serum albumin, 1 mM 1,10-phenanthrolone, 0.5 mg/L leupeptin, and 200 mg/L bacitracin. Cells were incubated with different concentrations of unlabeled test compounds and 125I labeled NDP-α-MSH (PerkinElmer, 100 000 cpm/well, 0.1386 nM) for 40 min at 37 °C. The medium was subsequently removed, and the cells were washed twice with the binding buffer. The cells were lysed by 50 μL of 0.1 mM NaOH and 50 μL of 1% Triton X-100. The plates were left to dry overnight under the hood, and 50 μL of scintillation fluid (OptiPhase SuperMix, PerkinElmer) was added in each well. The radioactivity was measured using MicroBeta 2 microplate counter (PerkinElmer). Data were analyzed using Graphpad Prism 6 (Graphpad Software, San Diego, CA).
Confocal Microscopy
One milliliter of A375 or HEK 293 cell suspension (∼5 × 104 cells) was seeded in a 35 mm No. 1.5 coverslip glass-bottomed microwell dish (MatTek) and cultured overnight. Cells were gently washed with PBS twice and treated with compound 2 (3 μM) with or without MT-II (30 μM) in phenol red-free, HEPES-supplemented DMEM. After incubation at 37 °C for an indicated time, cells were washed three times with PBS before imaging. Images were taken with Zeiss LSM880 inverted confocal microscope with either bright field or laser at 488 nM with 2% laser power and Plan-Apochromat 63x/1.4 Oil DIC M27 objective.
XTT Cell Viability Assay
A375 cells were seeded in 96 well plates at a density of around 105 cells per well in 95 μL of MEM medium and incubated for 24 h. Cells were treated with 5 μL of drugs in a final concentration of 0, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM, and 300 nM for 24 h. XTT solution was prepared by dissolving 4 mg of XTT in 4 mL of MEM medium. PMS solution was prepared by dissolving 3 mg of PMS in 1 mL of phosphate buffer saline (PBS). Ten microliters of PMS solution was added to 4 mL of XTT solution, and 25 μL of the PMS/XTT mixture was added to each well. The plate was incubated at 37 °C for another 2 h culture. Absorbance was measured at 450 nm using a μQuant Universal Microplate Reader (Bio-Tek instruments, Inc., Winooski, VT). Absorbance from a no-cells control was subtracted, and the percentage of cell viability was calculated as the percentage of absorbance relative to cells treated with medium.
Acknowledgments
This work was supported in part by grants from the U.S. Public Health Service, National Institutes of Health, Grants DK017420 and GM 108040, and Proof of Concept grant from the University of Arizona Tech Launch Transfer. We would like to thank the Marley Light Microscopy Facility at the University of Arizona for their assistance in the confocal microscopy studies.
Glossary
Abbreviations
- Boc
tert-butyloxycarbonyl
- Fmoc
fluorenylmethoxycarbonyl
- Trt
trityl
- Alloc
allyloxycarbonyl
- Allyl, ACN
acetonitrile
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- TIS
triisopropylsilane
- DMAP
4-dimethylaminopyridine
- SPPS
solid-phase peptide synthesis
- RP-HPLC
reverse phase high-performance liquid chromatography
- PD-1
programmed cell death protein 1
- PSMA
prostate-specific membrane antigen
- MC1R
melanocortin 1 receptor
- MSH
melanocyte stimulating hormone
- CPT
camptothecin
- MT-II
melanotan II
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00072.
Synthetic experimental details, related spectroscopic data of intermediate and target compounds, and biological assay protocols (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript. Y.Z. and M.C. designed the experiments. Y.Z., S.M.H., and L.W. synthesized the compounds. Z.L. performed NMR studies. Y.Z. performed the bioassays. Y.Z. and M.C. wrote the manuscript. M. C. and V.J.H. secured the funding.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. (2019) Cancer statistics, 2019. Ca-Cancer J. Clin. 69, 7–34. 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- Sandru A.; Voinea S.; Panaitescu E.; Blidaru A. (2014) Survival rates of patients with metastatic malignant melanoma. J. Med. Life 7, 572–576. [PMC free article] [PubMed] [Google Scholar]
- Sosman J. A.; Kim K. B.; Schuchter L.; Gonzalez R.; Pavlick A. C.; Weber J. S.; McArthur G. A.; Hutson T. E.; Moschos S. J.; Flaherty K. T.; Hersey P.; Kefford R.; Lawrence D.; Puzanov I.; Lewis K. D.; Amaravadi R. K.; Chmielowski B.; Lawrence H. J.; Shyr Y.; Ye F.; Li J.; Nolop K. B.; Lee R. J.; Joe A. K.; Ribas A. (2012) Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 366, 707–714. 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M.; Salangsang F.; Landman A. S.; Sellers W. R.; Pryer N. K.; Levesque M. P.; Dummer R.; McMahon M.; Stuart D. D. (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255. 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W.; Zaretsky J. M.; Sun L.; Song C.; Moreno B. H.; Hu-Lieskovan S.; Berent-Maoz B.; Pang J.; Chmielowski B.; Cherry G.; Seja E.; Lomeli S.; Kong X.; Kelley M. C.; Sosman J. A.; Johnson D. B.; Ribas A.; Lo R. S. (2016) Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis A. J.; Bivona T. G. (2019) Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 25, 185–197. 10.1016/j.molmed.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Manstein V.; Yang C. M.; Richter D.; Delis N.; Vafaizadeh V.; Groner B. (2014) Resistance of Cancer Cells to Targeted Therapies Through the Activation of Compensating Signaling Loops. Curr. Signal Transduction Ther. 8, 193–202. 10.2174/1574362409666140206221931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Singh R. K. (2011) Resistance to chemotherapy and molecularly targeted therapies: rationale for combination therapy in malignant melanoma. Curr. Mol. Med. 11, 553–563. 10.2174/156652411800615153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasarao M.; Galliford C. V.; Low P. S. (2015) Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discovery 14, 203–219. 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- Srinivasarao M.; Low P. S. (2017) Ligand-Targeted Drug Delivery. Chem. Rev. 117, 12133–12164. 10.1021/acs.chemrev.7b00013. [DOI] [PubMed] [Google Scholar]
- Chen S.; Zhao X.; Chen J.; Chen J.; Kuznetsova L.; Wong S. S.; Ojima I. (2010) Mechanism-based tumor-targeting drug delivery system. Validation of efficient vitamin receptor-mediated endocytosis and drug release. Bioconjugate Chem. 21, 979–987. 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Min S. H.; Wang Y.; Campanella E.; Low P. S.; Goldman I. D. (2009) A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J. Biol. Chem. 284, 4267–4274. 10.1074/jbc.M807665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leamon C. P.; Reddy J. A.; Vlahov I. R.; Westrick E.; Parker N.; Nicoson J. S.; Vetzel M. (2007) Comparative preclinical activity of the folate-targeted Vinca alkaloid conjugates EC140 and EC145. Int. J. Cancer 121, 1585–1592. 10.1002/ijc.22853. [DOI] [PubMed] [Google Scholar]
- Lin J. Y.; Fisher D. E. (2007) Melanocyte biology and skin pigmentation. Nature 445, 843–850. 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- Siegrist W.; Solca F.; Stutz S.; Giuffrè L.; Carrel S.; Girard J.; Eberle A. N. (1989) Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells. Cancer Res. 49, 6352–6358. [PubMed] [Google Scholar]
- Barkey N. M.; Tafreshi N. K.; Josan J. S.; De Silva C. R.; Sill K. N.; Hruby V. J.; Gillies R. J.; Morse D. L.; Vagner J. (2011) Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. J. Med. Chem. 54, 8078–8084. 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkey N. M.; Preihs C.; Cornnell H. H.; Martinez G.; Carie A.; Vagner J.; Xu L.; Lloyd M. C.; Lynch V. M.; Hruby V. J.; Sessler J. L.; Sill K. N.; Gillies R. J.; Morse D. L. (2013) Development and in vivo quantitative magnetic resonance imaging of polymer micelles targeted to the melanocortin 1 receptor. J. Med. Chem. 56, 6330–6338. 10.1021/jm4005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M.; Liu Z.; Qu H.; Fan H.; Zheng Z.; Hruby V. J. (2011) Utilize conjugated melanotropins for the earlier diagnosis and treatment of melanoma. Eur. J. Pharmacol. 660, 188–193. 10.1016/j.ejphar.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Gallazzi F.; Miao Y. (2012) Gallium-67-labeled lactam bridge-cyclized alpha-MSH peptides with enhanced melanoma uptake and reduced renal uptake. Bioconjugate Chem. 23, 1341–1348. 10.1021/bc300191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Miao Y. (2014) Introduction of an 8-aminooctanoic acid linker enhances uptake of 99mTc-labeled lactam bridge-cyclized α-MSH peptide in melanoma. J. Nucl. Med. 55, 2057–2063. 10.2967/jnumed.114.145896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafreshi N. K.; Huang X.; Moberg V. E.; Barkey N. M.; Sondak V. K.; Tian H.; Morse D. L.; Vagner J. (2012) Synthesis and characterization of a melanoma-targeted fluorescence imaging probe by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Bioconjugate Chem. 23, 2451–2459. 10.1021/bc300549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Obeidi F.; Castrucci A. M.; Hadley M. E.; Hruby V. J. (1989) Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: design based on molecular dynamics. J. Med. Chem. 32, 2555–2561. 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- Cai M.; Hruby V. J. (2016) Design of cyclized selective melanotropins. Biopolymers 106, 876–883. 10.1002/bip.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Cai M. (2017) Novel approaches to the design of bioavailable melanotropins. Expert Opin. Drug Discovery 12, 1023–1030. 10.1080/17460441.2017.1351940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudman D.; Hollins B. M.; Kutner M. H.; Moffitt S. D.; Lynn M. J. (1983) Three types of alpha-melanocyte-stimulating hormone: bioactivities and half-lives. Am. J. Physiol. 245, E47–54. 10.1152/ajpendo.1983.245.1.E47. [DOI] [PubMed] [Google Scholar]
- Ugwu S. O.; Blanchard J.; Nguyen L. D.; Hadley M. E.; Dorr R. T. (1994) A comparison of HPLC and bioassay methods for plasma melanotan-II (MT-II) determination: application to a pharmacokinetic study in rats. Biopharm. Drug Dispos. 15, 383–390. 10.1002/bdd.2510150505. [DOI] [PubMed] [Google Scholar]
- Cai M.; Stankova M.; Pond S. J.; Mayorov A. V.; Perry J. W.; Yamamura H. I.; Trivedi D.; Hruby V. J. (2004) Real time differentiation of G-protein coupled receptor (GPCR) agonist and antagonist by two photon fluorescence laser microscopy. J. Am. Chem. Soc. 126, 7160–7161. 10.1021/ja049473m. [DOI] [PubMed] [Google Scholar]
- Yang J.; Chen H.; Vlahov I. R.; Cheng J. X.; Low P. S. (2006) Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. U. S. A. 103, 13872–13877. 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kularatne S. A.; Venkatesh C.; Santhapuram H. K.; Wang K.; Vaitilingam B.; Henne W. A.; Low P. S. (2010) Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. J. Med. Chem. 53, 7767–7777. 10.1021/jm100729b. [DOI] [PubMed] [Google Scholar]
- Grieco P.; Gitu P. M.; Hruby V. J. (2001) Preparation of ’side-chain-to-side-chain’ cyclic peptides by Allyl and Alloc strategy: potential for library synthesis. J. Pept. Res. 57, 250–256. 10.1111/j.1399-3011.2001.00816.x. [DOI] [PubMed] [Google Scholar]
- Roy J.; Nguyen T. X.; Kanduluru A. K.; Venkatesh C.; Lv W.; Reddy P. V.; Low P. S.; Cushman M. (2015) DUPA conjugation of a cytotoxic indenoisoquinoline topoisomerase I inhibitor for selective prostate cancer cell targeting. J. Med. Chem. 58, 3094–3103. 10.1021/jm5018384. [DOI] [PubMed] [Google Scholar]
- Carotenuto A.; Merlino F.; Cai M.; Brancaccio D.; Yousif A. M.; Novellino E.; Hruby V. J.; Grieco P. (2015) Discovery of Novel Potent and Selective Agonists at the Melanocortin-3 Receptor. J. Med. Chem. 58, 9773–9778. 10.1021/acs.jmedchem.5b01285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog Jonsson A.; Granqvist A.; Elvin J.; Johansson M. E.; Haraldsson B.; Nystrom J. (2014) Effects of melanocortin 1 receptor agonists in experimental nephropathies. PLoS One 9, e87816. 10.1371/journal.pone.0087816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditto V. J.; Simanek E. E. (2010) Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol. Pharmaceutics 7, 307–349. 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Chen M.; Li S.; Ye R. D. (2019) Targeted Delivery of a Ligand-Drug Conjugate via Formyl Peptide Receptor 1 through Cholesterol-Dependent Endocytosis. Mol. Pharmaceutics 16, 2636–2647. 10.1021/acs.molpharmaceut.9b00188. [DOI] [PubMed] [Google Scholar]
- Barbero N.; Barni E.; Barolo C.; Quagliotto P.; Viscardi G.; Napione L.; Pavan S.; Bussolino F. (2009) A study of the interaction between fluorescein sodium salt and bovine serum albumin by steady-state fluorescence. Dyes Pigm. 80, 307–313. 10.1016/j.dyepig.2008.08.006. [DOI] [Google Scholar]
- Vlashi E.; Kelderhouse L. E.; Sturgis J. E.; Low P. S. (2013) Effect of folate-targeted nanoparticle size on their rates of penetration into solid tumors. ACS Nano 7, 8573–8582. 10.1021/nn402644g. [DOI] [PubMed] [Google Scholar]
- Sánchez-Laorden B. L.; Jiménez-Cervantes C.; García-Borrón J. C. (2007) Regulation of human melanocortin 1 receptor signaling and trafficking by Thr-308 and Ser-316 and its alteration in variant alleles associated with red hair and skin cancer. J. Biol. Chem. 282, 3241–3251. 10.1074/jbc.M606865200. [DOI] [PubMed] [Google Scholar]
- Vlahov I. R.; Santhapuram H. K.; Kleindl P. J.; Howard S. J.; Stanford K. M.; Leamon C. P. (2006) Design and regioselective synthesis of a new generation of targeted chemotherapeutics. Part 1: EC145, a folic acid conjugate of desacetylvinblastine monohydrazide. Bioorg. Med. Chem. Lett. 16, 5093–5096. 10.1016/j.bmcl.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Liu M.; Jia M.; Pan H.; Li L.; Chang M.; Ren H.; Argoul F.; Zhang S.; Xu J. (2014) Instrument response standard in time-resolved fluorescence spectroscopy at visible wavelength: quenched fluorescein sodium. Appl. Spectrosc. 68, 577–583. 10.1366/13-07236. [DOI] [PubMed] [Google Scholar]
- Chari R. V.; Miller M. L.; Widdison W. C. (2014) Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed. 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Housman G.; Byler S.; Heerboth S.; Lapinska K.; Longacre M.; Snyder N.; Sarkar S. (2014) Drug resistance in cancer: an overview. Cancers 6, 1769–1792. 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; An S.; Ward R.; Yang Y.; Guo X. X.; Li W.; Xu T. R. (2016) G protein-coupled receptors as promising cancer targets. Cancer Lett. 376, 226–239. 10.1016/j.canlet.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Liu X.; Yu J.; Song S.; Yue X.; Li Q. (2017) Protease-activated receptor-1 (PAR-1): a promising molecular target for cancer. Oncotarget 8, 107334–107345. 10.18632/oncotarget.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert T. L.; Bennett T. A.; Maestas D. C.; Cimino D. F.; Prossnitz E. R. (2001) Internalization of the human N-formyl peptide and C5a chemoattractant receptors occurs via clathrin-independent mechanisms. Biochemistry 40, 3467–3475. 10.1021/bi001320y. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Mowlazadeh Haghighi S.; Zoi I.; Sawyer J. R.; Hruby V. J.; Cai M. (2017) Design of MC1R Selective γ-MSH Analogues with Canonical Amino Acids Leads to Potency and Pigmentation. J. Med. Chem. 60, 9320–9329. 10.1021/acs.jmedchem.7b01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowlazadeh Haghighi S.; Zhou Y.; Dai J.; Sawyer J. R.; Hruby V. J.; Cai M. (2018) Replacement of Arg with Nle and modified D-Phe in the core sequence of MSHs, Ac-His-D-Phe-Arg-Trp-NH. Eur. J. Med. Chem. 151, 815–823. 10.1016/j.ejmech.2018.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlino F.; Zhou Y.; Cai M.; Carotenuto A.; Yousif A. M.; Brancaccio D.; Di Maro S.; Zappavigna S.; Limatola A.; Novellino E.; Grieco P.; Hruby V. J. (2018) Development of Macrocyclic Peptidomimetics Containing Constrained α,α-Dialkylated Amino Acids with Potent and Selective Activity at Human Melanocortin Receptors. J. Med. Chem. 61, 4263–4269. 10.1021/acs.jmedchem.8b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.